

Abstract
Developmental epigenetic changes, such as DNA methylation, have been recognized as potential pathogenic factors in inflammatory bowel diseases, the hallmark of which is an exaggerated immune response against luminal microbes. A methyl-donor (MD) diet can modify DNA methylation at select murine genomic loci during early development. The components of the MDs are routinely incorporated into prenatal human supplements. Therefore, we studied the effects of maternal MD supplementation on offspring colitis susceptibility and colonic mucosal DNA methylation and gene expression changes in mice as a model. Additionally, we investigated the offspring mucosal microbiomic response to the maternal dietary supplementation. Colitis was induced by dextran sulfate sodium. Colonic mucosa from offspring of MD-supplemented mothers following reversal to control diet at weaning was interrogated by methylation-specific microarrays and pyrosequencing at postnatal days 30 (P30) and P90. Transcriptomic changes were analyzed by microarray profiling and real-time reverse transcription polymerase chain reaction. The mucosal microbiome was studied by high throughput pyrosequencing of 16S rRNA. Maternal MD supplementation induced a striking susceptibility to colitis in offspring. This phenotype was associated with colonic mucosal DNA methylation and expression changes. Metagenomic analyses did not reveal consistent bacteriomic differences between P30 and P90, but showed a prolonged effect of the diet on the offspring mucosal microbiome. In conclusion, maternal MD supplementation increases offspring colitis susceptibility that associates with persistent epigenetic and prolonged microbiomic changes. These findings underscore that epigenomic reprogramming relevant to mammalian colitis can occur during early development in response to maternal dietary modifications.
INTRODUCTION
Epidemiologic studies show a significant rise in the incidence of many chronic diseases in the industrialized world, including inflammatory bowel diseases (IBD), which are comprised of Crohn's disease (CD) and ulcerative colitis (UC) (1). Critical components of IBD development are the immune system, the intestinal microflora and the gut mucosa (2,3). The disorders are recognized to be modulated by genetic predisposition and environmental factors (3,4). There are >60 susceptibility loci recognized for IBD (5), but epidemiologic and monozygotic twin studies suggest that environmentally sensitive mechanisms are also important for the development and progression of these diseases (6). However, it is still unclear which factor in the intricate network of the microbiota, mucosa and the host immune system is affected critically leading to disease progression. It is also unclear which characteristics of the key cellular components are modified by pathogenic environmental factors.
One of the molecular mechanisms that can dynamically respond to environmental and nutritional exposures in mammals is defined as epigenetic. Epigenetic changes are mitotically heritable molecular modifications that affect gene expression potential without alteration in the DNA sequence (7). Epigenetic derangements may be important in IBD (8,9). However, definite data supporting this connection remain limited.
The most stable epigenetic alteration is the methylation of cytosines at CpG [cytosine followed by a guanine and connected by a phosphodiesther bond (p)] dinucleotides, designated as DNA methylation, which commonly correlates with transcriptional silencing. This molecular modification is catalyzed by DNA methyltransferases that utilize the mammalian one-carbon pool, and it has been shown to be influenced by environmental and nutritional exposures (10). Maternal dietary supplementation of methyl-donor micronutrients has been found to be effective in modifying the developmental establishment of DNA methylation at unique genomic loci, leading to persistent phenotypic modification in mouse offspring (11). Such observations highlight the potential for nutritionally induced epigenetic modifications to play an important role in common human disease development (12). Importantly, methyl donors in the seminal nutritional studies were betaine, choline, vitamin B12 and folic acid (11,13). All of these compounds can be found in various prenatal vitamins and supplements, the consumption of which has become common in the developed world during pregnancy.
In this study, we addressed whether maternal supplementation of methyl-donors (MDs) might lead to altered offspring colitis susceptibility in mice. A striking augmentation of dextran sulfate sodium (DSS) colitis was observed in the MD offspring, which associated with altered colonic mucosal DNA methylation and expression at genes previously implicated in intestinal inflammation. Our study may impact future public health policy in regards to the judicious use of maternal dietary supplements.
RESULTS
Pediatric supplementation of MD does not alter colitis susceptibility in young adult mice
The peak incidence of IBD is in young adulthood (14). Consequently, there is an extended developmental period during which environmental influences may critically impact disease susceptibility. We first addressed the pediatric time period with respect to MD supplementation and colitis susceptibility using DSS. Molecular and histologic features of colitis correlate well with weight change and colonic length in this model (15–17). We have shown recently that relevant epigenetic changes can occur in murine colonic mucosa during pediatric development with respect to mucosal inflammatory responses [from postnatal days 30 (P30) to P90] (18). Nevertheless, animals supplemented with MD from P30 to P80 followed by 10-day reversal to control diet (COD) had similar responses to DSS as the control mice (data not shown). Therefore, MD supplementation during pediatric development did not induce persistent phenotype modification with respect to young adult murine DSS colitis.
Maternal MD supplementation persistently increases offspring colitis susceptibility
We next determined whether maternal MD supplementation may affect offspring colitis susceptibility. Offspring weight was not influenced by the maternal dietary exposure either at P21, P30 or P90 (data not shown). In the meantime, MD offspring had greater weight loss (necessitating euthanasia, i.e. mortality) and increased colonic shortening upon DSS exposure at both P30 and P90, compared with control (Fig. 1). This outcome was independent of sex. These results show that maternal MD supplementation induces long-lasting, strikingly augmented susceptibility to colonic mucosal injury in the pups, which lasts to young adulthood.
Figure 1.

Mortality curves and colonic lengths of DSS exposed P30 (above) and P90 animals (below). MD supplemented offspring had higher mortality and shorter colon lengths (P30: n= 8–9, P= 0.0008, P90: n= 7–11, P< 0.001) supporting a persistently and remarkably increased susceptibility to colitis even 69 days after weaning to COD.
