

Abstract
We recently identified a class of pyridyl aniline thiazoles (PAT) that displayed selective cytotoxicity for von Hippel-Lindau (VHL) deficient renal cell carcinoma (RCC) cells in vitro and in vivo. Structure-activity relationship (SAR) studies were used to develop a Comparative Molecular Field Analysis (CoMFA) model that related VHL-selective potency to the three dimensional arrangement of chemical features of the chemotype. We now report the further molecular alignment-guided exploration of the chemotype to discover potent and selective PAT analogues. The contribution of the central thiazole ring was explored using a series of 5- and 6-membered ring heterocyclic replacements to vary the electronic and steric interactions in the central unit. We also explored a positive steric CoMFA contour adjacent to the pyridyl ring using Pd-catalyzed cross-coupling Suzuki-Miyaura, Sonogashira and nucleophilic displacement reactions to prepare of a series of aryl-, alkynyl-, alkoxy- and alkylamino-substituted pyridines, respectively. In vitro potency and selectivity were determined using paired RCC cell lines: the VHL-null cell line RCC4 and the VHL-positive cell line RCC4-VHL. Active analogues selectively induced autophagy in RCC4 cells. We have used the new SAR data to further develop the CoMFA model, and compared this to a 2D-QSAR method. Our progress towards realizing the therapeutic potential of this chemotype as a targeted cytotoxic therapy for the treatment of RCC by exploiting the absence of the VHL tumor suppressor gene is reported.
Keywords: drug design, structure-activity relationships, renal cell carcinoma, autophagy, von Hippel Lindau, Suzuki-Miyaura, Sonogashira, QSAR
1. Introduction
Advanced clear cell renal cell carcinoma (RCC) is a disease typified by an aggressive phenotype and poor treatment outcomes.1,2 Mutation or inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene occurs in the majority of RCCs,3,4 and this is associated with tumour growth and invasiveness, resulting in a poor prognosis for patients. This gene encodes an E3 ubiquitin ligase that specifically regulates the protein stability of the Hypoxia-Inducible Factor (HIF) family of transcription factors.5 Loss of VHL results in constitutive expression of HIF leading to increased transcription of genes involved in metabolism, angiogenesis, invasion, metastasis, and proliferation leading to an aggressive phenotype.6–10 VHL is also known to be involved in a variety of HIF-independent processes.11–14
We recently used a synthetic lethality approach15,16 to identify the pyridyl anilino thiazole (PAT) chemotype 1 by high-throughput screening of commercial libraries (Figure 1A). The initial lead 2 was validated as a selective cytotoxin to VHL-deficient cells using XTT growth inhibition and clonogenic survival assays.17 As well as being selectively cytotoxic to VHL-deficient cells in vitro, compound 2 reduced VHL-deficient RCC growth in a xenograft tumor model in vivo. We demonstrated that the PATs cytotoxicity occurs in a HIF-independent manner through autophagy, however, the molecular target of the PAT class remains unknown. Induction of autophagy to selectively kill tumor cells represents a novel approach to anti-cancer therapy.18,19
Figure 1. PAT chemotype and biologically relevant alignment.
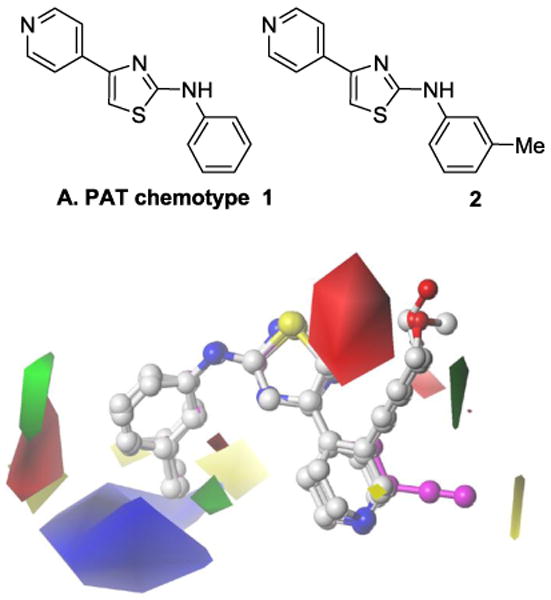
An initial exploration of structure-activity relationship (SAR) studies was carried out with the synthesis of a range of analogues bearing substitutions on each ring of the PAT chemotype as well as modifications of the linker units between the domains.20 A quantitative analysis of the SAR was used to determine a possible bioactive conformation for the PAT analogues using Comparative Molecular Field Analysis (CoMFA).
We now report further SAR studies in order to identify more potent and selective PAT analogues with increased potency and selectivity for VHL-deficient RCC. The in vitro activity of PATs as VHL-selective cytotoxins was evaluated using paired RCC cell lines; VHL-deficient RCC4 cell line and the matched line, RCC4-VHL, with the VHL function restored. Two features indentified in the original CoMFA contour maps are explored in this study (Figure 1B). The first feature is the orientation of the thiazole B-ring and the roles of the heteroatom substituents, while the second feature is the positive steric contour (green volume) adjacent to the pyridine ring 3-position which has a nearby disfavored electrostatic red region. We first investigated the influence of thiazole orientation predicted by the alignment model on the biological activity by replacing the thiazole central ring with various heterocycles (3–13). We then explored the steric region adjacent to the pyridine ring with a range of PAT derivatives bearing various functionalities at the 2- and 3-positions (compounds 14–54) of the pyridine ring. We also report on the impact of additional SAR data on the CoMFA model and compare its predictive ability to that of a 2D-QSAR method, Hologram QSAR (HQSAR).21
2. Results
2.1. Chemistry
The thiazole ring was replaced by a range of other 5-membered ring heterocycles. The imidazole analogue 3 was synthesized by condensation22 between bromoketone 5520 and 3-methylphenylguanidinium nitrate 5623 using KOH as base (Scheme 1). Similarly, condensation of bromoketone 55 and 3-methylphenylurea 57 afforded oxazole 4. Thiadiazole derivative 5 was synthesized from 4-pyridinecarbonitrile which was converted to the corresponding amidine 58 by treatment with NaOMe followed by NH4Cl addition. Subsequent DIAD-induced heterocyclization of amidine 58 with 3-methylphenylisothiocyanate gave thiadiazole 5. Methylation of thiourea 59,20 gave thiocarbamate 60 which underwent condensation with 4-pyridinecarbohydrazide to give triazole 6 as a mixture of regioisomers. Addition of MeNH2 to 3-methylphenylisothiocyanate gave methylated thiourea 61 which was converted to thiocarbamate 62 by alkylation with MeI, and subsequent condensation with 4-pyridinecarbohydrazide afforded triazole 7. 3-Methylphenylisothiocyanate was converted to the corresponding hydrazinecarbothioamide 63 by treatment with hydrazine hydrate in EtOH and subsequent condensation with isonicotinic acid in the presence of EDCI gave oxadiazole 8.
