

Abstract
Fibroblast growth factor 2 (FGF2) consists of multiple protein isoforms (low molecular weight, LMW, and high molecular weight, HMW) produced by alternative translation from the Fgf2 gene. These protein isoforms are localized to different cellular compartments, indicating unique biological activity. FGF2 isoforms in the heart have distinct roles in many pathological circumstances in the heart including cardiac hypertrophy, ischemia–reperfusion injury, and atherosclerosis. These studies suggest distinct biological activities of FGF2 LMW and HMW isoforms both in vitro and in vivo. Yet, due to the limitations that only the recombinant FGF2 LMW isoform is readily available and that the FGF2 antibody is nonspecific with regards to its isoforms, much remains to be determined regarding the role(s) of the FGF2 LMW and HMW isoforms in cellular behavior and in cardiovascular development and pathophysiology. This review summarizes the activities of LMW and HMW isoforms of FGF2 in cardiovascular development and disease.
Keywords: fibroblast growth factor, low and high molecular weight isoforms, ischemia–reperfusion injury, cardiac hypertrophy, vascular biology, cardiovascular development, myocardial development, epicardium, epicardium-derived cells, heart valves, coronary vessels, smooth muscle cells, aortic arch, cardiac fibroblasts, transgenic and knockout mouse models
FIBROBLAST GROWTH FACTOR 2 PROTEIN ISOFORMS
Fibroblast growth factor 2 (FGF2) consists of multiple protein isoforms resulting from different translational start sites from a single Fgf2 gene (Fig. 1A). One 18-kDa FGF2 isoform, termed low molecular weight (LMW), is translated from a conventional Kozak AUG start codon and consists of 155 amino acids, representing the core sequence common to all FGF2 isoforms. Several high molecular weight (HMW) isoforms of FGF2 have been identified in many species, including human, rat, bovine, guinea pig, and chicken (Sommer et al., 1987; Doble et al., 1990; Powell and Klagsbrun, 1991; Dono and Zeller, 1994). These HMW isoforms are amino terminal extensions of the LMW isoform and use upstream in-frame CUG codons as alternative translational start sites. In mouse, there are two HMW isoforms (20.5 and 21 kDa while in human, there are four HMW isoforms (22, 22.5, 24, and 34 kDa). The 34-kDa FGF2 HMW isoform, originally identified in HeLa cells, is translated from a fifth initiation (CUG) codon (Arnaud et al., 1999) and, in contrast to the other FGF2 HMW isoforms, permits NIH 3T3 cell survival in low-serum conditions (Arese et al., 1999; Arnaud et al., 1999).
Fig. 1.
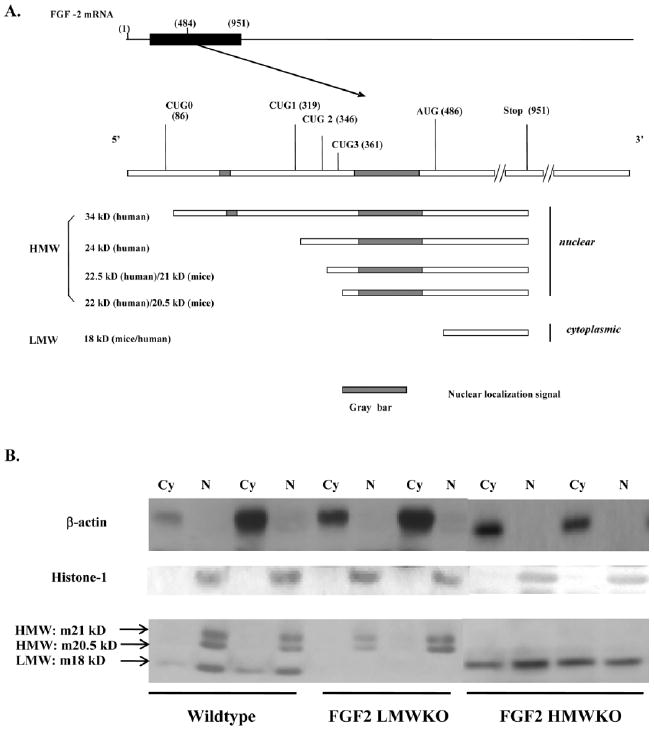
Low and high molecular weight isoforms of fibroblast growth factor 2 (FGF2). A: Schematic depicting the multiple FGF2 isoforms in mouse (m) or human (h) arising from different translational start sites. B: Representative Western blot of FGF2 isoform localization in nonischemic wild-type (Wt), FGF2 low molecular weight knockout (LMWKO) and FGF2 high molecular weight knockout (HMWKO) adult mouse hearts. In Wt hearts, the LMW, 18-kDa isoform was localized to the cytosolic (Cy) and nuclear (N) fractions of the heart; whereas, the HMW, 20.5 and 21 kDa, isoforms were nuclear localized. In the absence of the LMW isoform (FGF2 LMWKO), the HMW isoforms were only expressed in the nucleus. In the absence of the HMW isoforms (FGF2 HMWKO), the LMW isoform was localized in the cytoplasm and nucleus of the heart. β-actin is a cytosolic protein and used as a measure of cytosolic fraction enrichment. The cytosolic fraction depicts cytosolic FGF2 and extracellular FGF2. Histone-1 is a nuclear protein and used as a measure of nuclear fraction enrichment. n = 3 per group.
The three-dimensional structure of the LMW isoform of FGF2 has been determined by several groups (Eriksson et al., 1991; Zhu et al., 1991). The backbone of FGF2 can be described as a trigonal pyramidal structure with 12 antiparallel β-sheets. Helix-like structures have been identified at residues 131–136, the receptor-binding site, and residues 13–30, part of the heparan binding site (Feige and Baird, 1989; Moy et al., 1996). The human LMW FGF2 isoform contains two potential phosphorylation sites: serine 64, a protein kinase A (PKA) site, and threonine 112, a protein kinase C (PKC) site. The phosphorylation of FGF2 modifies the affinity of FGF2 to its receptor and assists in the release of FGF2 from the basement membrane (Plouet et al., 1988). Although FGF2 contains four cysteines, of which two are conserved within the FGF family, there are no intramolecular disulfide bonds (Thompson, 1992), indicating that the free cysteines will not form intermolecular disulfide bridges between two FGF2s. Two inverse sequence arginine-glycine-aspartate (RGD) sequences, proline-aspartate-glycine-arginine (PDGR) and glutamate-aspartate-glycine-arginine (EDGR) may be involved in the modulation of the mitogenic activity of the FGF2 LMW isoform, independent of FGF receptor activity (Presta et al., 1991). Currently, there is no evidence regarding the protein structure of FGF2 HMW isoforms. It is hypothesized that all the isoforms have the same protein structure because they all share the same structural residues. Whether this hypothesis is true remains to be determined.