Maternal MDs induce prolonged offspring colonic mucosal DNA methylation changes
DSS exposure is primarily a model for acute colonic mucosal injury and regeneration. Consequently, we tested the effects of our epigenetically active maternal dietary exposure on offspring colonic mucosal DNA methylation by methylation-specific amplification microarray (MSAM). Fifty-nine intervals with persistently increased methylation and 96 intervals with persistently decreased methylation were identified (Supplementary Material, Tables S1 and S2). Interestingly, >50% of the intervals with decreased methylation were X chromosomal. We confirmed maternal MD-dependent epigenetic changes at the Ptpn22-associated SmaI/XmaI interval by bisulfite pyrosequencing at both P30 and P90 (Fig. 2). Ptpn22 was chosen for validation because it has been implicated to have an important role in diseases, including type 1 diabetes, rheumatoid arthritis and lupus, making it a particular gene of interest in IBD (19). It is also among the 69 recently updated IBD susceptibility loci (www.ibdgenetics.org). Methylation of the Ptpn22-associated SmaI/XmaI interval showed a 0.753-fold decrease using MSAM and a 0.714-fold decrease with pyrosequencing in MD animals compared with control.
Figure 2.

Methylation of the Ptpn22-associated SmaI/XmaI interval at P30 and P90. Methylation decreased in MD supplemented offspring colonic mucosa in a persistent fashion both 9 days (P30) and 69 days (P90) after reversal (P30 n = 8–9, P = 0.0009; P90 n= 4–5; P < 0.0001).
Gene ontology (GO) analysis revealed that cellular component genes in the synapse part (GO: 0044456) were enriched, whereas genes related to non-membrane bound organelles (GO: 0043228) and organelle parts (GO: 0044422) were under-represented. These results support that the intervals epigenetically responsive to maternal MD supplementation in offspring colonic mucosa represent a select group of genomic loci.
Maternal MDs induce prolonged offspring colonic mucosal gene expression changes
Since epigenetic alterations can associate with gene expression changes, we assessed genome-wide transcriptomic alterations in the colonic mucosa of MD offspring. Four hundred seventy three transcripts with increased expression and 514 with decreased expression were identified at P90 (Supplementary Material, Tables S3 and S4). Therefore, ∼3% (987/33 074) of all murine transcripts were affected in a prolonged manner by prenatal/early developmental exposure to MDs. Interestingly, we noted Cpn2 on the list of genes that decreased in expression. Cpn2 has been previously implicated to play a role in the pathogenesis of both CD and UC (20). Therefore, we chose Cpn2 for validation of the expression microarrays in spite of a relatively low expression difference at this gene (Supplementary Material, Table S4). A 0.81-fold decrease in expression by microarray showed a 0.5-fold decrease in real-time reverse transcription polymerase chain reaction (RT–PCR) (Fig. 3).
Figure 3.

Expression of Cpn2 decreased in MD supplemented offspring colonic mucosa compared with control at P90 (n= 9, P= 0.021).
GO analysis was performed to compare all genes with modified expression to the remaining genome. Within the category of biologic processes, genes involved in immune responses (GO: 0006955) and biosynthetic processes (GO: 0009058) were enriched among the MD responsive group. Genes related to neurological processes (GO: 0050877) and cell communication (GO: 0007154) were underrepresented. Within the category of cellular components, genes associated with extracellular region part (GO: 0044421) and cell part (GO: 0044464) were both enhanced. Genes associated within the category of molecular function had notable results with enrichment in the following: oxidoreductase activity (GO: 0016491), structural constituent of ribosome (GO: 0003735), carrier activity (GO: 0005386), nucleic acid binding (GO: 0003676), peptide transporter activity (GO: 0015197), selenium binding (GO: 0008430), ion transporter activity (GO: 0015075), enzyme activator activity (GO: 0008047) and nucleoside binding (GO: 0001882). There was an underrepresentation within this category of genes involved in receptor activity (GO: 0004872). These findings support that maternal MD supplementation induces prolonged cellular, biologic and, in particular, molecular functional transcriptional reprogramming in offspring colonic mucosa. Importantly, the rearrangement in the expression of immunologically active genes (Table 1) can specifically play an important role in modifying colitis susceptibility.
Table 1.
List of enriched genes among the MD responsive group associated with immunologic processes according to Babelomics (GO: 0006955)
| Cd6 | Ccl19 | Gadd45g | Ccl6 |
| Samhd1 | H2-K1 | Cxcl13 | H2-Oa |
| Ccl5 | Thy1 | Was | Ly86 |
| Serpina3g | Ambp | Tap2 | Skap1 |
| Gpb3 | Ctsw | Tap1 | Cxcl10 |
| Lck | Gbp2 | Il18 | Ptpn22 |
| Mbp | Ccl9 | Mnda | Fth1 |
| Tnfrsf13c | Cd7 | Hc | Cd72 |
| H2-T3 | Mapk11 |
Epigenomic and transcriptomic overlaps in the colonic mucosa of MD offspring
We wished to determine the extent of overlaps between the MSAM and gene expression microarrays to assess the potential direct functional relevance of the epigenetic changes identified. Of the 155 genomic intervals with a change in methylation, 18 were associated with an expression change at a neighboring transcript (Table 2).
Table 2.