Scheme 1.

Reagents and conditions: (a) KOH, NEt3, EtOH, 80 °C; (b) NaOMe, MeOH, then NH4Cl; (c) 3-methylphenyl isothiocyanate, iPr2NEt, DMF, then DIAD; (d) MeI, EtOH; (e) 4-pyridinecarbohydrazide, pyridine; (f) MeNH2, EtOH; (g) NH2-NH2·H2O, EtOH; (h) isonicotinic acid, EDCI, DCM. $ = commercially available.
A series of 6-membered ring heterocyclic or carbocyclic analogues were then prepared to further investigate the geometry of the central B ring (Scheme 2). Pyrimidinamine 9 was prepared by condensation of the enaminoketone 64, obtained by reaction of 4-acetylpyridine with DMF-DMA, and subsequent reaction with guanidinium nitrate 56 in the presence of KOH.24 The 2,6-pyrimidine derivative 10 was synthesized from 2,6-dichloropyrimidine by Suzuki cross-coupling reaction with 4-pyridineboronic acid to afford chloropyrimidine 65, which was reacted with m-toluidine under Buchwald conditions to give pyrimidine 10. The pyrazine 11 and pyridine analogues 12 were prepared using similar synthetic routes. Suzuki reaction of 3-bromoaniline and 4-pyridineboronic acid gave intermediate 68, which underwent Buchwald reaction with m-bromotoluene to give the carbocyclic analogue 13.
Scheme 2.

Reagents and conditions: (a) DMF-DMA, reflux; (b) 56, NaOH, iPrOH, reflux; (c) Pd(PPh3)4, 4-pyridineboronic acid, K2CO3, 1,4-dioxane, 80 °C; (d) m-toluidine, p-TsOH, 1,4-dioxane, reflux; (e) m-toluidine, Pd(OAc)2, BINAP, NaOtBu, toluene, DMF, 90 °C; (f) m-bromotoluene, Pd(OAc)2, BINAP, NaOtBu, toluene, DMF, 90 °C. $ = commercially available.
We had previously demonstrated that the introduction of small, lipophilic moieties at the 2- and 3-position was tolerated, while 3-aryl (e.g., 3-Ph-4-Ac 27) were selective and 3-acetylenic groups (e.g., 3-C≡CH2OMe 29) gave increased potency without lowering selectivity leading to the positive CoMFA derived contour.20
To further explore this positive contour we initially targeted a series of fused heteroaryl rings (14-17, Scheme 3). Quinoline analogue 14 was prepared from quinoline-4-carboxylic acid which was converted to the corresponding Weinreb amide 69 by reaction with oxalyl chloride followed by treatment with N, O-dimethyl hydroxylamine. Subsequent Grignard reaction with MeMgBr to the ketone 70, then bromination with Br2/HBr gave the bromoketone 71 which underwent condensation with thiourea 58 to afford 14. Quinoline 15 was synthesized from 2-phenylquinoline-4-carboxylic acid by conversion to the acid chloride then treatment with TMSCH2N2 and the intermediate diazoketone was reacted with HBr to give the bromoketone 72, which was condensed with 58 to give 15. Imidazopyridine 16 was synthesized by bromination of methylimidazopyridinyl ethanone with Br2/HBr to give the bromoketone intermediate 72 and subsequent condensation with thiourea 58. Ketone 74, prepared by 1,3-dipolar cycloaddition reaction of 3-butyn-2-one and 1-aminopyridinium iodide,25 was treated with Br2/HBr to give bromoketone 75 which was condensed with 3-methylphenylthiourea to provide pyrazolopyridine 17.
Scheme 3.

Reagents and conditions: (a) (COCl)2, DMF, DCM; (b) Me(OMe)NH·HCl, NEt3, DCM; (c) MeMgBr, THF; (d) Br2, HOAc/HBr; (e) arylthiourea 58, EtOH; (f) TMSCH2N2, THF; then HBr. $ = commercially available.
Two sets of 3- and 2-substituted pyridyl analogues (18–38 and 39–54, respectively) were then synthesized to further explore the positive contour adjacent to the pyridine ring (Scheme 4). These were obtained by functionalization of the pyridine ring, starting with halopyridines (76–78)20 that were further elaborated using Pd-catalyzed Suzuki and Sonogashira cross-coupling reactions (in the case of bromo and iodo compounds) as well as nucleophilic substitution. In some instances, 3-iodopyridine 79, conveniently prepared from 3-bromopyridine 76 by Cu-catalyzed halogen exchange in the presence of NaI, was used due to the low reactivity of the corresponding 3-bromopyridine 76. Halopyridines (76, 77, 79) were reacted with a series of aryl boronic acids or boronate esters in the presence of PdCl2(dppf) to afford the Suzuki products 18–27 and 39–49. Sonogashira reactions of 3-bromopyridine 76 with propargyl alcohol, methyl propargyl ether, 1-butynol and 1-pentynol, using PdCl2(PPh3)2 and CuI as catalysts gave compounds 28, 29, 30 and 31, respectively. The terminal hydroxyl group was also replaced with polar amine groups to increase aqueous solubility. Boc protection of 76 using Boc2O in DCM gave bromide 80 which underwent Sonogashira cross-coupling with 1-butynol to produce intermediate alcohol 81 which was further functionalized by treatment with MsCl, displacement with morpholine or N-methylpiperazine, and removal of the Boc group with TFA to give analogues 32 and 33.
Scheme 4.

Reagents and conditions: (a) CuI, NaI, H2N(CH2)2NH2, 1,4-dioxane, 110 °C; (b) PdCl2(dppf), Ar-boronic acid or ester, K2CO3, Tol/EtOH/H2O/DMF, 90 °C; (c) PdCl2(PPh3)2, CuI, HC≡C-R, NEt3/DMF, 50–70 °C; (d) Boc2O, DMAP, DCM; (e) i) MsCl, NEt3, DCM, −20 °C, ii) morpholine or N-methylpiperazine, K2CO3, DMF, iii) TFA, DCM; (f) CuSO4, Na ascorbate, BnN3, DCM/EtOH/H2O, 20 °C; (g) ROH or RNH2, neat, Cs2CO3, 160–180 °C.
Triazole analogues 34 and 50 were synthesized using “Click” chemistry. Sonogashira cross-coupling reactions between bromides 76 and 77 and TMS acetylene in the presence of PdCl2(PPh3)2 and CuI, followed by removal of the TMS group using K2CO3 gave acetylenes 82 and 83.20 Reaction of 82 and 83 with benzyl azide in the presence of copper sulfate and sodium ascorbate gave benzyltriazole analogues 34 and 50, respectively.