SUBCELLULAR LOCALIZATION OF FGF2 LMW AND HMW ISOFORMS
The amino-terminal extensions of the HMW isoforms encoded by the Fgf2 gene contain several GR (Glu/Arg) repeats that act as nuclear localization sequences (NLS) for FGF2 (Bugler et al., 1991; Quarto et al., 1991; Dono et al., 1998a; Arese et al., 1999). The 34-kDa isoform contains an arginine-rich type of NLS in addition to the common NLS for other FGF2 HMW isoforms (Sorensen et al., 2006). This Arg-rich NLS is similar to that of the HIV type1 Rev (Regulator of Virion) protein, suggesting that the 34-kDa HMW isoform enters the nucleus by binding to the human nuclear import receptor, importin β (Sorensen et al., 2006). In contrast, the 18-kDa LMW isoform is mainly found in the cytoplasm and stored in the extracellular matrix (Renko et al., 1990).
Recent evidence indicates that the LMW and HMW isoforms are not always localized only to the cytoplasm and nucleus, respectively. The 18-kDa FGF2 isoform does contain a C-terminal NLS, consisting of Arg116 and Arg118, which is able to target itself or a fusion protein to the nucleus (Claus et al., 2003; Sheng et al., 2004). Like other polypeptides, such as insulin and interleukin-1, extracellular 18-kDa FGF2 can translocate into the nucleus after internalization (Bouche et al., 1994). This internalization, which can be abolished by heparanase treatment (Bouche et al., 1994), may be mediated by heparan sulfate proteoglycans (HSPGs). The endogenous 18-kDa FGF2 isoform can also directly translocate from endoplasmic reticulum (ER) to the nucleus (Choi et al., 2000). When LMW FGF2 remains in the nucleus, there is a slight stimulation of cell proliferation and a down-regulation of its receptor, FGFR1, suggesting that the intracellular biological activity of 18-kDa FGF2 is independent of its receptor signaling pathway. On the other hand, the HMW FGF2 isoforms can also be released from the cell through vesicle shedding (Taverna et al., 2003). Furthermore, there is speculation that HMW and LMW FGF2 isoforms reciprocally associate, either directly or indirectly, controlling each other’s biological activity in a concentration- and/or localization-dependent manner (Pintucci et al., 2005; Quarto et al., 2005).
The localization of the LMW and HMW isoforms of FGF2 in nonischemic wild-type, FGF2 LMW knockout (KO; only HMW isoforms present) and FGF2 HMWKO (only LMW isoform present) was identified in adult mouse hearts. Western immunoblot data showed that the HMW isoforms were localized only to the nucleus in Wt and LMWKO hearts; whereas, the LMW isoform was found in the cytoplasm (including extracellular) as well as in the nucleus in wild-type and FGF2 HMWKO hearts (Fig. 1). These results are consistent with those of other investigators, who demonstrated that the LMW isoform was present in both cytoplasm and nucleus, while HMW isoforms were localized only to the nucleus (Choi et al., 2000; Claus et al., 2003; Garmy-Susini et al., 2004; Sheng et al., 2004). The nuclear localization of the FGF2 LMW isoform may indicate that the FGF2 LMW isoform can elicit its biological activity not only through FGFR, but also by acting as a transcription factor or a co-factor in the nucleus to regulate gene expression under normal or stress conditions. Others have shown that, in adult cardiomyocytes, the FGF2 LMW isoform localizes with the external cell membrane surface, specialized intercellular junctions, and myofibril Z lines in the cytoplasm (Kardami et al., 1991a). Localization of FGF2 in the developing heart is shown in Figure 3 and discussed in “FGF2 ISOFORMS IN HEART VALVE DEVELOPMENT”, “FGF2 ISOFORMS IN EPICARDIAL, MYOCARDIAL, AND CORONARY VASCULAR DEVELOPMENT”, and “FGF2 ISOFORMS IN AORTIC ARCH ARTERY DEVELOPMENT” sections.
Fig. 3.
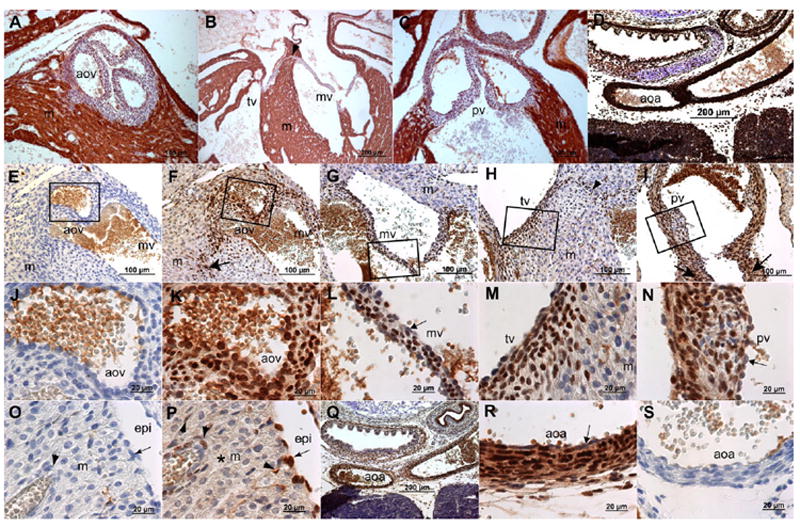
Immunohistochemical (IHC) localization of fibroblast growth factor 2 (FGF2) isoforms in wild-type fetal cardiovascular tissues at embryonic day (E) 18.5 day of gestation. A–C: Cardiac muscle actin (HHF-35) IHC. D: Smooth muscle (SM) actin (1A4) IHC. E,J,O,S: Rabbit IgG control antibody staining for the FGF2 IHC. F–I,K–N,P–R: FGF2 IHC in fetal cardiovascular tissues using a rabbit anti-FGF2 antibody. J–N: Magnified images from the boxes in E–I, respectively. IgG isotype control antibody staining reveals no significant background staining. Brown color in A–S indicates horse-radish peroxidase staining, and blue color shows nuclei stained with hematoxylin. Fibrous valve leaflets in A–C show marked absence of muscle actin, whereas ventricular myocardium is strongly stained with muscle actin marker. The walls of aortic arch are marked with significant SM actin expression (D). FGF2 is predominantly localized in the nucleus in mesenchymal cells of the aortic (F,K), mitral (G,L), tricuspid (H,M) and pulmonary (I, N) valves. Nuclear FGF2 staining in valve endothelium is reduced in comparison to the valvular mesenchyme (arrow in L,N). The fibrous continuity (arrowhead in B,H) of the arteriovenous septum and fibrous annulus of both aortic (arrow in F) and pulmonary (arrows in I) valves are also stained strongly with nuclear FGF2. The adjacent myocardium (F–H,M) exhibit predominantly cytoplasmic FGF2. Epicardium (arrow in P) exhibits strong nuclear FGF2 signal. Epicardium-derived cells (arrowheads in P) in ventricular myocardium and which surround the developing coronary vasculature are also predominantly stained with nuclear FGF2. The subcellular localization of FGF2 is predominantly cytoplasmic in the ventricular myocardium (asterisk in P). The wall of aortic arch shows significant FGF2 predominantly in the nucleus of SMC (Q,R,D), whereas FGF2 is not detectable in endothelium of the vascular wall of aortic arch (arrow in R). Scale bars (μm) are indicated in all micrographs. m, myocardium; aov, aortic valve; tv, tricuspid valve; mv, mitral valve; pv, pulmonary valves; aoa, aortic arch.