List of genes with overlap between SmaI/XmaI interval methylation and expression change identified with the microarrays
| Gene name | SmaI/XmaI Interval | TSS | TES | Methylation | Expression |
|---|---|---|---|---|---|
| Nr4a1 | chr15: 101084352–101084854 | +12673 | +20871 | ↓ | ↑ |
| Pgrmc1 | chrX: 33993965–33995933 | +143270 | +151125 | ↓ | ↑ |
| Ppara | chr15: 85501945–85502320 | +63861 | +135149 | ↓ | ↑ |
| Ptpn22 | chr3: 103706444–103706814 | +42414 | +9541 | ↓ | ↑ |
| Slc16a2 | chrX: 101016514–101018499 | −179 | −124753 | ↓ | ↑ |
| Sobp | chr10: 42894350–42894784 | −231 | −172262 | ↓ | ↑ |
| Lonrf3 | chrX: 33755133–33755875 | +112899 | +151347 | ↓ | ↓ |
| Ppp1r1b | chr11: 98189980–98191059 | +19533 | +28590 | ↓ | ↓ |
| Utp14b | chr1: 78654480–78655157 | +196 | +9357 | ↓ | ↓ |
| Vsig1 | chrX: 137346441–137347795 | +126893 | +126893 | ↓ | ↓ |
| Gsdmc | chr15: 63182682–63183481 | −457213 | −493650 | ↑ | ↓ |
| 201003K11Rik | chr19: 4608517–4608707 | −110029 | −111825 | ↑ | ↓ |
| Ndufb9 | chr15: 58829987–58830663 | −64961 | −59281 | ↑ | ↓ |
| Pcx | chr19: 4614094–4614913 | +20185 | +7249 | ↑ | ↓ |
| Rpl38 | chr11: 113673746–113674714 | +855627 | +859415 | ↑ | ↓ |
| Timp3 | chr10: 85828871–85828989 | −65774 | −16680 | ↑ | ↓ |
| Hyi | chr4: 117927340–117928820 | +104539 | +107269 | ↑ | ↑ |
| Rap1gap | chr4: 137279276–137279675 | −41857 | +6307 | ↑ | ↑ |
TSS: distance of SmaI/XmaI interval midpoint from transcription start site in bases relative to the gene; TES: distance of SmaI/XmaI interval midpoint from transcription end site in bases relative to the gene. Eighteen genes associated with 18 intervals.
The reason for this restricted overlap (∼12%) is likely due to the inherent limitations of the microarray methods employed (18). Nevertheless, Ppara, a gene previously implicated in murine colitis (21), was among the candidates identified. Ppara exhibited decreased methylation and increased expression with a significant inverse correlation (Fig. 4). We next analyzed Ptpn22, a gene already validated to associate with decreased methylation (Fig. 2). The overall expression of Ptpn22, however, was below the detection limit of our real-time RT–PCR assay, not allowing validation.
Figure 4.
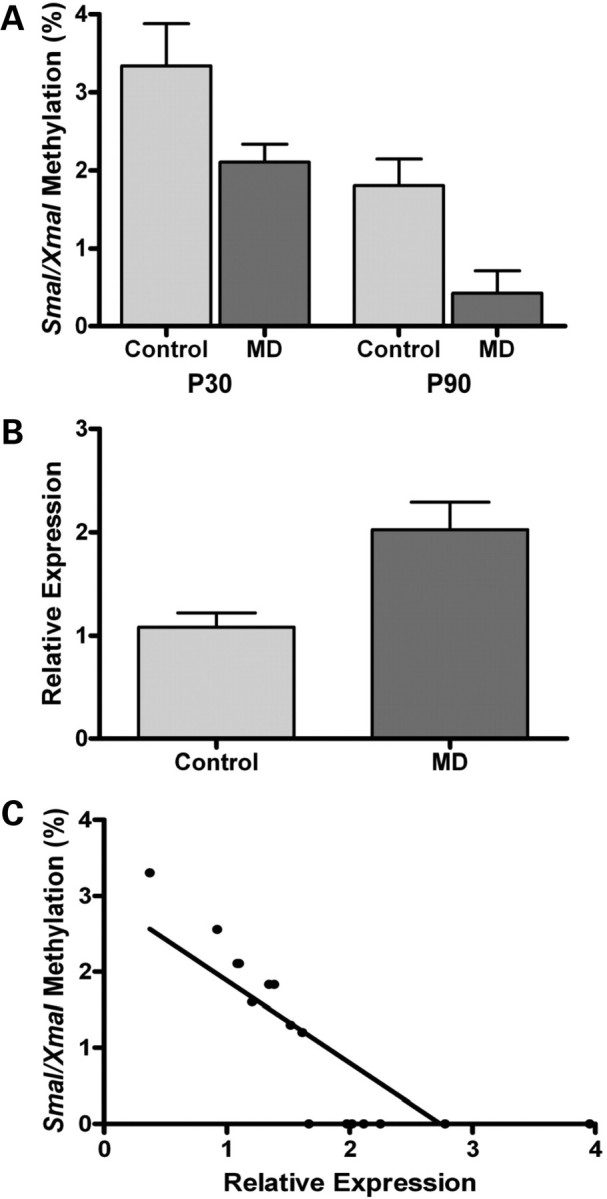
DNA methylation changes at Ppara correlate with its expression. (A) Methylation of the Ppara SmaI/XmaI interval decreased in MD supplemented offspring at P30 and P90 (P30: n= 10–10, P= 0.041; P90: n= 8–8, P= 0.0083). (B) Ppara expression increased in MD supplemented offspring colonic mucosa at P90 (n= 10–10, P= 0.008). (C) Methylation at Ppara inversely correlates with expression (r = 0.79; P= 0.000034).
Prolonged mucosal metagenomic changes in offspring
Dietary exposures can significantly affect the intestinal microbiota (22,23) and microbial alterations can prominently modify the severity of acute colitis in mice (24). Consequently, we evaluated the differences between the mucosa-associated microbiome in MD and control offspring. While there was a remarkable separation at the genus level between MD and control pups at P30, this was lost by P90 (Figs 5 and 6). Analysis of variance (ANOVA) in repeated measurements showed that at P30, the genera Clostridium, Bacteroides, Staphylococcus, Syntrophococcus, Coprococcus, Parabacteroides, Alistipes, Butirvibrio and Anaerovorax were overrepresented, and the genera Lactobacillus, Rikenella and Sporobacterium were underrepresented (P< 0.05) in the colonic mucosa of MD pups in relation to the control group. The most striking composition variation was for Clostridia (57% average in MD, and 27% in control; P= 0.00016) and Lactobacilli (2.5% average in MD, and 46.5% in control; P= 0.000019) at P30. Seven of the 12 (58%) genera with significant difference between MD and control mucosa were present in >1% of the microbial composition in at least one of the groups (Supplementary Material, Fig. S1). At P90, however, seven genera were influenced (P< 0.05) by maternal MD exposure in the offspring, only two (29%) of which involved genera present in more than 1% of the microbiome (Turicibacter, Roseburia) (Supplementary Material, Fig. S2). Importantly, there was no overlap between the significantly affected genera at P30 and P90.