In an effort to explore whether a flexible linker could access the steric contour as effectively as the equivalent soluble acetylenes 32 and 33, a range of alkylamines linked at the 2- and 3-positions via nitrogen and oxygen linker atoms were prepared. Nucleophilic substitution of 3-fluoropyridine 78 and 2-bromopyridine 77 with N-methylpiperazine, 2-morpholinoethanamine, 2-morpholinoethanol, and 3-(dimethylamino)-1-propanol gave the derivatives 35–38 and 51–54, respectively.
2.2. Biological assays
Cellular growth inhibition was determined by IC50 assays, using a 4-day drug exposure of RCC4 and RCC4/VHL cells in 96-well plates as described previously.17 The selectivity ratio was calculated as the intraexperiment ratio IC50(RCC4/VHL)/IC50(RCC4) (Tables 1–4). Several active analogues were also examined for their ability to induce formation of intracytoplasmic vacuoles to indicate whether they were killing cells by the induction of autophagy. Cells were incubated with drug (5 μM) for 24 h before phase contrast images were taken (Figure 3).
Table 1.
IC50 values and selectivity ratios for B-ring analogues.
 | |||||||
|---|---|---|---|---|---|---|---|
| No | X | Y | Z | W | RCC4 IC50 μM | RCC4/VHL IC50 μM | Ratioa |
| 1 | N | CH | S | 7 | >70 | >10 | |
| 2 | N | CH | S | 2.1 | 40 | 19 | |
| 3 | N | CH | NH | >40 | >40 | ND | |
| 4 | N | CH | O | >40 | >40 | ND | |
| 5 | N | N | S | 24.8 | >40 | >1.6 | |
| 6 | N | N | NH | 28.7 | 22.2 | 0.8 | |
| 7 | NMe | N | N | >40 | >40 | ND | |
| 8 | O | N | N | >40 | >40 | ND | |
| 9 | N | CH | N | CH | 6.5 | 8.6 | 1.3 |
| 10 | CH | CH | N | N | 23.1 | 28.1 | 1.2 |
| 11 | N | N | CH | CH | 8.5 | 31.8 | 3.7 |
| 12 | N | CH | CH | CH | 7.4 | >40 | >5.4 |
| 13 | CH | CH | CH | CH | 36.9 | 38.1 | 1.0 |
Ratio = IC50 (RCC4/VHL)/IC50 (RCC4)
Table 4.
PAT CoMFA Model.
| Model | q2a | SEPb | Comp.c | r2d | SEE e | Steric | Electrostatic |
|---|---|---|---|---|---|---|---|
| 15 | 0.352 | 0.431 | 3 | 0.818 | 0.229 | 0.618 | 0.382 |
| 2 | 0.270 | 0.449 | 4 | 0.852 | 0.202 | 0.549 | 0.451 |
| 3 | 0.345 | 0.428 | 3 | 0.809 | 0.231 | 0.607 | 0.393 |
| 4 | 0.246 | 0.475 | 4 | 0.831 | 0.225 | 0.597 | 0.403 |
| 5 | 0.423 | 0.429 | 5 | 0.878 | 0.197 | 0.552 | 0.448 |
| 6 | 0.313 | 0.463 | 6 | 0.893 | 0.182 | 0.510 | 0.490 |
Footnotes: Models 1–6 were constructed as described in the text and the compounds in each set are listed in the Supplementary data.
q2 is the cross validated r2 derived from Leave-One-Out SAMPLS (LOO SAMPLS) cross validation analysis as implemented in SYBYL8.2.
SEP is the Standard Error of Prediction reported for the LOO SAMPLS analysis.
Comp. is the number of components used in the model;
r2, non-cross-validated correlation coefficient.
SEE is the Standard Error of Estimate derived from the Non-Validated analysis.
Figure 3.
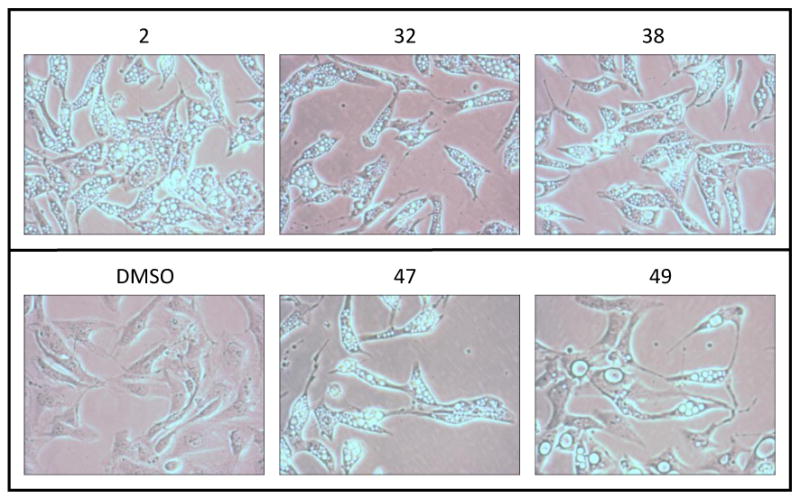
Induction of Autophagy by PATs in RCC4 Cells a
a RCC4 cells, 20×, after 5 μM drug exposure for 24 h.
2.3. Molecular modeling
We previously reported a CoMFA derived 3D-QSAR model that accounted for 34% (q2 = 0.344) of the variation in the selective cytotoxicity of the PAT series against a VHL deficient renal cell line, and provided a putative bioactive conformation for this compound series.20 This model (Figure 1B) was recreated by molecular alignment of the compound training set previously described against 28 (Table 4, Model 1) and differs marginally from that previously published. Here we examine the effect of new PAT analogues on this model (Table 4).
Incorporating Table 1 compounds of defined IC50 into Model 1 generated Model 2, and had a negative effect on the base model, reducing the q2 from 0.352 to 0.27. Two conformers for each of 14–16 (Table 2) were assessed against Model 1, these conformers explored different orientations of the pyridyl A-ring substitutions, and the orientations with the best q2 values were used to build Model 3, giving results similar to Model 1. Including 2-pyridyl substituents in model 1 had a negative effect on q2, decreasing it from 0.352 (Model 1) to 0.246 (Model 4), 3-pyridyl substituents improved q2 (0.423 vs 0.352). A set of compounds was then selected across all substitution patterns that, along with those used in Model 1, represent the complete PAT training set, and give Model 6 (See Supplementary data for composition of the training set). The q2 of this model was comparable, albeit slightly reduced, to Model 1 at 0.313, and is only able to account for 31% of the variance in biological activity.
Table 2.