POTENTIAL MECHANISMS OF FGF2 RELEASE
FGF1 and FGF2 are the predominant FGF family members expressed in the heart (Kardami and Fandrich, 1989; Kardami et al., 1991b) and FGF receptor (FGFR)-1 is the predominant receptor in murine hearts; whereas, both FGFR1 and 4 are markedly expressed in human heart tissue, with barely detectable FGFR2 and 3 (Sugi et al., 1995; Hughes, 1997). The activation of FGFR requires the binding of its ligand, most likely the LMW isoform of FGF2 However, one of the most perplexing properties of FGF2 is its lack of a consensus signal peptide for secretion (Abraham et al., 1986; Jaye et al., 1986). Typically, secreted proteins contain N-terminal signal peptides, which direct them to the ER (Walter et al., 1984). Secretory vesicles then carry the protein from the ER to the Golgi and finally to the cell surface. There, the secretory vesicles fuse with the plasma membrane, releasing their contents into the extracellular milieu (Rothman and Wieland, 1996; Mellman and Warren, 2000). Numerous studies have shown that FGF2 gets “released” from the cell, associating with the basement membrane, HSPG and FGFR and eliciting biological activity in vitro (Kessler et al., 1976; Folkman et al., 1988; Cooper and Barondes, 1990; Thompson et al., 1990). In fact, inhibitors of ER-Golgi-mediated secretion have no effect on FGF2 release (Rothman and Wieland, 1996). The mechanism of how the FGF2 is released remains unclear. However, there are several proposed mechanisms that involve an unconventional pathway for FGF2 secretion. In the late 1980s, studies demonstrated that FGF2 could be released by cell injury induced by high doses of endotoxin or irradiation (Gajdusek and Carbon, 1989; Witte et al., 1989). An initial study by Ito and colleagues (McNeil and Ito, 1989) drew speculation that in situ occurrence of plasma membrane wounding (transient, survivable disruption) followed by resealing might reflect a novel route for FGF2 trafficking in and out of the cytoplasm. Additionally, the export of FGF2 may also likely rely on plasma membrane-resident transport (Schafer et al., 2004) by means of Na+/K+-ATPase (Florkiewicz et al., 1998) or HSPGs (Zehe et al., 2006). Florkiewicz and colleagues (1998) suggested that the α/β heterodimers of Na+/K+-ATPase may form a higher-ordered complex that could catalyze LMW FGF2 export, while the Zehe group (Zehe et al., 2006) proposed that the cell surface receptor, HSPG, could form a molecular trap, driving net export of LMW FGF2. Furthermore, Mignatti and colleagues (1992) have proposed that LMW FGF2 secretion involves intracellular vesicles formed from endocytic membrane or internal vesicle fusion with the plasma membrane, because lowering temperature or administering methylamine, both of which would be expected to inhibit secretory vesicle fusion, inhibited LMW FGF2 secretion and FGF2-induced migration of 3T3 cells. However, most studies of the localization or secretion of FGF2 LMW and HMW isoforms occurred in vitro, limiting their biological interpretation (Bugler et al., 1991; Quarto et al., 1991; Dono et al., 1998a; Arese et al., 1999; Taverna et al., 2003). Still, in vivo, FGF2 can be detected in the urine and pericardial fluid from patients with myocardial ischemia (Fujita et al., 1996; Cuevas et al., 1997a; Hasdai et al., 1997). Our unpublished data indicate that it is the LMW isoform, and not the HMW isoforms, that is released from the adult mouse heart during ischemia–reperfusion injury and which can act upon FGFR1 to elicit cardioprotection, indicating that this isoform promotes autocrine or paracrine activity. Evidence suggests that FGF1 can form a complex with S100A13, which may assist in the release of FGF1 out of cells (Landriscina et al., 2001; Prudovsky et al., 2002, 2003). Because FGF1 and FGF2 share 73% similarity (Ornitz and Itoh, 2001; Itoh and Ornitz, 2004, 2008), an interaction with S100A13 may also facilitate the movement of FGF2.
GENE TARGETED ABLATION OR OVEREXPRESSION OF FGF2 ISOFORMS
Because differential functions have been attributed to LMW and HMW isoforms of FGF2 in various aspects of the cardiovascular system, significant efforts have been made to study their in vivo and ex vivo roles in mammals (House et al., 2003; Kardami et al., 2007; Liao et al., 2007b). By using the Tag and Exchange gene targeting technique (Askew et al., 1993), we have replaced the Hprt minigene of the original Fgf2 KO allele (Fgf2−/−; Zhou et al., 1998) with a mutant exon1 containing a mutation in the ATG encoding the LMW translational start site (Fig. 2). This new allele has resulted in a LMW FGF2 knockout mouse (FGF2lmw-ko/lmw-ko; Garmy-Susini et al., 2004). Recently, we have used a similar strategy to generate a HMW FGF2 knockout mouse strain (FGF2hmw-ko/hmw-ko; unpublished observations). Because FGF2 isoforms are differentially expressed in various developing cardiovascular tissues (Fig. 3), these three mouse strains will provide new avenues to definitively determine the developmental and adult cardiovascular functions of the different FGF2 isoforms.
Fig. 2.
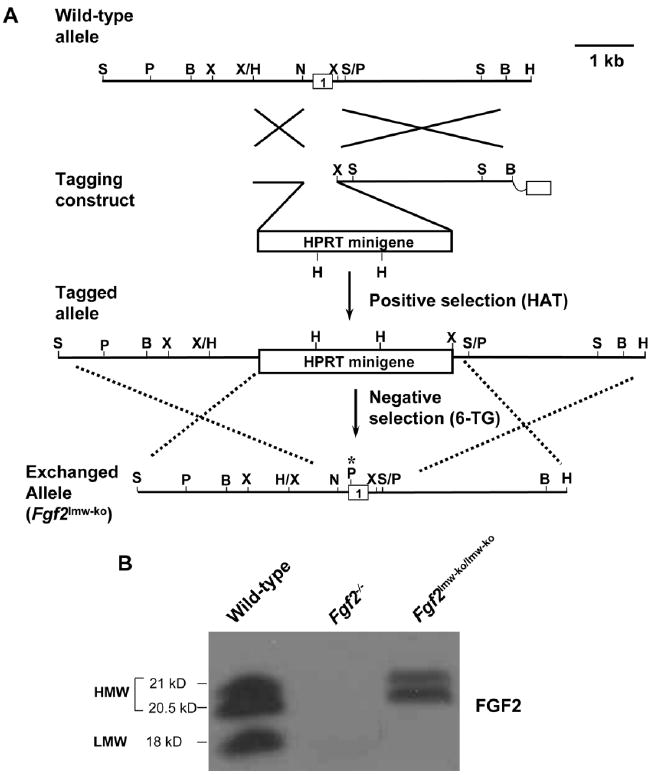
Gene targeting scheme for generating fibroblast growth factor 2 (FGF2) isoform knockout mice. A: Tag and Exchange gene targeting strategy for producing the Fgf2lmw-ko exchanged allele. The ATG site in the genomic DNA is mutated, which also results in a diagnostic PstI site. B: Representative Western blot showing the complete absence of all FGF2 isoforms in Fgf2−/− mice and of the low molecular weight (LMW) isoform in Fgf2lmw-ko/lmw-ko mice. B, BamHI; H, HindIII; N, NarI; P, PstI; S, SacI; Sm, SmaI; X, XbaI. Asterisk in the exchange construct and exchanged allele indicates the mutations and the resulting PstI site.
