

Abstract
Conformational selection is a primary mechanism in biomolecular recognition. The conformational ensemble may determine the ability of a drug to compete with a native ligand for a receptor target. Traditional docking procedures which use one or few protein structures are limited and may not be able to represent a complex competition among closely related protein receptors in agonist and antagonist ensembles. Here, we test a protocol aimed at selecting a drug candidate based on its ability to synergistically bind to distinct conformational states. We demonstrate, for the case of estrogen receptor α (ERα) and estrogen receptor β (ERβ), that the functional outcome of ligand binding can be inferred from its ability to simultaneously bind both ERα and ERβ in agonist and antagonist conformations as calculated docking scores. Combining a conformational selection method with an experimental reporter gene system in yeast, we propose that several phytoestrogens can be novel estrogen receptor β selective agonists. Our work proposes a computational protocol to select estrogen receptor subtype selective agonists. Compared with other models, present method gives the best prediction in ligands’ function.
Keywords: conformational ensemble, conformational selection, docking, phytoestrogen, SERMs, two-state theory
Proteins dynamics is a key factor in protein–ligand interactions. For the nuclear receptor, protein conformational changes coupled with ligand recognition and binding underlie signal transduction mechanisms (1). Currently, it is still a challenge to include dynamics in structure-based virtual screening. Molecular docking methods are routinely used to select drug candidates by predicting ligand binding affinities using some scoring functions (2, 3). Traditionally, one or several protein structures have been used to represent target structures. Docking scores fluctuate when different conformers are used to represent the same target receptor, which is known as the ‘cognate docking problem’ (4). This not only decreases prediction accuracy, but can also question the obtained biological insights. More accurate docking prediction may be achieved by using average scores from several structures (5) or allowing the receptor binding pocket to be ‘adaptive’ to ligand conformations through conformational selection (4).
Recently ensemble docking has become increasingly popular (6–10) with a quick search recording close to 200 such publications. These follow the pioneering work of Shoichet (11) over a decade ago, and the broad recognition that proteins exist in ensembles of conformational states. Among these, an encouraging number (close to 50) already considered conformational selection.
Still, the underlying biological significance of the sensitivity of the docking score to the target structure ensemble has been mostly overlooked. Receptors and ligands are dynamics molecules, and their interactions involve mutual conformational selection (12). Such scenarios can be well illustrated by ligand binding to the estrogen receptor (ER).
Estrogen receptor is an estrogen-inducible transcription factor that controls genes related to reproductive organs, the cardiovascular system, bone, and brain (13). Consequently, ER is an important therapeutic target. There are two subtypes of ER in the cytoplasm, ERα and ERβ, which share high sequence and structural similarity (14). Estrogen and related compounds are similarly responsive to ERα and ERβ. However, expression patterns (15, 16) and transcription activities (17) distinguish between the two subtypes. Structurally, ERα and ERβ have two distinct agonist and antagonist conformations, mainly differing around the helix 12 region (18) and the co-activator recruitment site (19). The conformation of the ligand-binding domain (LBD) may also change upon binding (20). A drug targeting ER to modulate following transcription has to compete with the native ligand estrogen. Side effects may arise if the drug has low selectivity toward the target (ERα or ERβ) in the right conformation (agonist or antagonist). Thus 17α-estradiol can have adverse side-effects [such as endometrial carcinoma and breast cancer (21, 22)]. Some ERβ-selective agonists (such as genistein) have lower adverse effects compared with 17β-estradiol but still appear to confer comparable benefits. Selective ERβ agonists, named selective estrogen receptor modulators (SERMs), can substitute for 17β-estradiol. Many phytoestrogens (23) were characterized as SERMs, such as genistein and daidzein (24); some can be used for inhibiting ER-dependent cancer-cell proliferation (25) and in bone and nerve system treatment (24). However, some phytoestrogens have side effects, interfering with development (26) and the immune system (27). Computational approaches to screen SERMs in silico have been reported, with different success rates (28–31). These works applied molecular dynamics, QSAR or the CoMFA algorithm to screen/design ligands specifically binding to either ERα or ERβ. However, they did not account for ER conformational variability of the functional states.
In this study, we pursued a novel computational screening approach for ERβ selective agonists. To find these, the ligand should not only bind favorably to ERβ in the agonist conformation, but also to the ERα antagonist conformer. We used such multi-targeting conformational selection scheme to screen 51 plant extracts. Based on the in silico prediction, candidate ligands were experimentally tested through a bipartite recombinant yeast reporter system and found to be ERβ selective agonists. Several phytoestrogen were successfully identified as ERβ agonist by computational and experimental studies. These studies not only suggest an explanation to ER selectivity of known compounds, but also an alternate strategy for drug design and screening.
Materials and Methods
Reagents
Salidroside, galanthamine, resveratrol, isoalantolactone, (+)-catechin, luteolin and curcumol were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Genistein and 17β-estradiol were purchased from Sigma-Aldrich (St. Louis, MO, USA). All estrogenic ligands stock solutions were prepared in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at 10−3 M before dilution by ddH2O. O-Nitrophenyl-β-d-galactopyranoside (ONPG) was from Amresco (Beijing branch). All other chemical supplies were of analytical grade. YEP-ERα, YEP-ERβ, YRPC2-2cERE plasmids, and BJ5409 yeast strain were kind donations from Dr. Dan Noonan (University of Kentucky).
Structure preparation
Schrodinger software package was used for receptor structure preparation and docking. Crystal structures of ERα and ERβ in agonist and antagonist conformations were downloaded from the Protein Data Bank (PDB IDs: 1a52, 1gwq, 1l2i, 2j7x, 1u3q, 1×7b, 1err, 2ouz, 3ert, 2fsz, 1l2j, and 1nde). Receptor data are given in Table S1. Missing atoms and residues were added and minimized in an OPLS-2005 force field using the program Maestro. Monomers of the LBD of the receptors were further minimized by deleting redundant water molecules and metal ions, except one retained water molecule which was structurally conserved in two receptors (18, 30). Receptor grids near the ligand-binding pocket were generated by the Glide® software (32). Co-crystallized ligands were used for receptor grids generation as the center. The smallest cubic containing the β-sheet region of the LBD is taken as the grid. No specific constraint is applied.
Ligands’ structures were constructed and minimized in MM94 force field with ChemBio3D Ultra 11.0 software. Poses were generated at variable pH, ion states and ring conformations with Epik®. The following Epik parameters were used: mode: predict states; H2O solvent; pH 7.0 ± 2.0. Possible generate tautomers were also generated with the Epik, and the maximum number of conformers generated for each compound was limited to 16.