IC50 values and selectivity ratios for fused A-ring analogues.
 | ||||
|---|---|---|---|---|
| No | R | RCC4 IC50 μM | RCC4/VHL IC50 μM | Ratioa |
| 14 |
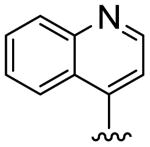
|
12.3 | 20.0 | 1.6 |
| 15 |
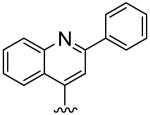
|
25.4 | >40 | >1.6 |
| 16 |
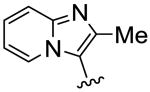
|
9.5 | 29.8 | 3.1 |
| 17 |
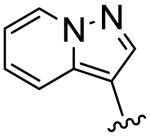
|
>40 | 15.3 | <0.4 |
Ratio = IC50 (RCC4/VHL)/IC50 (RCC4)
As one of the most important characteristics of a QSAR model is its ability to predict a target property for molecules not used in its development, the ability of Model 6 to predict IC50 values was tested against an external test set of compounds generated from Tables 1–3 along with additional compounds (from Reference 20) not used in the construction of Model 1 (Supplementary data, Table S1). Many of these compounds either displayed low activity, had IC50 values >40 μM, side chain substitutions with more than 1 rotatable bond, or unknown activity. Model 6 showed a linear relationship between experimental and predicted IC50 values for the training set with a non-validated r2 of 0.89, Standard Error of Estimation of 0.18 (Table 4 and Supplementary data, Figure S1), and a maximum difference between actual and predicted values (residual) of 0.44 (Supplementary data, Table S2A). While the log activity range of the Model 6 training set ranged from 4.3 to 6.7, most compounds (82%) were in the smaller range of 4.5 to 6 and this may contribute to the low q2 value. Comparison of actual and predicted activities for the test set does not show any correlation (r2 = 0.07) (Supplementary data, Figure S1), and many compounds with IC50 values >40 μM are incorrectly predicted, although an r2 of 0.10 when these compounds are excluded also indicates an absence of correlation for the remainder of the test set with defined IC50 values. The residual values ranged from 0.01 to 1.77 with approximately 55% of the test set predicted within 0.45 log units, the maximum residual value observed for the training set, and 60% predicted within 0.5 log units (Supplementary data, Table S2B). This low predictivity is consistent with the low q2 value. The CoMFA generated contour maps are illustrated in Figure 2.
Table 3.
IC50 values and selectivity ratios for substituted A-ring analogues.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No | 3-R | RCC4 IC50 μM | RCC4/VHL IC50 μM | Ratioa | No | 2-R | RCC4 IC50 μM | RCC4/VHL IC50 μM | Ratioa |
| 18 | Ph | 11.6 | 33.2 | 2.9 | 39 | 2-Ph | 19.1 | 40.0 | 2.1 |
| 19 | 4-pyridyl | 13.8 | 34.0 | 2.5 | 40 | Ph-4-Ac | 38.8 | 33.4 | 0.9 |
| 20 | Ph-4-CF3 | 12.8 | 22.6 | 1.8 | 41 | 4-pyridyl | 28.9 | 38.0 | 1.3 |
| 21 | Ph-4-iPr | >40 | >40 | ND | 42 | 2-pyridyl-6-F | >40 | >40 | ND |
| 22 | Ph-4-CONH2 | 4.1 | 1.6 | 1.6 | 43 | 5-pyrimidine | 26.4 | 37.0 | 1.4 |
| 23 | Ph-4-CONpiperazine-N-methyl | 12.8 | >40 | >3.1 | 44 | 4-(1H-indole) | 3.1 | 4.5 | 1.5 |
| 24 | Ph-4-CONH(CH2)3NMe2 | 4.0 | 3.9 | 1.0 | 45 | 3-thiophene | 8.7 | >40 | >4.6 |
| 25 | Ph-4-SO2CH3 | 1.6 | 13.4 | 8.4 | 46 | 4-(3,5-dimethyloxazole) | 3 | 36.5 | 12 |
| 26 | Ph-4-SO2NHtBu | 4.5 | 22.5 | 5.0 | 47 | 3-pyrazole | 0.48 | 20.0 | 14 |
| 27b | Ph-4-Ac | 3.9 | 35.1 | 9 | 48 | 4-(1-methylpyrazole) | 5.2 | 8.1 | 1.6 |
| 28b | C≡CCH2OH | 0.24 | 2.9 | 11.9 | 49 | 4-(1-benzylpyrazole) | 2.2 | 66.1 | 30 |
| 29b | C≡CCH2OMe | 0.62 | 27.9 | 45 | 50 | 4-(1-benzyltriazole) | 2.1 | 14.0 | 6.7 |
| 30 | C≡C(CH2)2OH | 1.1 | 2.8 | 2.5 | 51 | 4-methylpiperazine | 11.5 | 31.6 | 2.8 |
| 31 | C≡C(CH2)3OH | 6.3 | 0.3 | 0.1 | 52 | NH(CH2)2Nmorpholine | 3.7 | 8.0 | 2.2 |
| 32 | C≡CCH2Nmorpholine | 0.57 | 2.3 | 4.0 | 53 | O(CH2)2Nmorpholine | 7.0 | 21.1 | 3.0 |
| 33 | C≡CCH2Npiperazine-4-Me | 2.6 | 2.2 | 0.9 | 54 | O(CH2)3NMe2 | 6.0 | 8.8 | 1.5 |
| 34 | 4-(1-benzyltriazole) | >40 | >40 | ND | |||||
| 35 | 4-methylpiperazine | 2.6 | 6.6 | 2.5 | |||||
| 36 | NH(CH2)2Nmorpholine | 19.5 | 27.9 | 1.4 | |||||
| 37 | O(CH2)2Nmorpholine | 0.32 | 0.88 | 2.8 | |||||
| 38 | O(CH2)3NMe2 | 2.7 | 20.2 | 7.4 | |||||
Ratio = IC50 (RCC4/VHL)/IC50 (RCC4).
Data from reference 20.
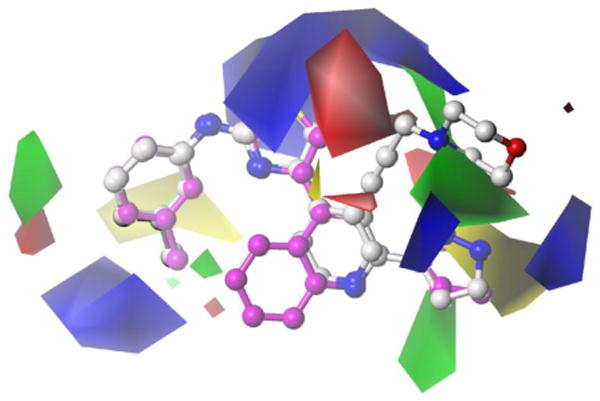
Figure 2. New biologically relevant alignment.