FGF2 isoform gain of function (transgenic overexpression) mouse models have also been developed by the Doetschman laboratory (Coffin et al., 1995; Davis et al., 1997) and others (Sheikh et al., 2001) to evaluate the functions of the low (LMW Tg) and high molecular weight (HMW Tg) protein isoforms of FGF2 in cardiovascular physiology and pathophysiology. Cardiovascular outcomes using LMW Tg and HMW Tg mice are next discussed in the contexts of ischemia–reperfusion injury, cardiac hypertrophy, and angiogenesis.
ROLE OF FGF2 ISOFORMS IN CARDIOVASCULAR DEVELOPMENT
Many studies suggest a major role for FGF2 in the developing and adult myocardium (Spirito et al., 1991; House et al., 2003; Pennisi et al., 2003; Kardami et al., 2007; Lavine and Ornitz, 2008). Fgf2−/− mice (129/Black-Swiss mixed genetic background with two renin genes) develop normally and have normal cardiac structure and mass. These mice exhibit decreased vascular tone, and display reduced cardiac hypertrophy in response to pressure overload (Zhou et al., 1998; Schultz et al., 1999). Of interest, other investigators have reported that Fgf2−/− mice on C57BL/6 (which contains one renin gene) develop dilated cardiomyopathy and that they exhibit an impaired hypertrophic response to angiotensin II (Pellieux et al., 2001). Taken together, these data indicate an important role of FGF2 in the maintenance of cardiovascular physiology in adult mice and raise the possibility that FGF2 HMW and LMW isoforms play an important role in cardiovascular development. Despite sufficient information on the function of FGF2 isoforms in adult cardiac remodeling, there has been no study that connects these different isoforms to myocardial growth and development in vivo.
Although in adult murine cardiovascular tissues, HMW FGF2 is found predominantly in the nucleus and LMW FGF2 is localized in the extracellular matrix and cytoplasm, the differential localization of FGF2 isoforms has not been demonstrated during cardiovascular development in mouse. Immunofluorescence studies in frozen adult bovine cardiac tissues have identified an FGF2-like reactivity in blood vessels, single (nonmuscle) connective tissue cells, and in nuclei and intercalated discs of muscle fibers (Kardami and Fandrich, 1989). Two studies have determined the developmental expression of FGF2 in the rat and chick cardiovascular system, but neither differentiated between FGF2 isoforms (Consigli and Joseph-Silverstein, 1991; Spirito et al., 1991). Consequently, it is important to revisit the subcellular distribution of LMW and HMW FGF2 isoforms in the cardiovascular system in the mouse using isoform-specific knockout strains.
FGF2 ISOFORMS IN HEART VALVE DEVELOPMENT
Endocardial cushions are the primordia of valves and septa and become mature structures through remodeling (embryonic day [E] 10.5–E18.5) and valve elongation and maturation (E14.5–1 week after birth; Person et al., 2005). Heart valve remodeling can be conceptually organized into multiple aspects, including mesenchymal expansion (E10.5–E12.5), differentiation (E12.5–E16.5) and condensation (E15.5–E18.5), all of which occur in an overlapping manner (Butcher and Markwald, 2007). FGF2 is implicated in congenital heart defects involving abnormal valvulogenesis in Noonan syndrome (Chen et al., 2000; Uhlen et al., 2006). It is used in various aspects of tissue valve engineering as a growth factor (Narine et al., 2006) and to stimulate growth and inhibit apoptosis of endocardial cushions in in vitro culture studies (Choy et al., 1996; Zhao and Rivkees, 2000). We have used a standard immunohistochemical procedure (LSAB+ kit, Dako, CA) to localize FGF2 in embryonic heart valves with a rabbit polyclonal FGF2 antibody (Liao et al., 2007b) that nonspecifically detects both LMW and HMW FGF2 (Fig. 3E–N). The data indicate that most valve mesenchyme in arteriovenous (mitral and tricuspid) and outflow tract (aortic and pulmonary) valves, that is not stained with a cardiac muscle actin marker antibody (Fig. 3A–C), exhibits intense and predominantly nuclear staining with FGF2 antibody (Fig. 3E–N). Additionally, there is no detectable staining of FGF2 in valve endothelium (arrow in Fig. 3L,N). These data are consistent with the known in vitro function of FGF2 in heart valve development (Zhao and Rivkees, 2000; Uhlen et al., 2006) and suggest that an intracrine FGF2 signaling plays an important role in developing heart valves.
FGF2 ISOFORMS IN EPICARDIAL, MYOCARDIAL, AND CORONARY VASCULAR DEVELOPMENT
Epicardium contributes a significant and critical proportion of nonmyocardial cells during heart development (Kruithof et al., 2006; Lie-Venema et al., 2007; Lavine and Ornitz, 2008). The epicardium is derived from the proepicardial organ and covers the heart by E10.5. Both myocardial proliferation and development and coronary vascular development occur concurrently during midgestation (E11.5–E16.5). Epicardial epithelial–mesenchymal transformation (EMT) gives rise to the epicardium-derived cells (EPDC) that form the subepicardial mesenchyme and subsequently migrate into the myocardium and differentiate into smooth muscle cells (SMC) and cardiac fibroblasts (Olivey et al., 2004). EPDC also contribute to the atrioventricular valves, cardiac fibroblasts and to the coronary vasculature and are required for the proper development of ventricular myocardium (Lie-Venema et al., 2007). FGF signaling is considered critical in epicardial and coronary vascular development (Lavine and Ornitz, 2008), and FGF2 positively regulates epicardial EMT in collagen gel assays of avian hearts (Morabito et al., 2001). Consistently, application of FGF2 supplemented beads results in enhanced coronary vasculature (Merki et al., 2005) and myocardial cell proliferation (Lavine et al., 2005). Also, it has been reported that an FGF-induced imbalance in myocardial cell proliferation at early stages of heart development results in cardiovascular anomalies during late embryogenesis (Franciosi et al., 2000). Furthermore, FGF2 inhibits apoptosis in cultured ventricular myocardium (Zhao and Rivkees, 2000). Epicardium is also required for the maintenance of the correct amount of myocyte proliferation in the compact myocardium through FGF2 expression levels in the myocardium of the embryonic chick heart (Pennisi et al., 2003).