Figure 5.
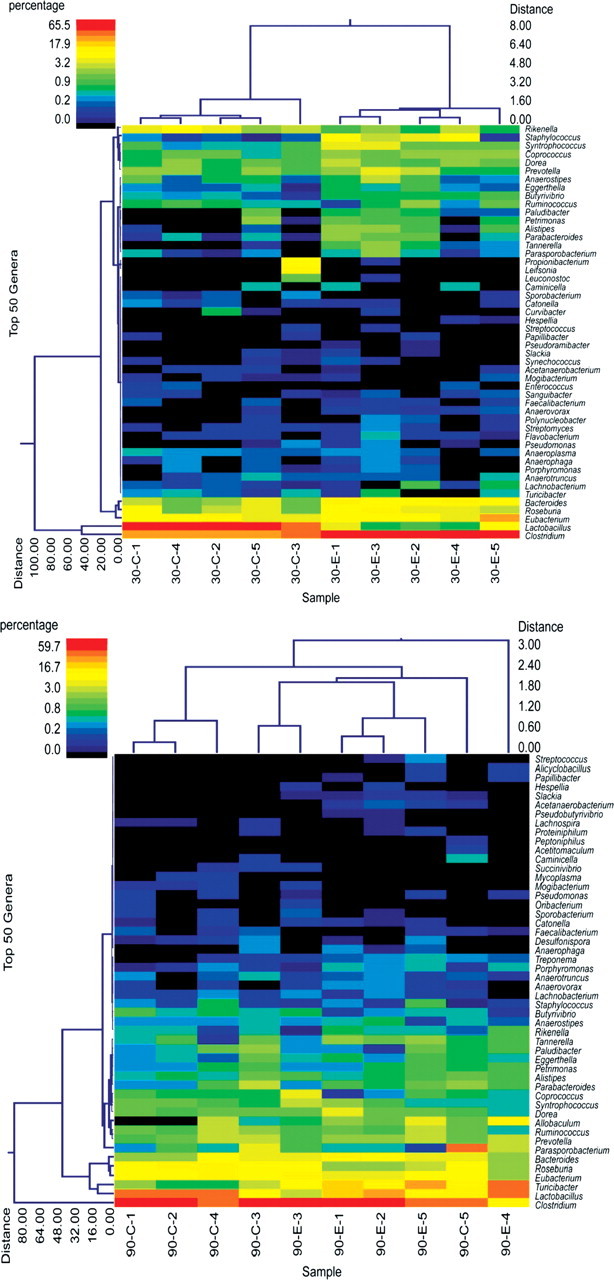
Dual hierarchical clustering dendograms of the most predominant and ubiquitous 50 genera among mucosal samples at P30 (upper) and P90 (lower). The heat maps depict the relative percentage of each genus for each sample. The color scales for the heat maps are shown in the upper left corners of each. There was a remarkable separation of MD offspring at P30 independent of litter clusters [control animals (C) were from two different litters: C1–4; C5, respectively; MD supplemented (experimental: E) were also from two different litters: E1–4; E5, respectively]. The separation between the experimental groups was overwhelmingly lost by P90.
Figure 6.

Principal component analysis (PCA) of UNIFRAC distance metric. This figure provides a three-dimensional visualization of the PCA analysis using the top three vectors. Green spheres represent the P30 MD exposed animals (two different litters: E1–4; E5, respectively). Blue spheres represent the P30 control animals (two different litters: C1–4; C5, respectively). Red spheres stand for the P90 MD exposed animals (three different litters: E1–2; E3–4; E5 respectively). Yellow spheres mark the P90 control animals (two different litters: C1–4; C5, respectively). Based on the distances, it can be concluded that interindividual (within the same litter) variation exceeded than that of inter-litter variation within the experimental groups. Therefore, the notable separation of the P30 groups and the less, but continued parting at P90 was a consequence of the maternal dietary intervention and not due to microbiomic separation of the litters.
A total of 335 different bacterial species were detectable in at least one sample from the P30 animals, while 256 were detectable from the P90 animals. In the meantime, only Staphylococcus saprophyticus had similar, but differing volume of variation between MD and control offspring at both P30 (4% average in MD, undetectable in control) and P90 (0.03% average in MD, undetectable in control). Therefore, we could not identify a prominent and persistent similarity between maternal MD exposure-dependent metagenomic variation at P30 and P90 either at the genus or the species level.
DISCUSSION
The potentially important role of environmentally responsive epigenetic changes has been recognized with respect to the fetal origins of adult disease (10,25,26). However, there are limited experimental data investigating the impact of prenatal and early developmental environmental exposures on consequent intestinal inflammatory responses. Early postnatal maternal deprivation resulted in increased offspring colitis susceptibility in young adult rats (27) and influenced colonic mast cell–nerve interactions (28). Additionally, the lipid composition of the maternal diet has been shown to modify colonic phospholipid constitution and young adult colitis susceptibility in rat offspring (29). However, the epigenetic consequences of early developmental environmental exposures have not been addressed in regards to mammalian intestinal inflammation. Here we studied the effects of an epigenetically active diet (11) on murine colitis susceptibility.