Docking procedure
Prepared ligands were screened using the standard SP Glide docking protocol to ERα or ERβ in agonist and antagonist conformations. The best pose of each ligand which survived in the screening was further calculated using the XP standard docking calculation. The following SP/XP docking parameters were selected: SP/XP precision, force field SP2005, water environment; variable bond movement was allowed without restraint; and no specific core, similarity group or constraint is applied.
Bipartite recombinant yeast reporter system
The bipartite recombinant yeast reporter system was constructed as described in our previous work (33). To test selected ligand’s abilities to activate ERα and/or β, BJ5409 yeast strain was transformed with a reporter plasmid containing a downstream reporter gene lacZ and plasmids which express ERα and β. To test ligands’ abilities to occlude estrogen’s activation of ERα, yeast strain expressing ERα and reporter gene was used. Upon reaching an absorbance for 600nm wavelength of 0.6, measured using a Universal Microplate Reader (ELX 800uv, BIO-TEK Instruments, Inc.), the bipartite recombinant yeast was incubated in 96-well plate at 30 °C for 24 h in the presence of copper ion and ligands. All chemicals were tested at a concentration of 10−5 m. For inhibition tests, 17β-estradiol was applied at final concentration of 10−8 m together with test ligands at various final concentrations. Following incubation, 100 µL of lyticase solution was added to each well, incubated for 2 h at 30 °C and absorbance of 405 nm wavelength was measured. β-galactosidase activity was calculated to represent relative activity.
Statistical analysis
Bipartite recombinant yeast reporter system data was analyzed by Origin 8 software (Origin Lab, Northampton, MA, USA) and expressed as mean±SD. The relative activities of each ligand underwent a two-sample t-test to compare the relative activities with and without ligand for each type of receptor. One-way anova was performed between different concentrations to distinguish between these activities (statistically significant (p < 0.05); very significant (p < 0.01)).
Results and discussion
Single-structure docking only predicts binding affinity
Single-structure docking approaches were first applied to ligand interactions with ERα and ERβ. A similar method for screening ERβ selective ligands was applied earlier by Wolohan and Reichet (31) to predict the relative binding affinity ratio (RBAratio) of a ligand to ERβ and to ERα. In their study, ligand RBAratio value was predicted directly by dividing its docking scores to ERβ by those to ERα. Higher RBAratio value indicates better affinity toward ERβ than ERα.
We first select two native ligand (17β-estradiol) ER-bounded structures as receptor structures (PDB codes: ERα 1a52 and ERβ 2j7x). These allow us to investigate the ability of a compound to replace the native 17β-estradiol, following experiments that measure the ligand’s ability to replace 17β-estradiol binding to ERα and ERβ (18). We collect 12 ERβ selective compounds with known experimental RBAratio values as a validation set (34–41). Instead of using the absolute docking scores of a ligand to ERα and ERβ, we predict the relative binding affinity ratio by comparing docking score to that of the native ligand (17β-estradiol) which mimics the competition experiment used for measuring binding affinity. Our predictions of RBA are based on equation 1:
| (1) |
where DS(ERα) and DS(ERβ) are the docking scores of a compound to ERα and ERβ, respectively, and the constants 13.92 and 13.75 obtained from the docking score of 17β-estradiol to ERα (−13.92) and ERβ(−13.75). The result of subtraction of ligand docking score to that of 17β-estradiol is supposed to be inversely proportional to the ligands’ binding affinity. The structure of each ligand and corresponding RBAratio values obtained from eqn 1 are listed in Table 1.
Table 1.
Validation set compounds used in single-structure model.
| Number | Structure | ERβ score/ERα score | Computational RBAratio | Experimental RBAratio | Reference |
|---|---|---|---|---|---|
| 1 | 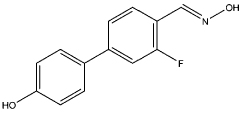 |
0.90 | 0.51 | 11 | compound 6e, (41) |
| 2 |  |
0.85 | 0.50 | 13 | compound 34 (38) |
| 3 | 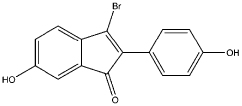 |
0.86 | 0.44 | 15 | compound 14 (38) |
| 4 |  |
0.87 | 0.49 | 29 | compound 90(37) |
| 5 | 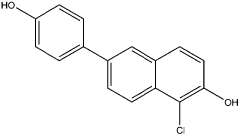 |
0.92 | 0.69 | 36 | compound 15 (39) |
| 6 |  |
0.90 | 0.67 | >43 | compound 6o (41) |
| 7 |  |
0.99 | 1.01 | 72 | compound 61 (36) |
| 8 |  |
0.97 | 0.95 | 72 | compound 16a (40) |
| 9 | 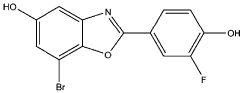 |
0.99 | 1.0 | 81 | compound 93 (37) |
| 10 |  |
1.02 | 1.12 | 102 | compound 12e (35) |
| 11 |  |
1.05 | 1.14 | 108 | compound 22 (34) |
| 12 |  |
1.12 | 2.81 | 226 | compound 117 (37) |
RBA, relative binding affinity.
Overall, our computed RBAratio were in line with the experimental RBAratio values. In Figure 1, we plot the linear regression of the experimental RBAratio values versus the computed RBAratio. The good correlation between predicted and experimental RBAratios (R2 = 0.95, Figure 1A) suggests that the docking procedure and parameters are reasonable candidates. However, in spite of the fit shown in Figure 1A, the current model cannot discriminate between a ligand agonist and antagonist which is what we are primarily interested in, because single-structure docking does not have additional scores to distinguish between these. Thus, the ligand’s efficacy cannot be predicted directly using the RBAratio. Consequently, we design a conformational ensemble model to predict the ligand’s efficacy.
Figure 1.

Docking scores to different conformers indicate ligands’ preferences towards different receptor conformation. (A) linear fitting of the computational RBAratio and experimental RBAratio values. The fitting coefficient was about 0.95, indicating a good fit to experimental RBAratio. (B) and (C) linear fitting of RBAratio (agonist), RBAratio (antagonist), and experimental RBAratios. The RBAratio (agonist) positively correlates with experimental RBAratio, while the RBAratio (antagonist) reversely correlates with experimental RBAratio. The edges of the scatter dots area are shown by dashed lines. (D) docking score ratios of some known SERMs. The docking scores reflect ligands efficacy and match with experiments in the literature (see text).