Although FGF2 expression is found in chick proepicardium (Kruithof et al., 2006), epicardium and compact myocardium (Consigli and Joseph-Silverstein, 1991; Spirito et al., 1991; Pennisi et al., 2003; Merki et al., 2005), the subcellular distribution of different isoforms of FGF2 in epicardium, myocardium and coronary vasculature in fetal hearts has been either missing or poorly described in the published literature, due in part to the unavailability of FGF2 isoform-specific antibodies. Still, to begin to address the issue of subcellular localization, we have used an FGF2 antibody, that identifies FGF2 when present and confirms the absence of FGF2 in Fgf2−/− mice (Fig. 2B), to determine the nuclear and/or cytoplasmic distribution of FGF2 in the epicardium, myocardium, and coronary vessels in wild-type fetal hearts at E18.5 of gestation. The data have demonstrated that FGF2 is predominantly found in the nucleus in epicardium (Fig. 3O,P, arrow). In compact myocardium FGF2 is predominantly present in the cytoplasm (Fig. 3P, asterisk). Moreover, the media of coronary vessels and EPDC that are present in the ventricular myocardium both exhibit nuclear staining for FGF2 (Fig. 3P, arrowheads). These data suggest an intracrine function of FGF2 in epicardium, EPDC, and coronary vascular development, and indicate a requirement for paracrine or autocrine FGF2 signaling in myocardial proliferation and differentiation. These data are consistent with previous information on the function of FGF2 in epicardial, myocardial and coronary vascular development. However, it will be essential to confirm the identity of these differentially localized isoforms in continued studies using FGF2 isoform-specific knockout mouse models.
FGF2 ISOFORMS IN AORTIC ARCH ARTERY DEVELOPMENT
Exogenous recombinant FGF2 treatment enhances angiogenesis and arteriogenesis in animal models of peripheral arterial occlusion (Baffour et al., 1992; Bush et al., 1998). Increased expression of FGF2 is associated with growing and newly formed microvascular segments in or around ischemic tissues (Walgenbach et al., 1995; Bush et al., 1998). Therefore, FGF2 is implicated in vascular growth and remodeling. Studies from Fgf2−/− mice have pointed out an important role of FGF2 in controlling the vascular tone in adult mice (Zhou et al., 1998). In addition, intracrine and autocrine function of overexpressed FGF2 protein isoforms in vascular SMC has been described in mice (Davis et al., 1997). Nuclear expression of FGF2 in blood vessels has been previously described in adult bovine cardiovascular tissues (Kardami and Fandrich, 1989). However, immunohistochemical localization in developing rat hearts has revealed cytoplasmic staining of FGF2 in SMC of the vascular wall of the aorta (Spirito et al., 1991). Collectively, although these studies suggest that FGF2 is important for development of the aortic arch artery system, a clear understanding of the subcellular distribution of LMW and HMW FGF2 has yet to emerge. Recent studies done in our laboratory have found that nuclear FGF2 predominates in SMC of the vascular walls of the aortic arch in E18.5 embryos (Fig. 3Q–S). Furthermore, this nuclear staining becomes less intense in vascular endothelium (Fig. 3R, arrow). Overall, these data are in concordance with the known function of FGF2 in adult vascular SMC in various experimental models, and the predominantly nuclear FGF2 localization observed suggests important functions of HMW FGF2 in development and remodeling of the great vessels.
IMPORTANCE OF FGF2 LMW AND HMW ISOFORMS IN ISCHEMIA–REPERFUSION INJURY AND CARDIOPROTECTION
Myocardial ischemia–reperfusion (I-R) injury represents a major clinical problem associated with cardiac dysfunction, arrhythmias, and irreversible cardiomyocyte damage (Dhalla et al., 2000). There is significant experimental evidence that growth factors, particularly fibroblast growth factors (FGF), elicit a long-term cardioprotective effect through their angiogenic properties (Frelin et al., 2000). Evidence also suggests that growth factors, including FGF2, can acutely protect the heart from I-R injury independent of their vascular actions (Padua et al., 1995; Cuevas et al., 1997b; Wang et al., 2000; House et al., 2003; Molin and Post, 2007; Nishijima et al., 2007). FGF2 protects the heart from I-R injury as demonstrated by the improvement in postischemic cardiac function and reduction in myocardial infarction (MI; Unger et al., 1994; Padua et al., 1995, 1998; Hasdai et al., 1997; Horrigan et al., 1999; Cuevas et al., 2000; Dhalla et al., 2000; Sheikh et al., 2001; House et al., 2003), both indices of cardioprotection. Currently, the roles of the LMW and HMW FGF2 isoforms in I-R injury and cardioprotection remain to be elucidated. Understanding the biological function(s) of the individual FGF2 isoforms in cardioprotection is of great clinical importance and may lead to the development of novel pharmacologic or gene therapy strategies for ischemic heart disease.
There is evidence that the LMW isoform of FGF2 participates in cardiac (Unger et al., 1994; Padua et al., 1995, 1998; Hasdai et al., 1997; Horrigan et al., 1999; Cuevas et al., 2000; Sheikh et al., 2001; Liao et al., 2007b), renal (Villanueva et al., 2006), intestinal (Fu et al., 2003), and cerebral (Jiang et al., 1996; Cheung et al., 2000; Zhang et al., 2005) I-R injury. These actions of the FGF2 LMW isoform may involve, but do not require, angiogenic activity. In a canine model of permanent coronary artery occlusion, intracoronary injection of recombinant LMW FGF2 reduces infarct size, which is associated with an increase in myocardial capillary density a week after infarction (Horrigan et al., 1999). Exogenous treatment with LMW isoform increases capillary density and blood flow in the ischemic myocardium by means of growth of new collateral vessels (Unger et al., 1994) and induces nephrogenic proteins after acute ischemic renal failure (Villanueva et al., 2006). In the brain, exogenous administration of FGF2 LMW isoform alleviates brain injury following global ischemia and reperfusion by down-regulating the expression of inflammatory factors and inhibiting their activities (Zhang et al., 2005). In the isolated adult rat or mouse heart model, recombinant rat FGF2 LMW isoform given by retrograde perfusion protects the heart from subsequent I-R–induced contractile dysfunction and myocardial cell damage and preserves cardiac energy metabolism (Padua et al., 1995). Also, addition of the FGF2 LMW isoform protects cultured neonatal cardiomyocytes from H2O2-induced cell injury and death (Cuevas et al., 1997a; Padua et al., 1998; Sheikh et al., 2001). Mouse models overexpressing the rat FGF2 LMW isoform (Sheikh et al., 2001) or lacking (Liao et al., 2007b) the murine LMW isoform demonstrate its importance in cardioprotection against I-R injury. Our laboratory demonstrated that the FGF2 LMW isoform is beneficial in protecting the heart from myocardial dysfunction, while the FGF2 HMW isoforms have a deleterious role with regard to protecting the heart from myocardial dysfunction after global, low-flow I-R injury (Liao et al., 2007a,b; unpublished data, Fig. 4). Yet, both LMW and HMW FGF2 isoforms are necessary in protecting the heart from myocardial cell injury/death (Liao et al., 2007a,b; and unpublished data). These actions of the FGF2 isoforms in I-R injury are independent of the angiogenic activity of this growth factor (Liao et al., 2007a,b; and unpublished data).
Fig. 4.