To determine if the CoMFA model was limited by the alignment strategy, a 2D-QSAR analysis was also performed using HQSAR.21 As HQSAR models are influenced by fragment size and distinction, a series of models were generated using a range of parameters (Table 5). Model 12 was the best fragment distinction combination, with a q2 of 0.361, and this was slightly improved to 0.375 when the fragment length was increased (Table 6, model 26 versus model 30). Interestingly, HQSAR Model 30 showed a reduced correlation between actual and predicted values compared to the CoMFA Model 6, while the Standard Error was slightly higher. The maximum difference between experimental and predicted activities for the training set was 0.58 (Supplementary data, Table S3A). External validation of Model 30 did not show any correlation between predicted and actual activities (r2=0.0009), and many compounds with poor activity (IC50 >40 μM) were incorrectly predicted (Supplementary data, Figure S2), and the r2 was not improved when these compounds were excluded. The residual values ranged from 0.06 to 1.44 with approximately 60% of the test set predicted within 0.58 log units, the maximum residual value observed for the training set (Supplementary data, Table S3B) and the range in difference outside this cutoff was 0.60 to 1.44.
Table 5.
Analysis of the effect of different fragment distinction sets on HQSAR models.
| Model | Fragment distinction a | q2b | SE c | r2d | SE c | Components | HL e |
|---|---|---|---|---|---|---|---|
| 7 | None | 0.193 | 0.502 | 0.408 | 0.430 | 6 | 353 |
| 8 | A | 0.046 | 0.529 | 0.256 | 0.467 | 6 | 353 |
| 9 | A B | 0.333 | 0.442 | 0.574 | 0.353 | 2 | 353 |
| 10 | A B C | 0.237 | 0.476 | 0.572 | 0.357 | 3 | 97 |
| 11 | A B C H | 0.250 | 0.484 | 0.705 | 0.303 | 6 | 151 |
| 12 | A B C D | 0.361 | 0.443 | 0.793 | 0.252 | 5 | 199 |
| 13 | A B C H D | 0.250 | 0.484 | 0.773 | 0.266 | 6 | 199 |
| 14 | A B H | 0.268 | 0.467 | 0.522 | 0.377 | 3 | 353 |
| 15 | A B H D | 0.212 | 0.496 | 0.794 | 0.254 | 6 | 151 |
| 16 | A B D | 0.082 | 0.535 | 0.788 | 0.257 | 6 | 257 |
| 17 | A C | 0.250 | 0.472 | 0.588 | 0.350 | 3 | 353 |
| 18 | A C H | 0.281 | 0.473 | 0.734 | 0.288 | 6 | 257 |
| 19 | A C H D | 0.201 | 0.491 | 0.659 | .0321 | 4 | 199 |
| 20 | A C D | 0.322 | 0.456 | 0.734 | 0.285 | 5 | 97 |
| 21 | A D | 0.028 | 0.551 | 0.683 | 0.314 | 6 | 257 |
| 22 | A H D | 0.199 | 0.488 | 0.472 | 0.396 | 3 | 199 |
Footnotes.
Fragment distinction; molecular features to be used in distinguishing among fragments, include A, atom, B, bond, C, connections, H, hydrogens, D, donor acceptor.
q2, cross validated r2.
SE, standard error.
r2, non-cross-validated correlation coefficient. All models were generated using a fragment atom count range of 4 to 7 and selected based on best cross-validated r2.
HL, Hologram Length.
Table 6.
Optimizing fragment size for the HQSAR analysis using fragment distinction (A B C D).
| Model | Fragment Size a | q2b | SE c | r2d | SE c | Components | HL e |
|---|---|---|---|---|---|---|---|
| 23 | 1-4 | 0.143 | 0.505 | 0.564 | 0.360 | 3 | 353 |
| 24 | 2-5 | 0.162 | 0.511 | 0.768 | 0.269 | 6 | 199 |
| 25 | 3-6 | 0.226 | 0.484 | 0.661 | 0.329 | 4 | 353 |
| 26 | 4-7 | 0.361 | 0.443 | 0.793 | 0.252 | 5 | 199 |
| 27 | 5-8 | 0.276 | 0.468 | 0.732 | 0.285 | 4 | 353 |
| 28 | 6-9 | 0.334 | 0.448 | 0.721 | 0.290 | 4 | 257 |
| 29 | 7-10 | 0.312 | 0.463 | 0.837 | 0.225 | 6 | 257 |
| 30 | 8-11 | 0.375 | 0.438 | 0.782 | 0.259 | 5 | 307 |
| 31 | 9-12 | 0.331 | 0.450 | 0.718 | 0.292 | 4 | 353 |
| 32 | 10-13 | 0.343 | 0.449 | 0.808 | 0.243 | 5 | 257 |
Footnotes.
Fragment size in atoms.
q2, cross-validated r2.
SE, standard error.
r2, non-cross-validated correlation coefficient. All models were generated using a fragment atom count range of 4 to 7 and selected based on best cross-validated r2.
HL, Hologram Length.
3. Discussion
A putative bioactive 3-dimensional conformation of the PAT series identified by CoMFA was used to guide synthesis of a set of PAT analogues which were designed to explore the central B-ring and its role in the alignment of the molecule and to probe the nature of the steric pocket adjacent to the pyridine A-ring. Previous exploration of the basic SAR around the aniline C-ring had shown that small, lipophilic substituents were optimal for activity hence we chose to use the 3-methylaniline ring.20
The ligand model gave a consistent curved orientation with the thiazole N placed in the inner surface and the thiazole S on the exposed rim (Figure 2). Replacement of the thiazole ring with any other 5-membered-ring heterocycle led to either a large drop in potency and selectivity or complete loss of activity (Table 1). Replacement of the thiazole S with another heteroatom (NH and O) led to inactive analogues (3 and 4). Incorporation of an additional N within the central ring gave much less potent derivatives (e.g., thiadiazole 5) while retaining a hint of selectivity, although lack of aqueous solubility prevent a full determination in the RCC4/VHL cell line (Table 1). Similarly, triazole analogues displayed low potency and selectivity. It was not possible to fully evaluate the influence of the thiazole N on the biological activity as our efforts towards the synthesis of a thiophene analogue were unsuccessful with the instability of the desired thiophene product precluding purification. We compared the effect of the substitution of O for S by overlaying minimised structures of thiazole 2, oxazole 4 and thiadiazole 8 and noted that only a small conformational shift was seen in the case of the oxazole (Supplementary data, Figure S3).
Evaluation of the 6-membered ring analogues showed that introduction of a phenyl ring totally abolished selectivity (e.g., 13). Retention of an N-atom in the 2-position of the central ring was necessary for activity against the VHL-deficient RCC4 line (e.g., 9, 11, 12), although it was not sufficient for selectivity with pyrimidine 9 being nonselective. There was no correlation between the pKa of either the pyridyl N, or the aniline NH, and cytotoxic potency across the series 1–13. Taken together, it seems likely that the thiazole N plays a key role in the binding to the target; isosteric replacement of the thiazole S with =CH is tolerated in six-membered rings, but other minor modifications of the central ring result in a loss in potency, thus these analogues were not pursued further.