The conformational ensemble model can assist in ER agonist-antagonist discrimination
The conformational ensemble of ERs can be divided into four groups: ERα in agonist conformation, ERβ in agonist conformation, ERα in antagonist conformation, and ERβ in antagonist conformation. Ideally, a ligand can selectively bind to a specific group; in reality, a ligand will bind all four ER groups with different populations.
In order to analyze the ligand affinity to different receptor conformations, we calculated the ligand-binding affinity to all ER structures in the four groups. We selected twelve ER structures to represent the four conformers, three structures for each group (Table S1). To avoid bias when the ensemble structures were used to screen phytoestrogens, we excluded several known structures of ER-phytoestrogens complexes (1×7r, 1qkm, and 1×7j for ER-Genistein complex). For each group, the average binding affinities was averaged using partition function to reflect possible Boltzman distribution:
| (2) |
The theoretical foundation of conformational ensemble model lies in the correlation between efficacy and binding affinity, which has been elaborately described in the ‘two-state model’ (42). Based on that model, efficacy can be interpreted as ligand’s binding affinity to active conformer. In real world, equilibrium of ligand population binding to different receptor conformers gives rise to apparent RBA. Costa et al. (43) and Kenakin et al. (44) demonstrated that in ligand-binding studies, ligands’ potencies (ability to bind) depend not only on ligand efficacy (ability to activate) but also on that of the tracer. In the case of ER, competition experiment favors agonist conformation since 17β-estradiol is a pure agonist. Thus experimental RBAratio should be correlated to agonism ratio and inversely correlated to antagonism ratio.
To test whether docking score represents ligand affinity to different conformers, two sets of RBAratio were derived from these data:
| (3) |
| (4) |
These procedures were applied to the twelve compounds and 17β-estradiol (Table S1 and Table 2). When the RBAratio(agonist) and RBAratio(antagonist) were compared with the experimental RBAratio (Figures 1B,1C), we found ‘medium’-level correlations: the RBAratio(agonist) positively correlated with experimental RBAratio (Figure 1B), while the RBAratio(antagonist) inversely correlated with experimental RBAratio (Figure 1C). This suggests that RBAratio(agonist) and RBAratio(antagonist) may be informative with respect to ligand preferences.
Table 2.
Analysis of 12 compounds and 17β-estradiol by conformational ensemble model. a
| Compounds | Eave-ERα-agonist | Eave-ERβ-agonist | Eave-ERα-antagonist | Eave-ERβ-antagonist | RBAratio (agonist)b | RBAratio (antagonist)b |
|---|---|---|---|---|---|---|
| 1 | −12.5 | −12.3 | −12.4 | −11.7 | 0.99 | 0.95 |
| 2 | −12.8 | −12.8 | −12.5 | −12.8 | 1.00 | 1.02 |
| 3 | −12.6 | −11.9 | −12.0 | −11.9 | 0.94 | 0.99 |
| 4 | −12.4 | −12.0 | −12.7 | −12.2 | 0.97 | 0.96 |
| 5 | −12.9 | −12.7 | −13.7 | −13.1 | 0.99 | 0.96 |
| 6 | −11.5 | −11.7 | −11.7 | −11.3 | 1.01 | 0.97 |
| 7 | −12.7 | −12.4 | −11.2 | −11.5 | 0.98 | 1.02 |
| 8 | −11.6 | −11.3 | −11.9 | −11.3 | 0.97 | 0.95 |
| 9 | −13.3 | −13.7 | −13.2 | −11.6 | 1.03 | 0.88 |
| 10 | −10.9 | −11.0 | −10.5 | −10.6 | 1.00 | 1.01 |
| 11 | −13.4 | −14.8 | −14.2 | −12.2 | 1.10 | 0.86 |
| 12 | −13.8 | −14.2 | −13.6 | −11.9 | 1.03 | 0.88 |
| 17β-estradiol | −13.7 | −13.6 | −12.2 | −12.3 | 0.99 | 1.01 |
| ERB 041 | −12.1 | −14.1 | −13.6 | −12.3 | 1.17 | 0.91 |
| Way 20070 | −10.7 | −11.1 | −11.3 | −11.0 | 1.03 | 0.97 |
| PPT | −16.5 | −4.1 | −8.8 | −7.6 | 0.25 | 0.87 |
| ICI 164384 | −8.9 | −7.1 | −12.0 | −8.4 | 0.79 | 0.70 |
| 12a | −5.2 | −0.7 | −10.4 | −15.4 | 0.14 | 1.48 |
In order to investigate whether ligand preferences to different receptor conformations were actually linked to ligand’s efficacy, we further tested several well-characterized SERMs. Six compounds [ERB-041, Way 20070 (45) (selective ERβ agonists), propyl pyrazole triol (46) (PPT, selective ERα agonist), 17β-estradiol (agonist for both subtype), ICI164384 and 12a (47) (antagonists for both subtypes)] were docked into the twelve receptor structural ensemble (Table S1). Take PPT as an example, its Eave for ERα agonist is −16.54, and ERα antagonist is −8.79. Thus, the score ratio of agonist to antagonist for ERα is 1.88, which suggests that PPT prefers to select the ERα agonist conformation. In the case of ERβ, PPT gave a ratio of 0.54 for agonist versus antagonist. As shown in Figure 1D, PPT can be located in the fourth quadrant, which may suggest that the ligand is a preferred ERα agonist. The docking scores of other ligands were similarly analyzed and plotted in Figure 1D, with all ligands tested located in the correct quadrant. Therefore, the results demonstrate a good match between score ratios and the ligands’ efficacy profiles.
Taken together, the results in Figure 1B,C,D show that the binding affinities to different structures correlate with the ligands activity, which may suggest that conformational selection among the ligands might be used to predict the ligand efficacy.
Conformational ensemble model can be used in predicting selective ERβ agonists
In order to select the ERβ agonist, we further normalize the docking scores using following equation:
| (5) |
The Selection ERβratio may reflect the preference of a compound to ERβ agonist conformations: conformations of ERβ-agonist and ERα-antagonist should be positively selected, while the ERα-agonist and ERβ-antagonist conformers should be discriminated. Intuitively, the higher the Selection ERβratio is, the more possible for the ligand to be a selective ERβ agonist. However, to use this index practically, a good threshold is needed, as the ligand-receptor interaction should not be interpreted as rigid activation or inactivation, but as a probability of an event happens in ligand–receptor population. A quantitative and statistical threshold can be set by a Monte Carlo simulation as described elsewhere.