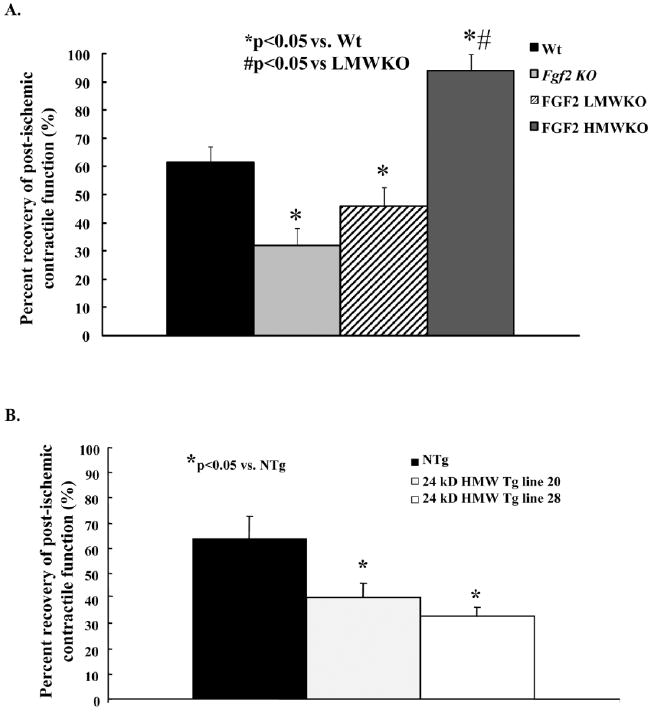
Percent recovery of postischemic contractile function in wild-type (Wt, black bar), Fgf2 knockout (KO; gray bar), fibroblast growth factor 2 (FGF2) low molecular weight (LMW) KO (striped bar), FGF2 high molecular weight (HMW) KO (dark gray bar), and NTg (black bar) and 24-kDa Tg (line 20: light gray bar, line 28: white bar) hearts following 60 min global low-flow ischemia and 120 min reperfusion. Recovery of cardiac function was calculated using +dP/dt at 120 min reperfusion as a percent of its baseline measure. A: There was a significant decrease in postischemic recovery of contractile function in Fgf2 KO and FGF2 LMW KO hearts; whereas, an increase in recovery of postischemic contractile function was observed in FGF2 HMW KO hearts compared with Wt hearts. B: There was a significant decrease in recovery of postischemic contractility in 24-kDa HMW Tg (line 20 and line 28) hearts compared with NTg hearts. n = 5 for Wt and FGF2 HMWKO hearts, n = 6 for Fgf2 KO and FGF2 LMW KO hearts, n = 7 for NTg hearts, n = 5 for 24 kDa HMW Tg line 20 hearts, and n = 6 for 24-kDa HMW Tg line 28 hearts. *P < 0.05 vs. Wt hearts.
Reperfusion of the ischemic myocardium, while necessary for restored function, also adds risk because it is associated with exacerbation of cell injury and death (Gross and Auchampach, 2007). Factors that can reduce damage during development of an MI and during reperfusion will attract much attention clinically (Garcia-Dorado et al., 2006; Hausenloy and Yellon, 2006). An in vivo study showed that the FGF2 LMW isoform, injected into the left ventricle of rats after coronary occlusion, exerts significant protection from tissue loss and contractile dysfunction (Jiang et al., 2002). Furthermore, an ex vivo study also demonstrated that perfusion with the recombinant rat FGF2 LMW isoform during reperfusion results in significantly improved contractile recovery and reduced apoptotic cell death (Jiang et al., 2004).
Several proposed mechanisms may lead to the protective effect of FGF2 LMW isoform in I-R injury. It is speculated that FGF2 LMW isoform-mediated cardioprotection requires activation of FGFR1. For example, Jiang and colleagues (Jiang et al., 2002) showed that the cardioprotective effect of exogenous treatment of recombinant rat LMW FGF2 to rat hearts during I-R injury involved FGFR1 activation. Our unpublished data indicate that the LMW isoform is released from the heart during I-R injury and elicits protection by interacting with FGFR1. In our study, an FGFR inhibitor (PD173074) caused a significant decrease in postischemic cardiac function in both wild-type and FGF2 HMW KO (presence of only the LMW isoform) mice. The role of human myocardial FGFR4 in I-R injury and cardioprotection remains to be elucidated.
There are three major signal transduction pathways of FGF, including PLC/PKC, Ras-Raf-MEK-MAP kinase, and PI3K/Akt, which are initiated upon FGFR activation (Friesel and Maciag, 1995; Bikfalvi et al., 1997; Padua et al., 1998; House et al., 2007). Overexpression of the rat FGF2 LMW isoform increases membraneassociated PKCα and cytosol-associated PKC∊ (Sheikh et al., 2001), which would be expected to elicit cardioprotection. Our laboratory has shown that FGF2-induced cardioprotection (by means of cardiac-specific overexpression of the human Fgf2 gene) is mediated by a PKC-dependent pathway and that the PKC and MAPK signaling cascades are integrally connected downstream of FGF2 (House et al., 2007). Chelerythrine blocks the protective effect when exogenous recombinant rat FGF2 LMW isoform is administrated to isolated hearts (Padua et al., 1998) or cardiomyocytes (Jiang et al., 2002). Findings from our laboratory also demonstrate that FGF2 elicits its cardioprotective effect against postischemic cardiac dysfunction and myocardial infarction by activating ERK and inactivating p38 (House et al., 2005). Particularly, the ERK pathway is involved in FGF2 LMW isoform-mediated cardioprotection by acting as an upstream activator of PKC (Sheikh et al., 2001). Furthermore, evidence from our lab also indicates that endogenous ablation of the FGF2 LMW isoform results in a poorer recovery of cardiac function after ischemia–reperfusion injury and the protective effect mediated by LMW isoform of FGF2 involves inactivation of the MKK7/JNK/c-Jun pathway (Liao et al., 2007b). In addition, exogenous administration of the recombinant rat FGF2 LMW isoform causes a hyper-phosphorylation of connexin (Cx)-43 in vitro (Doble et al., 1996, 2004) and in the adult perfused heart (Srisakuldee et al., 2006) by means of the PKC pathway. Altered localization of Cx-43 from gap junctions to mitochondria and overall reduction in Cx-43 levels are common features of ischemic heart disease (Smith et al., 1991; Peters and Wit, 2000). FGF2 LMW isoform causes a decrease in mitochondrial coupling by phosphorylation of Cx-43 (Doble et al., 1996). Administration of recombinant rat or human FGF2 LMW isoform during reperfusion can also activate several intracellular signals (PKC, ERK, Akt), which are expected to mediate its beneficial effects (Horrigan et al., 1999).