The putative space around the pyridine ring was explored with analogues bearing fused rings to provide an extended aromatic region. We had previously demonstrated20 that a 4-pyridine was the only regioisomer that retained activity and so the heterocycles were designed to place an N-atom in a similar spatial location. These rigid, bulky aromatic systems generally gave low potency and selectivity (Table 2), suggesting that optimising the steric interaction might require a more directed approach.
Hence, a set of aryl, alkynyl alkyloxy and alkylamino analogues were synthesized by elaboration of the pyridine with a variety of functional groups conveniently introduced by use of Suzuki and Sonogashira reactions, Click chemistry, as well as direct nucleophilic displacement of aryl halides. Functionalization of the pyridine at the 3-position was examined first with simple aryl 18 and pyridyl 19 substituents resulting in decreased potency and selectivity relative to 2. Addition of a range of lipophilic substituents at the 4-position of the phenyl group (20–27) gave a range of activities and suggested the potential for a lipophilic H-bond acceptor such as sulphone 25 or methyl ketone 27 to provide potency in the low micromolar range and modest selectivity. In contrast, more polar H-bond acceptor/donor carboxamides 22 and 24 were less potent and non-selective.
We had previously demonstrated the 3-propargyl analogues 28 and 29 displayed increased potency while maintaining selectivity,20 thus the higher homologues 30 and 31 were prepared. However, increasing alkyl chain length was not favourable and resulted in decreasing potency and selectivity and increasing the terminal bulk was also disfavoured, with only the morpholine 32 being comparable to 28 and 29. Incorporation of the benzyltriazole unit 34 abolished activity.
Pyridines bearing flexible solubilising basic amine side chains generally retained potency but displayed low selectivity (e.g., 35–38). Curiously, the oxygen-linked amines 37 and 38 were superior to the nitrogen-linked amines 35 and 36. The data on the 3-series show that while substituted phenyl groups at the 3-position of the pyridine gave some active compounds, the most potent analogues were obtained with the introduction of a small H-bond acceptor (e.g., CH2OH, CH2OMe) linked via a rigid acetylene.
We finally examined substitutions at the 2-position of the pyridine to see if these could also provide positive steric interactions. In contrast with the 3-series, use of a variety of 6-membered rings (40–44), gave compounds with low potency and selectivity. Interestingly, the introduction of a range of 5-membered ring heterocycles (45–50) gave analogues that generally were active and selective with the only anomaly being the 1-methyl pyrazole 48. The 1-benzylpyrazole derivative 49 had low micromolar activity and a high selectivity ratio of 30, which was similar to the benzyltriazole analogue 50, whereas the unsubstituted pyrazole analogue 47 showed a four-fold increase of potency while preserving a modest selectivity of 14-fold. Together the data suggests the presence of a second domain adjacent to the 2-position of the pyridine ring which can accommodate a five-membered ring. The exact definition of this domain and the associated H-bond contacts remain to be fully characterized. Introduction an alkyl chain at the 2-position with solubilising tertiary amines gave compounds (51–54) with low selectivity.
A series of active compounds (2, 32, 38, 47, and 49) were examined to confirm that the mechanism of cell death involved the induction of autophagy. Treatment of RCC4 cells with 5 μM of each drug for 24 h induced the formation of intracytoplasmic vesicles confirming VHL-selective induction of autophagy resulting in cell death (Figure 3).
CoMFA was used with the data from this and an earlier study to better understand the effects of substitutions around the PAT core on activity. The 3D-QSAR model generated could only explain approximately 30% of the variance in the IC50 data, and this was reflected in its lack of predictivity of an external test set of compounds. A similar result was also found when a 2D-QSAR method (HQSAR) was used, indicating that the alignment strategy used to generate the CoMFA model was appropriate, and that the SAR data of the PAT series with respect to autophagy may be quite complex.
Examination of the CoMFA contour maps generated from Model 6 using the complete training set (Figure 2) shows that, within the training set, the contouring around the aniline C-ring is similar to that previously published. The new PAT CoMFA model exhibits a large region adjacent to the sulfur of the thiazole B-ring of 28 that suggests a positive electrostatic effect will favor activity. Interestingly, this region has three distinct, yet interconnected regions. Two of these are found around either side of the B-ring while the third is more accessible by substitution from the pyridyl ring 3-position substitution, as substitutions on this ring were all seen to reduce activity. Around the pyridyl group, increased steric bulk is positively associated with activity around the 2- and 3-positions, consistent with the previous model. From the compounds characterized in Table 3, this region is better accessed by substitutions at the 3-position as suggested by the higher potency across a number of homologues. Two distinct regions around the pyridyl 2-position and nitrogen atom indicate contrasting effects of steric bulk on activity. A region adjacent to the 2-position indicates that increased steric bulk is associated with decreased activity (yellow contour); although this is contrasted by the 2-pyrazole that has increased bulk at this position yet has the strongest activity recorded for a 2-position substitution. This region of disfavored steric bulk is also accessed by the fused ring compounds (Table 2) e.g. 15. The pyrazole moiety may contribute to an additional favorable steric bulk zone adjacent to the N-atom (green contour).
The further development of the CoMFA alignment model has not produced gains in the ability of the model to explain the QSAR observed. Despite generating six different models we are unable to explain more than 31% of the variance observed and did not significantly improve on this using the alternative HQSAR approach. This low predictivity may be a consequence of the complexity of the phenotypic readout which reports the product of many combined parameters rather than just the interaction of the PAT with the molecular target. Although we have further explored the SAR around the pyridyl A-ring and B-ring, the relatively small gains in potency and selectivity seen with the 2- and 3-substituted pyridyl analogues, coupled with the relatively flat SAR profile observed in our previous study, suggest that the PAT series may occupy a local minimum and a large random leap may be necessary to improve potency and selectivity substantially. We will endeavour to use the alignment model generated in this study to conduct a virtual screen in an effort to identify new chemotypes with VHL-selective activity. In another approach, we plan to exploit the steric tolerance adjacent to the pyridyl ring to introduce linker moieties to assist in the preparation of molecular probes to isolate and identify the molecular target of this PAT chemotype.
4. Experimental Section
Chemistry 4.1
4.1.1. General method A. Suzuki reaction of 2,6-dichloropyrimidine, 2,6-dichloro pyrazine, 2,6-dichloropyridine and 3-bromoaniline (Compounds 65–68)
Boronic acid or ester (10.0 mmol), K2CO3 (25.0 mmol) in H2O (10 mL) and Pd(PPh3)4 (0.3 mmol) were added sequentially to a solution of aryl halide (10.0 mmol) in 1,4-dioxane (40 mL). The reaction mixture was stirred at 90 °C for 2–20 h and was then cooled to 20 °C and partitioned between EtOAc (200 mL) and H2O (100 mL). The organic phase was washed with water (3 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate gradient of EtOAc/pet. ether, to give the Suzuki product.