MD supplementation during childhood did not influence colitis susceptibility following 10 days of reversal in young adult animals. Consequently, we studied the effect of maternal MD supplementation on offspring intestinal inflammation and found a profound and prolonged sensitization to DSS in the pups resulting from the dietary intervention. The increased susceptibility to chemically induced colitis associated with low volume, but persistent DNA methylation changes at a functionally select, small number of genomic loci. Although we expected mostly increased levels of DNA methylation to result from the supplementation of MDs, both increases and decreases took place. Therefore, developmental reprogramming of DNA methylation appeared to occur in the offspring colonic mucosa as a result of the maternal dietary modification. Interestingly, the X chromosome was particularly sensitive to the MD supplementation with decreased levels of methylation independent from gender.
The MD maternal diet also persistently modified gene expression in the offspring, affecting ∼3% of all murine transcripts. Importantly, genes involved in immune responses (Table 1) were enriched in the MD responsive group. This observation supports the developmental reprogramming of colonic mucosal immune functions resulting from fetal MD exposure. In addition to the ontologically identified immune-related genes (Table 1), we found decreased expression of Cpn2 (Fig. 3). CPN2 encodes the large subunit of carboxypeptidase N (CPN) and stabilizes the smaller and catalytically active subunit, CPN1. CPN participates in the degradation of biologically active peptides. The targeted disruption of Cpn1 causes increased susceptibility to anaphylatoxin-mediated shock in mice (30). Consequently, the decreased expression of Cpn2 may lead to decreased amounts of active Cpn1 during the experimentally induced inflammatory cascade of DSS colitis and may increase MD offspring vulnerability. In the meantime, CPN1 and 2 were increased in the colonic mucosa of patients suffering from IBD compared with healthy controls (20). It is not known, however, whether the CPN response in IBD patients is dampened compared with acute forms of colitis, or what the baseline levels of CPN1 and 2 are from non-inflamed areas of the mucosa in IBD. Nevertheless, our Cpn2 results further support the persistent reprogramming of colonic mucosal immune responses.
We were able to delineate overlaps between DNA methylation and gene expression changes at a limited number of genomic intervals (18). The identified inverse correlation between methylation and expression of Ppara highlights the functionally important DNA methylation changes induced by the fetal exposure to MD supplementation. The observed increased expression of Ppara in the MD offspring would indicate a relative protection against chemically induced colitis based on previous studies (31). Therefore, the induced epigenetic change at Ppara may serve as a protective mechanism to lessen pro-inflammatory alterations (such as decreased Cpn2 expression for instance) and contribute to the grossly normal phenotype of the MD offspring.
Persistent microbiomic alterations in the colonic mucosa of MD offspring were not identified at the genus level. Only one species, Staphylococcus saprophyticus, had similar changes in composition at P30 and P90, but not at the same volume. Staphylococcus saprophyticus has been associated with sporadic diarrhea in children (32), but its relevance in murine colitis models has not been determined. Nevertheless, our study implicates that there is a significant, maternal diet driven prolonged mucosal microbial composition difference in mammalian offspring, regardless of the same dietary and housing environment following weaning. This maternal diet-induced variation is most pronounced in infancy, but persists into young adulthood. While none of the genera differences was preserved from P30 to P90, there were still seven genera that were significantly influenced by MD supplementation of the maternal diet in the offspring colonic mucosa at P90. These particular genera may be important with respect to colitis susceptibility. For example, Roseburia was decreased in the MD offspring at P90 (Supplementary Material, Fig. S2) which has been previously observed in patients with predominantly ileal Crohn's disease (33). The longstanding influence of maternal diet on the offspring microbiota is surprising considering that enteral microbial composition can shift significantly within a day following dietary changes in gnotobiotic murine models (22). Therefore, it is plausible that the prenatal reprogramming of mucosal immunology upon maternal MD supplementation creates a persistent effect on the enteral microbiome by inducing a longstanding modification of its physiologic pediatric development. Consequently, an overall inflammation prone interaction is created within the key elements (mucosal microbiome, mucosa, host immunity) of IBD pathology.
This study includes the first epigenetically focused developmental dietary intervention attempting to unravel key environmental factors for the rising incidence of IBD. Because the utilized MDs include many common components of prenatal dietary supplements (folate, methionine, betaine, vitamin B12), the results of this experiment could be of great significance. Conclusive results from subsequent studies may support the development of guidelines for the judicious use of MD supplements in future public health policies.
MATERIALS AND METHODS
Animals and tissue collection
Mice were purchased from the Jackson Laboratories, Bar Harbor, ME, USA. For the pediatric developmental studies, P21 male C57BL/6 mice received standard rodent diet (2920X, Harlan-Teklad, Madison, WI, USA) until P30, then were randomly assigned to NIH-31 control diet (COD) (TD #95262, Harlan-Teklad, 2 litters) or to a MD supplemented diet consisting of NIH-31 supplemented with 5 mg/Kg folic acid; 0.5 g/kg vitamin B12; 5 g/kg betaine and 5.76 g/kg choline (TD#01308, Harlan-Teklad, 2 litters) (10). The experimental diets were provided until P80, and then the MD group was reversed to COD for 10 days (P90).
For the maternal dietary supplementation studies, C57BL/6 female mice, 8 weeks of age, were housed with age-matched C57BL/6 males. Ten females were randomly assigned to COD and 10 to MD, respectively, for 2 weeks prior to mating, and throughout gestation and lactation. At P21, the pups were weaned to the control diet until P30 (9 day reversal) or P90 (69 day reversal). Consequently, only persistent effects of maternal MD supplementation were tested. At P30 and P90, mice were killed by CO2 asphyxiation between 1 and 3 PM without previous food restriction. Colonic mucosa of both males and females was collected exactly as described before (18). We demonstrated this method to result in >90% colonic epithelial cells in the scrapings, thereby providing an overwhelmingly homogeneous cell population of endodermal origin. The protocol was approved by the Institutional Animal Care Committee for Baylor College of Medicine.