Eave in each conformer group is in range of the highest and lowest docking score in that group. Random-assigned Eave within the range gives result for the distribution of Selection ERβratio. As different function groups have different Eave ranges, four Selection ERβratio distributions can be obtained for statistical analysis. Ligands used in Figure 1D have good representatives from each function groups and is again used in this Monte Carlo simulation. A total of 10 000 interactions for each group is calculated and plotted in Figure 2. It clearly shows that Selection ERβratio distributes significantly different from each function group with the given Eave ranges. The only complication for predicting selective ERβ agonist is the overlapped part between ERβ selective agonist and both agonist group. To distinguish these two groups from each other, the upper limit for both agonist group is set at 1.11 (one side 95% confidence). With this threshold, ligands with Selection ERβratio higher than 1.11 is believed to have a better chance to be selective ERβ agonists, while lower ones might be in one of the three function groups with corresponding depending on the value.
Figure 2.

Threshold Selection ERβratio value by Monte Carlo simulation. A total of 10 000 random Eave values for each group were used in Selection ERβratio calculation as described in text. ERβ selective agonist group is quite distinguishable from ERα selective agonist and both antagonist group but has some overlap with both agonist group. Red dash line shows the one side 95% confidence value of both agonist group. And blue dash line shows the lower limit of ERβ selective agonist group (95% confidence).
In accordance with our model, the Selection ERβratio for 17β-estradiol is 0.98, which falls into the both agonist group. The value for ERB 041 is 1.29, showing a good ERβ selectivity. But it should be noted that the lower limit of selective ERβ group extends to 0.99 (95% confidence), where three function groups (except for selective ERα) all have an unneglectable probability. So when ligand’s Selection ERβratio lies in 0.99–1.11, its functional property cannot be determined by our analysis. Nevertheless, as described in the following section, conformational ensemble model gives accurate predictions about phytoestrogens’ interactions with ERs.
Selection ERβratio may predict phytoestrogens properties as selective ERβ agonists and low ERα activation
We initially screened 51 plant extracts (see Appendix S1) using a single structure model (one ERα crystal structure: 1a52; and one ERβ crystal structure: 2j7x). Twenty compounds with relatively large score ratios of ERβ to ERα were selected as ERβ selective binder candidates and were used to test the effect of different receptor conformations (Figures 3 and 4). Their initial RBA varied from about 0.45 to 1.60.
Figure 3.

Schematic flowchart of the conformation ensemble model used to screen phytoestrogens. Receptor conformations were extracted from RCSB-PDB. PDB ID of each conformation used is noted.
Figure 4.

Twenty phytoestrogens with relatively large docking score ratios assumed to be ERβ selective binders were further calculated.
The predicted ERβratios are reported in Table 3. Ligands are grouped into positive prediction (higher than 1.11), neutral prediction (between 0.99 to 1.11) and negative prediction (lower than 0.99). Note that Cordycepin was marked as negative although it has higher Selection ERβratio, because of its very low overall docking scores (Table S2). Among those negative predictions, some ligands were already investigated. Phenylephrine was reported to have a pure antagonist activity (48), and Tanshinone II A was demonstrated not to be selective between two ER subtypes (49). Bergenin is not an effective agonist for ERβ, as it had a low cytotoxicity on Murine Breast Cancer Cell Line, FM3A (50). Our lab further confirmed that Bergenin hardly exhibited protective effect on amyloid-beta-treated neuroblastoma cell line, while many ERβ selective agonists did (data not shown). Cordycepin did not affect MCF7 cell—line proliferation, indicating a lack of ER activation (51). Among the ligands within neutral prediction group, some were confirmed as ERβ agonist previously, such as Apigenin and Naringenin (52). Osthole could significantly prevent cancellous bone loss without the change of uterus weight compared with 17β-estradiol (53), suggesting ERβ activation. And in positive prediction group, Daidzein (52) and Glycitein (54) have already been tested as ERβ selective agonists.
Table 3.
Twenty phytoestrogens’ results from Conformational ensemble model compared with other ones.
| Computational RBAratioa |
RBAratio (agonist) |
RBAratio (antagonist) |
Energyratio (MD-SA) |
Selection ERβratio |
Experiment/ reference |
|
|---|---|---|---|---|---|---|
| Glycitein | 0.84 | 0.93 | 0.62 | 1.09 | 1.50 | + |
| Trans Resveratrol | 0.93 | 0.97 | 0.67 | 1.04 | 1.45 | + |
| Cis Resveratrol | 1.60 | 1.05 | 0.86 | 1.09 | 1.23 | ?b |
| Daidzein | 1.08 | 1.00 | 0.87 | 1.07 | 1.15 | + |
| Huperzine | 0.53 | 0.77 | 0.67 | 0.99 | 1.14 | ? |
| (+)-Catechin | 1.16 | 0.98 | 0.86 | 1.10 | 1.14 | + |
| Luteolin | 0.94 | 0.88 | 0.78 | Nullc | 1.12 | + |
| Apigenin | 1.05 | 1.08 | 0.99 | 1.07 | 1.09 | + |
| Galanthamine | 0.55 | 0.64 | 0.60 | 0.92 | 1.06 | + |
| Naringenin | 1.24 | 0.91 | 0.87 | 1.07 | 1.04 | + |
| Salidroside | 1.22 | 1.03 | 1.01 | 0.98 | 1.01 | − |
| Osthole | 0.84 | 0.90 | 0.91 | 1.01 | 0.99 | + |
| 17β-estradiol | 1.00 | 0.99 | 1.00 | 0.99 | 0.98 | Cut-offd |
| Phenylephrine | 1.07 | 0.95 | 0.97 | 1.06 | 0.97 | − |
| Isoalantolactone | 0.92 | 0.83 | 0.87 | 0.99 | 0.96 | − |
| Xanthotoxol | 0.67 | 0.82 | 0.87 | 1.56 | 0.94 | ? |
| Genistein | 0.45 | 0.94 | 1.01 | 1.07 | 0.93 | − |
| Tanshinone II A | 0.92 | 0.95 | 1.15 | 1.03 | 0.83 | − |
| Bergenin | 1.25 | 1.07 | 1.34 | 1.06 | 0.80 | − |
| Curcumol | 0.72 | 0.77 | 1.16 | 1.09 | 0.66 | − |
| Cordycepin | 1.07 | 0.97 | 0.97 | 1.01 | 1.00 | − |
RBA, relative binding affinity.
The ratio was calculated in single-structure model.
“+” indicates that the ligand was experimentally confirmed as potent ERβ selective agonist. '−' indicates that the ligand was not ERβ selective agonist shown by experiments. '?' indicated lack of experimental evidence or reference for the ligand’s selective efficacy.
The parameters needed for molecular dynamics calculation cannot be generated for Luteolin.