4.1.2. General method B. Buchwald reaction (Compounds 11–13)
Pd(OAc)2 (0.37 mmol), BINAP (0.37 mmol) and NaOtBu (10.30 mmol) were sequentially added to a stirred, degassed solution of aryl halide (3.68 mmol) and amine (7.36 mmol) in a mixture of toluene (50 mL) and DMF (5 mL) and the mixture was heated at 90 °C for 4–5 h. The mixture was cooled to 20 °C, diluted with Et2O (200 mL), washed with water (3 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate blend of EtOAc/pet. ether, to give the Buchwald product.
4.1.3. General method C. Preparation of pyridyl 2-bromoethanones (Compounds 71, 73, 75)
Br2 (80 mmol) was added dropwise to a stirred solution of acetylpyridine (80 mmol) in 30% HBr/HOAc (100 mL) at 15 °C. The mixture was stirred at 40 °C for 1 h and then 75 °C for 1 h. The mixture was cooled to 20 °C, diluted with Et2O (400 mL) and stirred for 30 min. The precipitate was filtered, washed with Et2O (25 mL) and dried under vacuum to give the bromoethanone as the hydrobromide salt.
4.1.4. General method D. Preparation of thiazoles (Compounds 14–17)
A mixture of bromoketone hydrobromide (3 mmol) and phenylthiourea (3 mmol) in EtOH (20 mL) was stirred at reflux temperature for 1 h. The mixture was cooled to 20 °C, diluted with water (50 mL), the pH adjusted to ca. 8 with aqueous NH3 and the mixture stirred at 20 °C for 2 h. The precipitate was filtered, washed with water (5 mL) and dried. The solid was purified by column chromatography, eluting with an appropriate blend of EtOAc/pet. ether, to give the thiazole.
4.1.5. General method E. Suzuki reactions with bromo- and iodopyridines (Compounds 18–27 and 39–49)
PdCl2(dppf) (0.03 mmol) was added to a stirred, degassed solution of bromo- or iodopyridine (0.29 mmol), boronic acid or ester (0.45 mmol) and K2CO3 (3.00 mmol) in a mixture of toluene/EtOH/H2O/DMF (5:3:1.5:4, 13.5 mL) and the mixture was heated at 90 °C for 16 h. After cooling to 20 °C, the mixture was partitioned between EtOAc (100 mL) and water (50 mL), the combined organic phase was washed with H2O (3 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate blend of EtOAc/pet. ether or aqueous NH3/MeOH/DCM, to give the Suzuki product.
4.1.6. General method F. Sonogashira reactions with bromopyridine (Compounds 30, 31 and 81)
PdCl2(PPh3)2 (0.03 mmol) was added to a stirred, degassed solution of bromopyridine (0.58 mmol), alkyne (0.70 mmol) and CuI (0.03 mmol) in a 1:1 mixture of DMF and NEt3 (4 mL), and the reaction mixture was stirred at 50 °C for 16 h then at 70 °C for 4 h. After cooling to 20 °C, the mixture was partitioned between EtOAc (150 mL) and water (50 mL), the organic phase was washed with water (3 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate blend of EtOAc/pet. ether, to give the alkyne.
4.1.7. General method G. Elaboration of propargyl alcohol (Compounds 32 and 33)
MsCl (0.28 mmol) was added to a solution of alcohol 81 (0.24 mmol) and NEt3 (0.36 mmol) in anhydrous DCM (10 mL) at -20 °C and the reaction mixture was stirred at -20 °C for 1 h. The mixture was diluted with DCM (100 mL), washed with water (3 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. Amine (2.4 mmol) was added to a solution of the crude mesylate in anhydrous THF (5 mL), and the mixture was heated at 50 °C for 2 h. The mixture was cooled to 20 °C, partitioned between EtOAc (100 mL) and water (50 mL), the organic layer was washed with aqueous NaHCO3 (50 mL), washed with brine (50 mL), dried and the solvent was evaporated. A mixture of crude carbamate in DCM (10 mL) and TFA (1 mL) was then stirred at 20 °C for 1 h. The solvent was evaporated and the residue was purified by column chromatography, eluting with appropriate blend of aqueous NH3/MeOH/DCM, to give the alkyne.
4.1.8. General method H. Click reaction (Compounds 34 and 50)
CuSO4·5H2O (0.02 mmol) and sodium ascorbate (0.04 mmol) were added to a solution of acetylene (0.18 mmol) and benzyl azide (0.20 mmol) in a 1:1:1 mixture of DCM/EtOH/H2O (3 mL), and the mixture was stirred at 20 °C for 20 h. The mixture was partitioned between EtOAc (100 mL) and water (50 mL), extracted with EtOAc (2 × 70 mL), the combined organic phase was washed with water (2 × 30 mL), washed with brine (30 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate blend of MeOH/DCM, to give the triazole.
4.1.9. General method I. Fluoride and bromide displacement reactions (Compounds 35–38 and 51–54)
A mixture of 3-fluoro or 2-bromopyridine (0.35 mmol) in the appropriate amine or alcohol (2 mL) was heated in a sealed glass pressure vessel at 100–180 °C for 16-24 h (in some cases, the reaction was done in the presence of Cs2CO3). The solution was cooled to 20 °C, partitioned between EtOAc (100 mL) and water (50 mL), the organic phase was washed with water (2 × 50 mL), washed with brine (50 mL), dried and the solvent was evaporated. The residue was purified by column chromatography, eluting with an appropriate blend of aqueous NH3/MeOH/DCM to give the substitution product.
4.2. Cell Viability Assays
For 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide (XTT) assays, five thousand cells were plated in 96-well plates. The next day, vehicle (DMSO) or drug was added by serial dilution. Four days later the media was aspirated, XTT solution (0.3 mg/ml of XTT (Sigma), 2.65 mg/ml N-methyl dibenxopyrazine methyl sulfate (Sigma) in phenol red-free media) was added, and the plates were incubated at 37 °C for 1-2 hours. Metabolism of XTT was quantified by measuring the absorbance at 450 nm. IC50 values were calculated using linear interpolation.
4.3. QSAR modeling
4.3.1. Alignment Generation
A minimum energy conformation of 2 was generated using OMEGA2 (Openeyes Software, USA) with default parameters using the MMFF94s force field and the rms torsion driving parameter set at 0.6. All other structures were generated from this conformation using the SKETCHER (SYBYL8.0, TRIPOS) and minimised using MAXMIN2 (SYBYL8.0) with the Tripos Force field and Gastieger-Huckel charges. Minimisation was performed using 1000 steps of steep decents followed by conjugate gradients until convergence at 0.05 kcal/(molÅ). A distance dependent dielectric function was used with a dielectric constant of 80. Molecular alignments were generated by superimposition of all compounds onto 28 using ROCS (Openeye Software, USA) with default parameters using both the shape only and shape in combination with color force field method. While the shape only alignment was used for CoMFA, those compounds poorly aligned as assessed after visual inspection, were replaced by improved alignments from the shape and colour force field combination alignment if available. Alternate proton positions were explored for compound 6, while alternative conformers based on rotating the B-ring relative to the C-ring were also explored.