No direct evidence suggesting the involvement of FGF2 HMW isoforms in I-R injury was available until recently, when the Kardami group (Jiang et al., 2007) showed that intramyocardial injection of the recombinant rat 23-kDa FGF2 HMW isoform caused an improvement in cardiac function and a decrease in myocardial infarct size 24 hr after MI; however, 7–8 weeks after MI, the improvement in cardiac function and decrease in myocardial infarct size disappeared, possibly due to the postischemic hypertrophic effect of FGF2 HMW isoforms. Yet, the endogenous role of the FGF2 HMW isoforms in I-R injury has not been elucidated. Data from our lab indicate that mice lacking the endogenous FGF2 HMW isoforms had a significant improvement in postischemic cardiac function after I-R injury (unpublished data, Fig. 4). Conversely, we show that in mice overexpressing the 24-kDa human FGF2 HMW isoform, postischemic cardiac function was significantly decreased after I-R injury (unpublished data, Fig. 4). During cardiac I-R, the LMW isoform was detected in coronary effluent, while the HMW isoforms were not released from the heart. Overall, our evidence indicates that FGF2 HMW isoforms have a deleterious effect in cardiac I-R injury. Furthermore, recent evidence suggests that FGF2HMWmay be cytotoxic by increasing cytochrome C release and levels of Bax, a pro-apoptotic protein, in human embryonic kidney cells (Ma et al., 2007).
Coronary artery disease is the leading cause of death in the US and other industrialized countries (Association, 2007). Recent studies suggest the possibility of a novel therapeutic approach to protect the heart from I-R injury that relies on stimulation of collateral blood vessel growth by means of FGF2 (Sellke et al., 1998; Laham et al., 1999; Rajanayagam et al., 2000; Udelson et al., 2000; Unger et al., 2000; Post et al., 2001; Simons et al., 2002). Current clinical trials use recombinant human FGF2 (LMW isoform only) in the treatment of coronary artery disease (Sellke et al., 1998; Laham et al., 1999, 2000; Udelson et al., 2000; Simons et al., 2002). These studies are evaluating the utility of the angiogenic properties of FGF2 for treating patients with coronary heart disease. Most of the completed studies focused on testing the safety and efficacy of FGF2 in patients with coronary artery disease (Sellke et al., 1998; Laham et al., 1999, 2000; Udelson et al., 2000). With the treatment, patients in the FGF2 group had no angina 3 months after bypass surgery, while the placebo group had recurrent angina (Sellke et al., 1998; Laham et al., 1999). These effects are dose-dependent compared with the placebo-controlled group (Laham et al., 1999). Further studies show that the delivery of FGF2 results in attenuation of stress-induced ischemia and an improvement in resting myocardial perfusion injury up to 180 days after treatment (Udelson et al., 2000), but with a dose-dependent hypertensive effect (Udelson et al., 2000). However, a recent study failed to show improvement of cardiac function after exercise (Simons et al., 2002), indicating inconsistent efficacy and potential side effects of FGF2. The chronic angiogenic effect of FGF2 is a feasible therapy for patients with coronary heart disease and this treatment modality could even be used in patients at risk for coronary artery disease. However, when a patient has an acute ischemic event, the angiogenic effect of FGF2 is not immediate. Therefore, the nonangiogenic cardioprotective actions of FGF2 are also crucial for its potential therapeutic value in patients with acute ischemic heart disease.
FGF2 ISOFORMS IN CARDIAC HYPERTROPHY
The role of FGF2 in the hypertrophic response in the heart has been well established, since it was first noted that FGF2 may up-regulate proteins corresponding to those induced by increased hemodynamic load (Parker et al., 1990). In a rat model of hypertrophy using aortic ligation, both FGF2 and FGFR-1 were found to be up-regulated (Hellman et al., 2008). Similarly, a mouse model of pressure-overload hypertrophy showed an increase in Fgf2 mRNA (Spruill et al., 2008). This up-regulation of FGF2 has also been confirmed in humans with studies examining mRNA levels in patients with ventricular hypertrophy (He et al., 2005). The dependence of pressure-induced hypertrophy on FGF2 has been confirmed in our laboratory, which showed that a mouse model lacking the Fgf2 gene, subjected to pressure overload by means of aortic coarctation, had a reduced hypertrophic response (Schultz et al., 1999). In addition to pressure-induced hypertrophy, biochemical signals associated with hypertrophy also increase FGF2 production, with higher levels of FGF2 protein and mRNA found in rats injected with a high dose of isoproterenol (Padua and Kardami, 1993). Additionally, angiotensin II fails to induce compensatory hypertrophy in mice lacking FGF2 (Pellieux et al., 2001).
Many of these investigations, while confirming FGF2’s role in hypertrophy, do not distinguish the roles of individual isoforms of FGF2. In vitro studies of neonatal cardiomyocytes show that overexpression of both LMW and HMW FGF2 isoforms increase cell proliferation, but overexpression of only the FGF2 HMW isoforms cause binucleation independent of FGFR pathways, possibly by directly affecting chromatin structure (Pasumarthi et al., 1996; Sun et al., 2001). The expression of FGF2 LMW isoform is up-regulated in hearts subjected to hemodynamic stress and undergoing hypertrophy (Kardami et al., 2004; House, 2005). Administration of the recombinant human FGF2 LMW isoform to ventricular myocytes causes hypertrophy in the absence of mechanical stress (Kaye et al., 1996) and reactivation of fetal forms of contractile proteins (Parker et al., 1990). Data also indicate that HMW FGF2 overexpression in NIH-3T3 cell line can stimulate the production of IL-6 cytokines (Delrieu et al., 1998), which can mediate a hypertrophic response (Molkentin and Dorn, 2001), with a corresponding down-regulation of IL-6 by LMW FGF2 administration. Furthermore, evidence by Kardami and colleagues (Kardami et al., 2004) suggests that cardiac hypertrophy is only associated with HMW FGF2, and not the 18-kDa LMW isoform, as administration of HMW FGF2 alone results in a 40% increase in cell size of cultured rat myocytes. Additionally, the Kardami group (Jiang et al., 2007) found that subjecting rat hearts that have undergone left coronary artery ligation to exogenous HMW and LMW FGF2 demonstrates that high, but not low, molecular weight isoforms of FGF2 are responsible for hypertrophic remodeling after MI. In contrast, LMW FGF2 administration did not correspond to an increase in cellular protein synthesis or left ventricular (LV) mass/body mass ratio, but did contribute to an increase in small vessel density in hearts after MI. This study also investigated potential pathways for HMW FGF2-enhanced cardiac hypertrophy, examining prohypertrophic cytokines of the IL-6 family. It was found that cardiotrophin-1 and gp130, both of which have been implicated in the hypertrophic response, are found in higher levels in all compartments of the heart when HMW FGF2 is administered (Aoyama et al., 2000; Freed et al., 2003). Our data (unpublished) are consistent with those of the Kardami group, such that no spontaneous cardiac hypertrophy is observed in FGF2 HMW KO hearts (only the endogenous LMW FGF2 isoform is present). Interestingly, our studies (unpublished) using nonischemic 24-kDa HMW Tg hearts demonstrate no spontaneous cardiac growth, indicating that neither the murine FGF2 LMW isoform nor the human FGF2 24-kDa HMW isoform caused spontaneous hypertrophy in our mouse models. This inconsistency in the effect of FGF2 HMW isoforms on cardiac hypertrophy may be due to differences in the models. In the studies by Kardami and investigators (Kardami et al., 2004; Jiang et al., 2007), the FGF2 HMW isoforms were given exogenously in the short-term (days) to cardiomyocytes or whole heart, while in our mouse models, the human 24-kDa FGF2 HMW isoform was overexpressed chronically from birth. This observed difference may be due to either the timeframe when the measurements are taken or the difference between exogenous treatment and endogenous activity. Because the HMW FGF2 isoforms and the FGF2 LMW isoform bind to heparan with similar affinity, it is possible that the exogenously administered FGF2 HMW isoforms can act by means of a paracrine/autocrine mode of action with the FGFR, while the FGF2 HMW isoforms overexpressed endogenously work solely in an intracrine manner. These findings may also indicate that other growth factors absent from the in vitro system, such as TGFβ (Schultz et al., 2002; Azhar et al., 2003), may mediate the hypertrophic response in vivo, or that hypertrophy is not triggered in the myocardium until an ischemic or hypoxic event activates growth factor release and hypertrophic signaling (Speir et al., 1992). The stimulus by which HMW FGF2 induces hypertrophy is not yet clear. In addition, the precise mechanism by which FGF2 HMW isoforms are able to stimulate up-regulation of pro-hypertrophic cytokines is assumed to be intracrine, due to the nuclear localization of the isoforms, but has yet to be determined.