Except in conformational ensemble model, the value where 17β-estradiol lies labels the cut-off value. Higher than this value indicates a positive prediction, meaning the ligand is supposed to be ERβ agonist; lower value means a negative prediction. Gray boxes show wrong predictions as compared to experimental results showed in the most right column.
We selected three ligands from the seven positive predictions, two from five neutral predictions and three from eight negative predictions to investigate their transcription activity with a bipartite recombinant yeast reporter system. 17β-estradiol was also tested as positive control (Figure 5A). The relative activities were calculated using the measured absorption data according to our previous work (33). The relative activity of 17β-estradiol was in accordance with our previous work, confirming the reporter systems’ stability. Whereas ERα maximal activation by 17β-estradiol reached about 200 normalized units, ERβ reached about 120 normalized units. Three ligands, which were predicted as ERβ selective agonists, including trans-Resveratrol, (+)-Catechin and Luteolin were experimentally confirmed as ERβ-selective agonists. Galanthamine and Salidroside (from neutral prediction) also activated ERβ. The activation of ERβ reached statistical significance of p < 0.01. Isoalantolactone, Genistein, and Curcumol were predicted as impotent ERβ selective agonists. Isoalantolactone and Curcumol did not show any estrogenic activity as predicted. Genistein was experimentally confirmed as a known ERβ-selective agonist.
Figure 5.

Transcription activity of the tested ligands through different estrogen receptors by a biparticle recombinant yeast reporter gene system. (A) 17β-estradiol was used as control. Resveratrol, (+)-Catechin, Galanthamine, Luteolin and Salidroside selectively activated gene expression through estrogen receptor β as predicted. On the other hand, Curcumol and Isoalantolactone did not activate gene expression as predicted. Only genistein showed response different from that predicted. (B) Inactivation profile of these ligands. Yeast cells expressing ERα are treated with 17β-estradiol and one of the ligands at the same time. Only results at ligand final concentrations in 10−5M are shown, because all the curves are monotonic that highest concentration showed biggest effect. (+)-Catechin, Galanthamine, and Isoalantolactone significantly inhibit receptor activation. ** indicates the results are statistically significant (p < 0.01).
We further investigated ligands’ ERα antagonism. Water was used as control and its result is set as 100%. As shown in Figure 5B, among three positive prediction ligands, (+)-Catechin showed significant inhibition; Resveratrol and Luteolin did not change much of the estrogen’s effect. In neutral prediction group, Galanthamine showed potent inhibition but Salidroside had some agonism effect when treated together with 17β-estradiol. Two of the negative prediction ligands, Genistein and Curcumol, synergized 17β-estradiol effect, while Isoalantolactone significantly reduced activation. The results show that higher ERβ selectionratio would be better interpreted as low ERα activation but not necessarily as ERα antagonism.
Our models indicate that Genistein may not be selective ERβ agonist, in contrast to several reported assays and our own activation assay. However, it is known that Genistein can bind both ERα agonist and ERβ partial agonist, as indicated by three crystals structures of ER—Genistein complexes, i.e., ERα agonist 1×7r and two ERβ partial agonists, 1qkm and 1×7j. Several experiments clearly indicate that Genistein can be ERα-selective in different environments, in agreement with our inhibition assay. Barkhem et al. (55) found that genistein has an ERβ selective affinity and potency but an ERα-selective efficacy. It was also found recently that in cells with a predominance of ERα, genistein acts as an agonist to ERα, and in cells with ERβ alone, genistein most likely acts as an antiestrogen (56). Di et al. (57) also found that a low concentration of genistein induces ERα signaling.
Several compounds are not available (Huperzine and Xanthotoxol) or not the most stable isomers (cis-Resveratrol); thus, there is no information about their experimental activities. Combining activation and inhibition assay results, all ligands exhibit properties as predicted by conformational ensemble model. Salidroside and Genistein are marked negative because of their agonist actions when treated together with 17β-estradiol. From these results, we demonstrate that Selection ERβratio makes good prediction for specific ERβ agonists. Compared with the quadrant plot used in Figure 1D, the single parameter is easier to compare. Selection ERβratio would actually be more accurate in prediction for ERβ agonist, as the Monte Carlo simulation gave statistically significant threshold.
The conformational ensemble model may give more functional information and show higher success rate than other models in efficacy prediction
Conformation ensemble model is more powerful than traditional single-structure model in predicting ERβ selective agonists. Using data from literature and our present work, all ligands exhibit the predicted action in vivo. If we use ligand’s docking score to 2j7x versus to 1a52, as a simple representation of single-structure model, and set cut-off value using that of 17β-estradiol’s, the success rate will be 8 of 17 (Table 3, 47%). RBAratio(agonist) does not show good correlation with ERβ activation neither, with the same success rate. And RBAratio(antagonist) has the worst success rate as 4 of 17 (24%).
Table 3 also presents prediction results from a flexible docking model (58). For each do cked structure, we run 20 molecular dynamics simulated annealing (MD-SA) calculations, with same protocol described in the aforesaid references. The Charmm parameters for ligands were estimated using program. Antechamber was running at CHARMMGUI web-server (http://www.charmm-gui.org). After MD-SA, the ligand—protein interaction energies were calculated with distance-dependent dielectric constant (4r). We selected ERα agonist (pdb: 1a52) and ERβ agonist (pdb 2j7x) for the calculation (see Table S3). Energyratio(MD-SA) was the ratio of docking energy 2j7x to energy 1a52. The MD-SA method shows similar prediction as our conformation ensemble model in positive and neutral prediction groups (100% hit). However, it does not work well in negative prediction group, giving an overall success rate of 59%. The main reason is that the protein flexibility in the MD-SA method is still limited, not enough to cover large conformational change. In future, the combination of the MD-SA method with multiple protein structures should be examined.
These predictions are summarized in Figure 6, with two groups of experimental results. Selection ERβratio model gives a clear border between these two groups while other models, even with different structures average, would not be able to distinguish the two.
Figure 6.

Comparison of conformational ensemble model to other kinds of models. Each column of the figure describes a model. Sixteen ligands with experimental information (except for Cordycepin) are plotted. Black dots indicate the ligand is confirmed by reference or yeast test as selective ERβ agonist, while empty dots shows they are not. The bar in each column shows the value of 17β-estradiol calculated in that model, as the cut-off value. For conformational ensemble model, thresholds are marked at 1.11 and 0.99 as described in text. The figure shows that only the conformational ensemble model gives the closest prediction to experimental data.
Conclusions and future directions
Estrogen receptors are classical examples of coupling between protein conformational change and selective transcription activation. This study suggests that ligands can interact with similar targets in different conformations, and that the biological outcomes like ERβ selective agonist depend on the relative affinities of a ligand to ensembles of protein conformations. While docking screening for SERMs are becoming more popular, the present work illustrates the usefulness of an ensemble of crystallized ligand—receptor complexes. The variability in these crystal structures represent ensemble snapshots and provide information relating to protein dynamics.