4.3.2. 3D-QSAR Analysis
CoMFA was used to determine a relationship between the compounds tested and in vitro cytotoxicity as previously described.20 The SAMPLS Leave One Out (LOO) protocol was used to determine the cross-validated r2 (q2) as a indicator of the predictivity of the 3D-QSAR model along with the optimum number of components, which was subsequently used in a conventional non-validated PLS analysis. Only compounds that had defined IC50 data and cytotoxicity ratios, and substitutions including up to one rotatable bond were used. A base CoMFA 3D-QSAR model was created using a previously described compound set (Model 1)20 and compounds from each substitution pattern were added into this generating Models 2-5, and a compound set representing all PAT analogues was used to generate Model 6. The compound sets used to generate each model are listed in the Supplementary data.
4.3.3. 2D-QSAR Analysis
The 2D-Hologram QSAR (HQSAR) method21 as implemented in SYBYL×1.2 was used to eliminate any dependency of the CoMFA QSAR model on compound 3D structure, conformation and alignment strategy. In this method each compound is broken down into all possible linear, branched and cyclic structural fragments and these are counted into arrays of fixed length, a molecular hologram. The bin occupancies associated with the molecular hologram are descriptor variables that can be related to biological activity by Partial Least Squares analysis. As HQSAR models can be affected by hologram length, fragment size and distinction, a series of models characterising variation in these parameters were generated to find the optimal QSAR model for the PAT series as determined by the highest q2 value. Using the default fragment size of 4–7 atoms, the effect of different fragment distinctions on q2 was explored. The optimal fragment distinction combination was then used to characterise the effect of fragment size on HQSAR model q2.
Supplementary Material
Acknowledgments
The authors thank Dr Maruta Boyd, Dr Shannon Black, Stefanie Maurer and Sisira Kumara for technical assistance and acknowledge the Association for International Cancer Research 10-0042 (MB), the Maurice Wilkins Centre for Biodiscovery (MPH, JUF), US NCI-CA-82566 (AJG, MPH), NCI-CA-123823 (DAC), NCI-T32-CA-121940 (EWL) for funding.
Abbreviations
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- CoMFA
comparative molecular field analysis
- DCM
dichloromethane
- DIAD
diisopropyl azodicarboxylate
- DMF-DMA
dimethylformamide-dimethylacetal
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- EDCI
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- HQ-SAR
hologram quantitative structure activity relationship
- HIF
hypoxia-inducible factor
- PAT
pyridyl anilinothiazoles
- VHL
von Hippel-Lindau
- RCC
renal cell carcinoma
- SAR
structure-activity relationship
- XTT
2, 3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide
Footnotes
Supplementary data: Supplementary data, including full experimental descriptions of compound synthesis, model descriptions and tables of actual and predicted activities and additional compounds, associated with this article can be found, in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. J Clin Oncol. 1999;17:2530. doi: 10.1200/JCO.1999.17.8.2530. [DOI] [PubMed] [Google Scholar]
- 2.Murai M, Oya M. Curr Opin Urol. 2004;14:229. doi: 10.1097/01.mou.0000135078.04721.f5. [DOI] [PubMed] [Google Scholar]
- 3.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, Lubensky I, Duan DR, Florence C, Pozzatti R, Walther MM, Bander NH, Grossman HB, Brauch H, Pomer S, Brooks JD, Isaacs WB, Lerman MI, Zbar B, Linehan WM. Nature Genet. 1994;7:85. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 4.Kim WY, Kaelin WG., Jr J Clin Oncol. 2004;22:4991. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 6.Gnarra JR, Zhou S, Merrill MJ, Wagner JR, Krumm A, Papavassiliou E, Oldfield EH, Klausner RD, Linehan WM. Proc Natl Acad Sci USA. 1996;93:10589. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci USA. 1996;93:10595. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knebelmann B, Ananth S, Cohen HT, Sukhatme VP. Cancer Res. 1998;58:226. [PubMed] [Google Scholar]
- 9.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Nature. 2003;425:307. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 10.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Nature. 2006;440:1222. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 11.Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Jr, Iliopoulos O. Mol Cell. 1998;1:959. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- 12.Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Nat Cell Biol. 2003;5:64. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- 13.Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, Minato N, Ohno S, Shuin T. J Biol Chem. 2001;276:43611. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- 14.Na X, Duan HO, Messing EM, Schoen SR, Ryan CK, di Sant'Agnese PA, Golemis EA, Wu G. EMBO J. 2003;22:4249. doi: 10.1093/emboj/cdg410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaelin WG., Jr Nature Rev Cancer. 2005;5:689. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 16.Chan DA, Giaccia AJ. Cell Cycle. 2008;7:2987. doi: 10.4161/cc.7.19.6776. [DOI] [PubMed] [Google Scholar]
- 17.Turcotte S, Sutphin PD, Chan DA, Hay MP, Denny WA, Giaccia AJ. Cancer Cell. 2008;14:90. doi: 10.1016/j.ccr.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turcotte S, Giaccia AJ. Curr Opin Cell Biol. 2010;22:246. doi: 10.1016/j.ceb.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleming A, Noda T, Yoshimori T, Rubinstein DC. Nature Chem Biol. 2011;7:9. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 20.Hay MP, Turcotte S, Flanagan JU, Bonnet M, Chan DA, Sutphin PD, Nguyen P, Giaccia AJ, Denny WA. J Med Chem. 2010;53:787. doi: 10.1021/jm901457w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heritage TW, Lowis DR. Molecular Hologram QSAR. In: Parrill AL, Reddy MR, editors. Rational Drug Design Novel Methodology and Practical Applications. Vol. 719. American Chemical Society; Washington, DC: 1999. pp. 212–225. (ACS Symposium Series). [Google Scholar]
- 22.Mahboobi S, Sellmer A, Eswayah A, Elz S, Uecker A, Böhmer FD. Eur J Med Chem. 2008;43:1444. doi: 10.1016/j.ejmech.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 23.Tavares FX, Boucheron SH, Griffin RJ, Preugschat F, Thomson SA, Wang TY, Zhou HQ. J Med Chem. 2004;47:4716. doi: 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- 24.Adams H, Batten SR, Davies GM, Duriska MB, Jeffery JC, Jensen P, Lu J, Motson GR, Coles SJ, Hursthouse MB, Ward MD. Dalton Trans. 2005:1910. doi: 10.1039/b502892b. [DOI] [PubMed] [Google Scholar]
- 25.Miki Y, Nakamura N, Hachiken H, Takemura S. J Het Chem. 1989;26:1739. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.