DSS exposure
Three percent (wt/vol) DSS (MW = 36 000–50 000, MP Biomedicals, LLC, Solon, OH, USA) was dissolved in the drinking water of the animals and provided ad libitum for 5 days, followed by 9 days of regular drinking water. This molecular weight of DSS has been shown to induce colonic inflammation in previous work (34). Mice were weighed daily until day 14 when they were asphyxiated and colonic lengths were measured except for animals that lost >20% of their starting weight. The later animals were euthanized early according to our protocol and designated as the mortality group.
Isolation and manipulation of nucleic acids
All procedures were performed on colonic mucosal scrapings. Genomic DNA was isolated with the QIAamp DNA Mini Kit (51304, Qiagen, Valencia, CA, USA) and bisulfite converted with EZ DNA Methylation-Gold Kit (D5006, Zymo Research, Orange, CA, USA). Total RNA was isolated with the RNeasy Mini Kit (74106, Qiagen) and stored at −80°C. Reverse transcription was executed with the Taqman Reverse Transcription Kit (N808-0234, Applied Biosystems, Branchburg, NJ, USA).
Methylation-specific amplification microarray (MSAM)
MSAM was carried out as previously described (35); two (one male/male and one female/female) MD versus COD offspring microarrays for both P30/P90 animals were performed (http://www.ncbi.nlm.nih.gov/geo/, accession number: GSE24354). Males and females were used equally in the microarray analyses to avoid potential gender bias. MSAM is based upon serial digestion of genomic DNA with the methylation sensitive/insensitive isoschizomers SmaI and XmaI, followed by ligation-mediated PCR. We designed a custom microarray based on the above mechanisms (Agilent Technologies, Santa Clara, CA, USA). SmaI/XmaI intervals between 100 and 2200 bp were identified in the mm9 assembly excluding chromosome Y. The resulting 41 237 intervals were uploaded to Agilent eArray (https://earray.chem.agilent.com/earray/), and HD-ChIP Database Probes overlapping the intervals were identified allowing up to four probes per interval. A total of 95 386 probes covering 26 740 (65%) of the 100–2200 bp intervals and providing an average of 3.6 probes per interval were included in the array. Standard Agilent control probes (1565), along with 8121 probes overlapping 10–15 kb SmaI/XmaI intervals to act as further controls, completed the 2 × 105K array design. The average signal intensity within each SmaI/XmaI interval was calculated. An interval was considered a ‘hit’ if it showed a <0.8 or >1.25 change in all four microarrays (P< 0.05). We used a list of all SmaI/XmaI intervals (148 total) that did not change in methylation (0.9> or <1.1 ratio in all arrays) for control purposes.
Pyrosequencing
Bisulfite conversion was performed with the EZ DNA Methylation-Gold Kit (D5006, Zymo Research). The genes were amplified with traditional primer biotinylation. A quantitative bisulfite pyrosequencing protocol was used for all methylation analyses with the utilization of the Pyro Q CpG program (QIAGEN GmbH, QIAGEN Strasse 1. 40724 Hilden, Germany). Methylation measurements at both of the SmaI/XmaI sites of the gene-associated intervals were performed in the case of protein tyrosine phosphatase, non-receptor type 22 (Ptpn22) and peroxisome proliferator-activated receptor alpha (Ppara) (Supplementary Material, Table S5).
Whole genomic expression microarray
Four MD versus COD (two male and two female comparisons to eliminate gender bias) P90 offspring whole genomic expression microarray (Agilent Technologies: 4 × 44k Whole Mouse Genome microarray, Quick-Amp two color labeling kit) comparisons were performed (http://www.ncbi.nlm.nih.gov/geo/, accession number: GSE24354). Probe intensities for each transcript were averaged and MD versus CD offspring intensity ratios were calculated. Transcripts were considered a ‘hit’ if those showed a >1.45-fold change in at least two microarray comparisons out of the four, with the remaining arrays not contradicting the increase or decrease in expression (i.e.: 1 or above 1 in case at least two other >1.45; 1 or less than 1 in case at least two other <0.69).
Quantitative analysis of gene expression
Quantitative real-time PCR was performed using the following TaqMan gene expression assays: protein tyrosine phosphatase, non-receptor type 22, Ptpn22 (Mm 00501246_m1); beta actin: B-actin (Mm00607939_s1); CPN subunit 2: Cpn2 (Mm 01169716_m1) and peroxisome proliferator-activated receptor alpha, Ppara (Mm 00440939_m1). Expression changes were determined as described before (18).
DNA extraction for microbial studies
Mucosal scrapings were centrifuged at 14 000 rpm for 30 s and re-suspended in 500 μl RLT buffer (Qiagen) (with β- mercaptoethanol). Sterile 5 mm steel beads (Qiagen) and 500 μl sterile 0.1 mm glass beads (Scientific Industries, Inc., NY, USA) were added for complete bacterial lyses in a Qiagen TissueLyser (Qiagen), run at 30 Hz for 5 min. Samples were centrifuged briefly and 100 μl of 100% ethanol was added to a 100 μl aliquot of the sample supernatant. This mixture was added to a DNA spin column, and DNA recovery protocols were followed as instructed in the QIAamp DNA Mini Kit (Qiagen) starting at step 5 of the Tissue Protocol. DNA was eluted and diluted to a final concentration of 20 ng/μl.
Massively parallel bTEFAP
Bacterial tag-encoded FLX-Titanium amplicon pyrosequencing (bTEFAP) was performed as described previously (36). bTEFAP utilizes Titanium reagents and procedures, and a one-step PCR, mixture of Hot Start and HotStar high fidelity taq polymerases, and amplicons originating from the 27F region numbered in relation to E. coli rRNA. The bTEFAP procedures were performed at the Research and Testing Laboratory (Lubbock, TX, USA) (www.researchandtesting.com).