Current study has some interesting findings in phytoestrogens. Resveratrol was shown to have higher affinity to ERα than to ERβ (59), but our yeast experiments indicate that it stimulates ERβ more than ERα (Figure 5A). A similar result was reported earlier (52). Furthermore, trans-Resveratrol was found to be an anti-breast cancer agent (50), which may suggest that the ligand could be a selective ERβ agonist. Our results may provide insights into the apparent contradiction between binding affinity and transcriptional activity: as shown in Table S2, the docking score indeed indicates that trans-Resveratrol has higher affinity towards ERα agonist and antagonist conformations. However, trans-Resveratrol has similar binding affinities toward both ERα agonist and antagonist conformations. In contrast, trans-Resveratrol has much higher affinity to ERβ-agonist conformations than to ERβ-antagonist; consequently, trans-Resveratrol is expected to select the ERβ agonist conformation over the ERβ antagonist. Thus, trans-Resveratrol activates ERβ more than ERα. It is interesting to note that cis-Resveratrol, which is thermodynamically less stable than the trans-isomer, was also predicted to have high ERβ agonist selectivity.
Another interesting finding was that Galanthamine and Huperzine were also predicted to be ERβ agonists. Both are well-known acetylcholinesterase inhibitors (AChEIs) (60, 61) and used in treatment of Alzheimer’s disease (62). They were believed to act through pathways irrelevant to ER. However, Galanthamine was confirmed to be an ERβ agonist in our yeast system; it could be an effective multi-target ligand and provide a scaffold for medicinal design.
Our approach is also in consistent with a previous finding that THC has different activation profiles on two ERs (63). Consistent with the conformational change of the THC-ERα and the THC-ERβ complexes, THC acts as an ERα agonist and an ERβ antagonist. In order to find ERβ selective agonist, the ligand should not only bind well to ERβ, but also have sufficiently favorable affinities towards agonist-induced ERβ and antagonist-induced ERα.
Even though our model generally agrees with experimental data, the conflicting computational and experimental results for genistein selectivity prompt us to revisit the “genistein discrepancy”. While the “genistein discrepancy” may correctly reflect the sensitivity of ERs in complex with genistein to different environments, it nevertheless emphasizes some limitations of the current conformational ensemble model: (i) twelve ensemble structures are too few to accurately reflect the high conformational flexibility of ERs (64); (ii) ER binding does not exactly match functional activity (65); (iii) allosteric effects may reduce the accuracy of the current model. ER—ligand interactions (as all binding events) are allosterically controlled (66); (iv) the model may not provide a sufficiently accurate prediction. This is because SERMs exhibit distinctive effects in different tissues, selectively activating/deactivating estrogen receptor, which may also relate to allosteric effects. Cell variability and auxiliary (co-factor) molecules need to be considered. The present model only considers functional similarity. Finally, (v) the scoring function needs improvement.
In the present study, we test ligands that were not only predicted to activate ERβ, but also expected to not activate/inactivate ERα. Synergetic multiple-targeting docking could be an alternate approach for drug screening and design. Even though the ERα and ERβ are closely related, they represent different targets because of their different biological functions. Multi-target drugs were suggested to be more efficient than a drug that hits only one target (67). Computational multi-target screening was proposed to be a novel paradigm for drug discovery (68). Indeed, experimental evidence has shown that simultaneous targeting of multiple opioid receptors could improve the side-effects profile (69). Our current study combined multiple-targeting and conformational ensembles. Whether this principle can be applied in other receptors beyond ER needs further investigation.
Supplementary Material
Acknowledgements
We are grateful to Dr. Dan Noonan for generously providing yeast strains and plasmids. This work was supported by the National Basic Science Foundation for Talent Education (J0630648) and National Natural Science Foundation of China (30670647 and 30970914). This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Original docking scores of training set compounds and 17β-estradiol. The co-complexed ligands in each structure were also listed.
Table S2. Original docking scores of twenty phytoestrogens to twelve structures.
Table S3. Docking energies calculated by MD-SA model.
Appendix S1. The 51 phytoestrogen used in initial screen.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Ma B, Nussinov R. Amplification of signaling via cellular allosteric relay and protein disorder. Proc Natl Acad Sci U S A. 2009;106:6887–6888. doi: 10.1073/pnas.0903024106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jain AN. Ligand-based structural hypotheses for virtual screening. J Med Chem. 2004;47:947–961. doi: 10.1021/jm030520f. [DOI] [PubMed] [Google Scholar]
- 3.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 4.Jain AN. Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation. J Comput Aided Mol Des. 2009;23:355–374. doi: 10.1007/s10822-009-9266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salisbury JP, Williams JC., Jr Docking study of triphenylphosphonium cations as estrogen receptor alpha modulators. Bioinformation. 2009;3:303–307. doi: 10.6026/97320630003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrusier N, Mashiach E, Nussinov R, Wolfson HJ. Principles of flexible protein-protein docking. Proteins. 2008;73:271–289. doi: 10.1002/prot.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulsen JL, Anderson AC. Scoring ensembles of docked protein:ligand interactions for virtual lead optimization. J Chem Inf Model. 2009;49:2813–2819. doi: 10.1021/ci9003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerek ZN, Ozkan SB. A flexible docking scheme to explore the binding selectivity of PDZ domains. Protein Sci. 2010;19:914–928. doi: 10.1002/pro.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park SJ, Kufareva I, Abagyan R. Improved docking, screening and selectivity prediction for small molecule nuclear receptor modulators using conformational ensembles. J Comput Aided Mol Des. 2010;24:459–471. doi: 10.1007/s10822-010-9362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zacharias M. Accounting for conformational changes during protein-protein docking. Curr Opin Struct Biol. 2010;20:180–186. doi: 10.1016/j.sbi.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gustafsson JA. Estrogen receptor beta–a new dimension in estrogen mechanism of action. J Endocrinol. 1999;163:379–383. doi: 10.1677/joe.0.1630379. [DOI] [PubMed] [Google Scholar]
- 14.Witkowska HE, Carlquist M, Engstrom O, Carlsson B, Bonn T, Gustafsson JA, et al. Characterization of bacterially expressed rat estrogen receptor beta ligand binding domain by mass spectrometry: structural comparison with estrogen receptor alpha. Steroids. 1997;62:621–631. doi: 10.1016/s0039-128x(97)00047-0. [DOI] [PubMed] [Google Scholar]
- 15.Kuiper GG, Shughrue PJ, Merchenthaler I, Gustafsson JA. The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol. 1998;19:253–286. doi: 10.1006/frne.1998.0170. [DOI] [PubMed] [Google Scholar]
- 16.Moore JT, McKee DD, Slentz-Kesler K, Moore LB, Jones SA, Horne EL, et al. Cloning and characterization of human estrogen receptor beta isoforms. Biochem Biophys Res Commun. 1998;247:75–78. doi: 10.1006/bbrc.1998.8738. [DOI] [PubMed] [Google Scholar]
- 17.Stossi F, Barnett DH, Frasor J, Komm B, Lyttle CR, Katzenellenbogen BS. Transcriptional profiling of estrogen-regulated gene expression via estrogen receptor (ER) alpha or ERbeta in human osteosarcoma cells: distinct and common target genes for these receptors. Endocrinology. 2004;145:3473–3486. doi: 10.1210/en.2003-1682. [DOI] [PubMed] [Google Scholar]
- 18.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, et al. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mak HY, Hoare S, Henttu PM, Parker MG. Molecular determinants of the estrogen receptor-coactivator interface. Mol Cell Biol. 1999;19:3895–3903. doi: 10.1128/mcb.19.5.3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/co-activator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 21.Haimov-Kochman R, Lavy Y, Hochner-Celinkier D. Review of risk factors for breast cancer–what's new? Harefuah. 2002;141:702–708. [PubMed] [Google Scholar]
- 22.Shah NR, Borenstein J, Dubois RW. Postmenopausal hormone therapy and breast cancer: a systematic review and meta-analysis. Menopause. 2005;12:668–678. doi: 10.1097/01.gme.0000184221.63459.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Usui T. Pharmaceutical prospects of phytoestrogens. Endocr J. 2006;53:7–20. doi: 10.1507/endocrj.53.7. [DOI] [PubMed] [Google Scholar]
- 24.Cos P, De Bruyne T, Apers S, Vanden Berghe D, Pieters L, Vlietinck AJ. Phytoestrogens: recent developments. Planta Med. 2003;69:589–599. doi: 10.1055/s-2003-41122. [DOI] [PubMed] [Google Scholar]
- 25.Mense SM, Hei TK, Ganju RK, Bhat HK. Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ Health Perspect. 2008;116:426–433. doi: 10.1289/ehp.10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet. 1997;350:23–27. doi: 10.1016/S0140-6736(96)09480-9. [DOI] [PubMed] [Google Scholar]
- 27.Yellayi S, Naaz A, Szewczykowski MA, Sato T, Woods JA, Chang J, et al. The phytoestrogen genistein induces thymic and immune changes: a human health concern? Proc Natl Acad Sci U S A. 2002;99:7616–7621. doi: 10.1073/pnas.102650199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salum LB, Polikarpov I, Andricopulo AD. Structure-based approach for the study of estrogen receptor binding affinity and subtype selectivity. J Chem Inf Model. 2008;48:2243–2253. doi: 10.1021/ci8002182. [DOI] [PubMed] [Google Scholar]
- 29.Sugiyama H, Kumamoto T, Suganami A, Nakanishi W, Sowa Y, Takiguchi M, et al. Insight into estrogenicity of phytoestrogens using in silico simulation. Biochem Biophys Res Commun. 2009;379:139–144. doi: 10.1016/j.bbrc.2008.12.046. [DOI] [PubMed] [Google Scholar]
- 30.van Lipzig MM, ter Laak AM, Jongejan A, Vermeulen NP, Wamelink M, Geerke D, et al. Prediction of ligand binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear interaction energy method. J Med Chem. 2004;47:1018–1030. doi: 10.1021/jm0309607. [DOI] [PubMed] [Google Scholar]
- 31.Wolohan P, Reichert DE. CoMFA and docking study of novel estrogen receptor subtype selective ligands. J Comput Aided Mol Des. 2003;17:313–328. doi: 10.1023/a:1026104924132. [DOI] [PubMed] [Google Scholar]
- 32.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 33.Liang K, Yang L, Xiao Z, Huang J. A bipartite recombinant yeast system for the identification of subtype-selective estrogen receptor ligands. Mol Biotechnol. 2009;41:53–62. doi: 10.1007/s12033-008-9097-9. [DOI] [PubMed] [Google Scholar]
- 34.Collini MD, Kaufman DH, Manas ES, Harris HA, Henderson RA, Xu ZB, et al. 7-Substituted 2-phenyl-benzofurans as ER beta selective ligands. Bioorg Med Chem Lett. 2004;14:4925–4929. doi: 10.1016/j.bmcl.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 35.De Angelis M, Stossi F, Carlson KA, Katzenellenbogen BS, Katzenellenbogen JA. Indazole estrogens: highly selective ligands for the estrogen receptor beta. J Med Chem. 2005;48:1132–1144. doi: 10.1021/jm049223g. [DOI] [PubMed] [Google Scholar]
- 36.Edsall RJ, Jr, Harris HA, Manas ES, Mewshaw RE. ERbeta ligands. Part 1: the discovery of ERbeta selective ligands which embrace the 4-hydroxy-biphenyl template. Bioorg Med Chem. 2003;11:3457–3474. doi: 10.1016/s0968-0896(03)00303-1. [DOI] [PubMed] [Google Scholar]
- 37.Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, Collini MD, et al. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-beta ligands. J Med Chem. 2004;47:5021–5040. doi: 10.1021/jm049719y. [DOI] [PubMed] [Google Scholar]
- 38.McDevitt RE, Malamas MS, Manas ES, Unwalla RJ, Xu ZB, Miller CP, et al. Estrogen receptor ligands: design and synthesis of new 2-arylindene-1-ones. Bioorg Med Chem Lett. 2005;15:3137–3142. doi: 10.1016/j.bmcl.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 39.Mewshaw RE, Edsall RJ, Jr, Yang C, Manas ES, Xu ZB, Henderson RA, et al. ERbeta ligands. 3. Exploiting two binding orientations of the 2-phenylnaphthalene scaffold to achieve ERbeta selectivity. J Med Chem. 2005;48:3953–3979. doi: 10.1021/jm058173s. [DOI] [PubMed] [Google Scholar]