Bacterial diversity data analysis
All failed sequence reads, low-quality sequence ends and tags, and sequences shorter than 300 bp were removed, and any non-bacterial ribosome sequences and chimeras were depleted using custom software described previously (36) and the Black Box Chimera Check software B2C2 (described and freely available at http://www.researchandtesting.com/B2C2.html). This process provided 4303–8871 filtered sequences in the individual mucosal DNA samples, which were queried using a distributed BLASTn .NET algorithm (37) against a database of high-quality 16S rRNA bacterial sequences derived from NCBI. Database sequences were characterized as high-quality based upon similar criteria as described for RDP ver 9 (http://rdp.cme.msu.edu/) (38). Using a .NET and C# analysis pipeline, the resulting BLASTn outputs were validated using taxonomic distance methods, and compiled as described previously (36). Sequences with identity scores >97% (<3% divergence) were resolved at the species level, between 95 and 97% at the genus level, between 90 and 95% at the family level, between 85 and 90% at the order level, between 80 and 85% at the class level and below this to the phylum (77 and 80%). Statistical tests including principle component analysis using Unifrac-based distances, ANOVA with Tukey–Kramer post hoc and hierarchal clustering (Wards minimum variance clustering with Manhattan distances) to evaluate microbiome results were evaluated using NCSS 2007 (Kaysville, UT, USA).
Statistical and bioinformatic analysis
For the bioinformatic analysis of the microbiome data, please see the paragraphs above. Unpaired, two-tailed t-tests, correlation and odds ratio calculations were utilized in the group comparisons where statistical significance was declared at P< 0.05. Error bars represent standard error of the mean (SEM). Fatigo functional enrichment analysis (http://babelomics3.bioinfo.cipf.es/) was used to identify GO classifications significantly over- or underrepresented (adjusted P< 0.05 in this case) among the gene lists.
SUPPLEMENTARY MATERIAL
FUNDING
R.K. was supported by a young investigator joint award from the Crohn's and Colitis Foundation of America-Children's Digestive Health and Nutrition Foundation/North American Society of Pediatric Gastroenterology Hepatology and Nutrition (CCFA Ref #2426), and the Broad Medical Research Program, the Broad Foundation (IBD-0252). C.W.S. was supported by USDA/ARS CHRC, CRIS Project Grant (6250-51000-055). DNaseI mapping data were funded through NHGRI ENCODE grant U54HG004592 to John Stamatoyannopoulos, University of Washington.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Alfred Balasa for technical support and Russell Kratzer for manuscript preparation assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bernstein C.N. New insights into IBD epidemiology: are there any lessons for treatment. Dig. Dis. 2010;28:406–410. doi: 10.1159/000320394. [DOI] [PubMed] [Google Scholar]
- 2.Packey C.D., Sartor R.B. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J. Intern. Med. 2008;263:597–606. doi: 10.1111/j.1365-2796.2008.01962.x. [DOI] [PubMed] [Google Scholar]
- 3.Kellermayer R. ‘Omics' as the filtering gateway between environment and phenotype: the inflammatory bowel diseases example. Am. J. Med. Genet. A. 2010;152A:3022–3025. doi: 10.1002/ajmg.a.33726. [DOI] [PubMed] [Google Scholar]
- 4.Haller D. Nutrigenomics and IBD: the intestinal microbiota at the cross-road between inflammation and metabolism. J. Clin. Gastroenterol. 2010;44(Suppl. 1):S6–S9. doi: 10.1097/MCG.0b013e3181dd8b76. [DOI] [PubMed] [Google Scholar]
- 5.Thompson A.I., Lees C.W. Genetics of ulcerative colitis. Inflamm. Bowel Dis. 2010 doi: 10.1002/ibd.21375. Nov 12. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Halfvarson J., Bodin L., Tysk C., Lindberg E., Jarnerot G. Inflammatory bowel disease in a Swedish twin cohort: a long-term follow-up of concordance and clinical characteristics. Gastroenterology. 2003;124:1767–1773. doi: 10.1016/s0016-5085(03)00385-8. [DOI] [PubMed] [Google Scholar]
- 7.Wolffe A.P., Matzke M.A. Epigenetics: regulation through repression. Science. 1999;286:481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 8.Petronis A. Epigenetics and twins: three variations on the theme. Trends Genet. 2006;22:347–350. doi: 10.1016/j.tig.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Petronis A., Petroniene R. Epigenetics of inflammatory bowel disease. Gut. 2000;47:302–306. doi: 10.1136/gut.47.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waterland R.A., Michels K.B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 11.Waterland R.A., Jirtle R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolinoy D.C., Jirtle R.L. Environmental epigenomics in human health and disease. Environ. Mol. Mutagen. 2008;49:4–8. doi: 10.1002/em.20366. [DOI] [PubMed] [Google Scholar]
- 13.Wolff G.L., Kodell R.L., Moore S.R., Cooney C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12:949–957. [PubMed] [Google Scholar]
- 14.Stappenbeck T.S., Rioux J.D., Mizoguchi A., Saitoh T., Huett A., Darfeuille-Michaud A., Wileman T., Mizushima N., Carding S., Akira S., et al. Crohn disease: a current perspective on genetics, autophagy and immunity. Autophagy. 2010;7 doi: 10.4161/auto.7.4.13074. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albert E.J., Marshall J.S. Aging in the absence of TLR2 is associated with reduced IFN-gamma responses in the large intestine and increased severity of induced colitis. J. Leukoc. Biol. 2008;83:833–842. doi: 10.1189/jlb.0807557. [DOI] [PubMed] [Google Scholar]