- 40.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-beta potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–4251. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 41.Yang C, Edsall R, Jr, Harris HA, Zhang X, Manas ES, Mewshaw RE. ERbeta ligands. Part 2: synthesis and structure-activity relationships of a series of 4-hydroxy-biphenyl-carbaldehyde oxime derivatives. Bioorg Med Chem. 2004;12:2553–2570. doi: 10.1016/j.bmc.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 42.Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- 43.Costa T, Ogino Y, Munson PJ, Onaran HO, Rodbard D. Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol Pharmacol. 1992;41:549–560. [PubMed] [Google Scholar]
- 44.Kenakin TP, Morgan PH. Theoretical effects of single and multiple transducer receptor coupling proteins on estimates of the relative potency of agonists. Mol Pharmacol. 1989;35:214–222. [PubMed] [Google Scholar]
- 45.Harris HA. Preclinical characterization of selective estrogen receptor beta agonists: new insights into their therapeutic potential. Ernst Schering Found Symp Proc. 2006;1:149–161. doi: 10.1007/2789_2006_021. [DOI] [PubMed] [Google Scholar]
- 46.Harrington WR, Sheng SB, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003;206:13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 47.Zhou HB, Comninos JS, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Synthesis and evaluation of estrogen receptor ligands with bridged oxabicyclic cores containing a diarylethylene motif: estrogen antagonists of unusual structure. J Med Chem. 2005;48:7261–7274. doi: 10.1021/jm0506773. [DOI] [PubMed] [Google Scholar]
- 48.Arbo MD, Franco MT, Larentis ER, Garcia SC, Sebben VC, Leal MB, et al. Screening for in vivo (anti)estrogenic activity of ephedrine and p-synephrine and their natural sources Ephedra sinica Stapf. (Ephedraceae) and Citrus aurantium L. (Rutaceae) in rats. Arch Toxicol. 2009;83:95–99. doi: 10.1007/s00204-008-0324-8. [DOI] [PubMed] [Google Scholar]
- 49.Fan GW, Gao XM, Wang H, Zhu Y, Zhang J, Hu LM, et al. The anti-inflammatory activities of Tanshinone IIA, an active component of TCM, are mediated by estrogen receptor activation and inhibition of iNOS. J Steroid Biochem Mol Biol. 2009;113:275–280. doi: 10.1016/j.jsbmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 50.Schlachterman A, Valle F, Wall KM, Azios NG, Castillo L, Morell L, et al. Combined resveratrol, quercetin, and catechin treatment reduces breast tumor growth in a nude mouse model. Transl Oncol. 2008;1:19–27. doi: 10.1593/tlo.07100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomadaki H, Tsiapalis CM, Scorilas A. Polyadenylate polymerase modulations in human epithelioid cervix and breast cancer cell lines, treated with etoposide or cordycepin, follow cell cycle rather than apoptosis induction. Biol Chem. 2005;386:471–480. doi: 10.1515/BC.2005.056. [DOI] [PubMed] [Google Scholar]
- 52.Harris DM, Besselink E, Henning SM, Go VL, Heber D. Phytoestrogens induce differential estrogen receptor alpha- or Beta-mediated responses in transfected breast cancer cells. Exp Biol Med (Maywood) 2005;230:558–568. doi: 10.1177/153537020523000807. [DOI] [PubMed] [Google Scholar]
- 53.Li XX, Hara I, Matsumiya T. Effects of osthole on postmenopausal osteoporosis using ovariectomized rats; comparison to the effects of estradiol. Biol Pharm Bull. 2002;25:738–742. doi: 10.1248/bpb.25.738. [DOI] [PubMed] [Google Scholar]
- 54.Choi SY, Ha TY, Ahn JY, Kim SR, Kang KS, Hwang IK, et al. Estrogenic activities of isoflavones and flavones and their structure-activity relationships. Planta Med. 2008;74:25–32. doi: 10.1055/s-2007-993760. [DOI] [PubMed] [Google Scholar]
- 55.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–112. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 56.Rajah TT, Du N, Drews N, Cohn R. Genistein in the presence of 17beta-estradiol inhibits proliferation of ERbeta breast cancer cells. Pharmacology. 2009;84:68–73. doi: 10.1159/000226123. [DOI] [PubMed] [Google Scholar]
- 57.Di X, Yu L, Moore AB, Castro L, Zheng X, Hermon T, et al. A low concentration of genistein induces estrogen receptor-alpha and insulin-like growth factor-I receptor interactions and proliferation in uterine leiomyoma cells. Hum Reprod. 2008;23:1873–1883. doi: 10.1093/humrep/den087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang Z, Wong CF. Conformational selection of protein kinase A revealed by flexible-ligand flexible-protein docking. J Comput Chem. 2009;30:631–644. doi: 10.1002/jcc.21090. [DOI] [PubMed] [Google Scholar]
- 59.Mueller SO, Simon S, Chae K, Metzler M, Korach KS. Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor alpha (ERalpha) and ERbeta in human cells. Toxicol Sci. 2004;80:14–25. doi: 10.1093/toxsci/kfh147. [DOI] [PubMed] [Google Scholar]
- 60.Liang YQ, Tang XC. Comparative effects of huperzine A, donepezil and rivastigmine on cortical acetylcholine level and acetylcholinesterase activity in rats. Neurosci Lett. 2004;361:56–59. doi: 10.1016/j.neulet.2003.12.071. [DOI] [PubMed] [Google Scholar]
- 61.Sramek JJ, Frackiewicz EJ, Cutler NR. Review of the acetylcholinesterase inhibitor galanthamine. Expert Opin Investig Drugs. 2000;9:2393–2402. doi: 10.1517/13543784.9.10.2393. [DOI] [PubMed] [Google Scholar]
- 62.DeLaGarza VW. Pharmacologic treatment of Alzheimer's disease: an update. Am Fam Physician. 2003;68:1365–1372. [PubMed] [Google Scholar]
- 63.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, et al. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 64.Nettles KW, Bruning JB, Gil G, O'Neill EE, Nowak J, Guo Y, et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–568. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, Harris HA, et al. Structure-based design of estrogen receptor-beta selective ligands. J Am Chem Soc. 2004;126:15106–15119. doi: 10.1021/ja047633o. [DOI] [PubMed] [Google Scholar]
- 66.Nettles KW, Sun J, Radek JT, Sheng S, Rodriguez AL, Katzenellenbogen JA, et al. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: implications for other nuclear receptors. Mol Cell. 2004;13:317–327. doi: 10.1016/s1097-2765(04)00054-1. [DOI] [PubMed] [Google Scholar]
- 67.Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci. 2005;26:178–182. doi: 10.1016/j.tips.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 68.Jenwitheesuk E, Horst JA, Rivas KL, Van Voorhis WC, Samudrala R. Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol Sci. 2008;29:62–71. doi: 10.1016/j.tips.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, Lambert DG. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br J Anaesth. 2009;103:38–49. doi: 10.1093/bja/aep129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.