- 16.Tao R., de Zoeten E.F., Ozkaynak E., Chen C., Wang L., Porrett P.M., Li B., Turka L.A., Olson E.N., Greene M.I., et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 17.Maslowski K.M., Vieira A.T., Ng A., Kranich J., Sierro F., Yu D., Schilter H.C., Rolph M.S., Mackay F., Artis D., et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kellermayer R., Balasa A., Zhang W., Lee S., Mirza S., Chakravarty A., Szigeti R., Laritsky E., Tatevian N., Smith C.W., et al. Epigenetic maturation in colonic mucosa continues beyond infancy in mice. Hum. Mol. Genet. 2010;19:2168–2176. doi: 10.1093/hmg/ddq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y.H., Rho Y.H., Choi S.J., Ji J.D., Song G.G., Nath S.K., Harley J.B. The PTPN22 C1858T functional polymorphism and autoimmune diseases—a meta-analysis. Rheumatology (Oxford) 2007;46:49–56. doi: 10.1093/rheumatology/kel170. [DOI] [PubMed] [Google Scholar]
- 20.Sommer H., Schweisfurth H., Schulz M. Serum angiotensin-I-converting enzyme and carboxypeptidase N in Crohn's disease and ulcerative colitis. Enzyme. 1986;35:181–188. doi: 10.1159/000469341. [DOI] [PubMed] [Google Scholar]
- 21.Knoch B., Barnett M.P., Cooney J., McNabb W.C., Barraclough D., Laing W., Zhu S., Park Z.A., Maclean P., Knowles S.O., et al. Molecular characterization of the onset and progression of colitis in inoculated interleukin-10 gene-deficient mice: a role for PPARalpha. PPAR Res. doi: 10.1155/2010/621069. doi:10.1155/2010/621069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turnbaugh P.J., Ridaura V.K., Faith J.J., Rey F.E., Knight R., Gordon J.I. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009;1 doi: 10.1126/scitranslmed.3000322. 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tannock G.W., Savage D.C. Influences of dietary and environmental stress on microbial populations in the murine gastrointestinal tract. Infect. Immun. 1974;9:591–598. doi: 10.1128/iai.9.3.591-598.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng T., Wang L., Schoeb T.R., Elson C.O., Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J. Exp. Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warner M.J., Ozanne S.E. Mechanisms involved in the developmental programming of adulthood disease. Biochem. J. 2010;427:333–347. doi: 10.1042/BJ20091861. [DOI] [PubMed] [Google Scholar]
- 26.Suter M.A., Aagaard-Tillery K.M. Environmental influences on epigenetic profiles. Semin. Reprod. Med. 2009;27:380–390. doi: 10.1055/s-0029-1237426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barreau F., Ferrier L., Fioramonti J., Bueno L. Neonatal maternal deprivation triggers long term alterations in colonic epithelial barrier and mucosal immunity in rats. Gut. 2004;53:501–506. doi: 10.1136/gut.2003.024174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barreau F., Salvador-Cartier C., Houdeau E., Bueno L., Fioramonti J. Long-term alterations of colonic nerve-mast cell interactions induced by neonatal maternal deprivation in rats. Gut. 2008;57:582–590. doi: 10.1136/gut.2007.126680. [DOI] [PubMed] [Google Scholar]
- 29.Innis S.M., Dai C., Wu X., Buchan A.M., Jacobson K. Perinatal lipid nutrition alters early intestinal development and programs the response to experimental colitis in young adult rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2010 doi: 10.1152/ajpgi.00258.2010. doi:10.1152/ajpgi.00258. [DOI] [PubMed] [Google Scholar]
- 30.Mueller-Ortiz S.L., Wang D., Morales J.E., Li L., Chang J.Y., Wetsel R.A. Targeted disruption of the gene encoding the murine small subunit of carboxypeptidase N (CPN1) causes susceptibility to C5a anaphylatoxin-mediated shock. J. Immunol. 2009;182:6533–6539. doi: 10.4049/jimmunol.0804207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Esposito E., Mazzon E., Paterniti I., Dal Toso R., Pressi G., Caminiti R., Cuzzocrea S. PPAR-alpha contributes to the anti-inflammatory activity of verbascoside in a model of inflammatory bowel disease in mice. PPAR Res. 2010 doi: 10.1155/2010/917312. doi:10.1155/2010/917312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Efuntoye M.O., Adetosoye A.I. Enterotoxigenicity and drug sensitivity of staphylococci from children aged five years and below with sporadic diarrhoea. East Afr. Med. J. 2003;80:656–659. doi: 10.4314/eamj.v80i12.8784. [DOI] [PubMed] [Google Scholar]
- 33.Willing B.P., Dicksved J., Halfvarson J., Andersson A.F., Lucio M., Zheng Z., Jarnerot G., Tysk C., Jansson J.K., Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010 doi: 10.1053/j.gastro.2010.08.049. doi:10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 34.Kitajima S., Takuma S., Morimoto M. Histological analysis of murine colitis induced by dextran sulfate sodium of different molecular weights. Exp. Anim. 2000;49:9–15. doi: 10.1538/expanim.49.9. [DOI] [PubMed] [Google Scholar]
- 35.Waterland R.A., Kellermayer R., Rached M.T., Tatevian N., Gomes M.V., Zhang J., Zhang L., Chakravarty A., Zhu W., Laritsky E., et al. Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum. Mol. Genet. 2009;18:3026–3038. doi: 10.1093/hmg/ddp241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey M.T., Walton J.C., Dowd S.E., Weil Z.M., Nelson R.J. Photoperiod modulates gut bacteria composition in male Siberian hamsters (Phodopus sungorus) Brain Behav. Immun. 2010;24:577–584. doi: 10.1016/j.bbi.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 37.Dowd S.E., Zaragoza J., Rodriguez J.R., Oliver M.J., Payton P.R. Windows .NET Network Distributed Basic Local Alignment Search Toolkit (W.ND-BLAST) BMC Bioinformatics. 2005;6:93. doi: 10.1186/1471-2105-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole J.R., Wang Q., Cardenas E., Fish J., Chai B., Farris R.J., Kulam-Syed-Mohideen A.S., McGarrell D.M., Marsh T., Garrity G.M., et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.