ROLES OF FGF2 ISOFORMS IN VASCULAR ALTERATION AND ANGIOGENESIS
The process of angiogenesis, in which new blood vessels form from preexisting vessels by a process of proliferation and migration of endothelial and vascular smooth muscle cells, holds promise as a means of revascularizing the diseased and infarcted heart in a noninvasive manner (Yanagisawa-Miwa et al., 1992; Unger et al., 1994). Because FGF2 is a known activator of mesoderm migration (Montesano et al., 1986) and angiogenesis has been implicated as a major exacerbating factor in human cancer, widespread investigation into the role of FGF2 and its isoforms in this process has already taken place.
An early in vitro study of human FGF2 isoform overexpression in cultured aortic endothelial cells began to delineate the different roles of LMW FGF2 and HMW FGF2 in angiogenesis or vasculogenesis (Davis et al., 1997). While all FGF2 isoforms increased DNA synthesis, the nuclear FGF2 (presumed HMW) isoforms did so to a greater extent, suggesting their role in endothelial proliferation. Although the predominantly cytosolic, secreted, LMW isoform was responsible for increased endothelial cell growth, intracrine-acting (HMW) isoforms were able to amplify the proliferative effects of other growth factors, including PDGF and vascular endothelial growth factor. The Levin group (Piotrowicz et al., 2001) expanded on these findings, observing that the various N-terminal extensions found exclusively in FGF2 HMW isoforms inhibit migration, while the domains common to all FGF2 isoforms stimulate growth. This finding was confirmed in vivo, where expression of a truncated form of HMW (human 24 kDa) FGF2 consisting only of its 86 N-terminal amino acids inhibited tumor growth before vascular infiltration as well as tumor vascularization itself, but not chemotaxis of immune cells (Levin et al., 2004). Additionally, the occasional appearance of LMW FGF2 in the nucleus was traced to a C-terminal sequence that was postulated to interact with the remaining, nonmethylated, N-terminus of that isoform (Foletti et al., 2003). Thus, all FGF2 isoforms became candidate modulators of transcriptional activity.
To gain insight into which components of angiogenesis are promoted by the different FGF2 isoforms, DNA microarray technology has been used to examine changes in gene transcription instigated by expression of either LMW or HMW FGF2 in clones of an endothelial cell line (Quarto et al., 2005). LMW FGF2, as expected, increased transcript levels of VEGF-independent angiogenic markers RPS5 and ANGPT1-4. However, HMW FGF2 increased transcription of growth arrest marker NF1X and tumor suppressor ST5, while decreasing proliferative markers PCNA and Egr1 and ribosomal activity marker MAPK6. The results of the study suggested that, at least in basal conditions, the intracrine effects of HMW FGF2 may suppress angiogenic signals until being overwhelmed by dominant, receptor-mediated, autocrine/ paracrine LMW FGF2 proliferative signals.
Studies of FGF2 signaling induced by estrogen provide significant insight into the roles of FGF2 isoforms in the migratory and proliferative components of angiogenesis. Estradiol (E2) was previously known to stimulate vascular MAPK signaling in vitro by means of an FGF2 autocrine loop (Kim-Schulze et al., 1998). Using cultured endothelial cells obtained from the Fgf2 KO (all FGF2 isoforms ablated) and the LMW FGF2 isoform KO (only HMW isoforms present) mouse models developed by the Doetschman group, the Arnal group (Garmy-Susini et al., 2004) showed that up-regulation of intact FGF2 HMW isoforms by E2 promotes cell migration in culture regardless of LMW FGF2 expression. On the other hand, DNA synthesis could not be induced by E2, but was induced by exogenous recombinant LMW FGF2 administration, in both Fgf2 KO and LMW KO cells, suggesting that LMW FGF2 is required for E2-dependent proliferation observed in wild-type endothelial cells. Signaling through the estrogen α receptor accomplished a switch in predominant FGF2 protein isoform expression from LMW to HMW, concurrent with a 2.5-fold decrease in FGF2 mRNA expression, perhaps by increasing reliance on 3′ untranslated region elements for translation at upstream CUG sites. Estrogen also increased expression of high-affinity FGFR1IIIc receptors whose subsequent loss did not affect cell migration, suggesting the possibility of an altered response to extracellular LMW FGF2. A previously unknown FGF2-interacting factor was also identified in the study, which is up-regulated by estrogen receptor signaling and required for the nuclear effects of the remaining FGF2 HMW isoforms in LMW KO mice. Overall, these results are consistent with a requirement for all protein isoforms of FGF2 in estrogen-dependent angiogenesis.
Conflicting evidence regarding the synergistic and antagonistic actions of LMW and HMW FGF2 isoforms raised interest in the spatiotemporal profile of the angiogenic actions of FGF2. Furthermore, previous models did not directly account for the context of tissue injury, such as that encountered during atherosclerosis and MI. Thus, an in vivo model of perivascular electric injury was used in conjunction with reciprocal bone marrow transplantation involving Fgf2 KO (all isoforms ablated) and wild-type mice. The Arnal group (Fontaine et al., 2006) determined that Fgf2 gene expression was required in bone marrow, but not in the remainder of the animal, for vascular repair. Furthermore, they found that estrogen signaling increased expression of all FGF2 protein isoforms in the bone marrow, but only HMW FGF2 in aortic-derived endothelial cells cultured from the mice. These findings suggest that FGF2 expression by endothelial progenitor cells from the bone marrow are responsible for formation of new vessels, while FGF2 in the damaged or stressed existing vessel may promote recruitment of the progenitors. Additionally, the Pintucci group (Yu et al., 2008) demonstrated cleavage of HMW FGF2 by thrombin to a LMW-like form with a short N-terminal extension, ELF-2. The stimulation of endothelial cell migration and proliferation by ELF-2 suggests a possible FGF2 HMW isoform-mediated recruitment mechanism.
Findings regarding FGF2 isoform action in the context of cancer may also provide insight for developing effective methods to induce reparative angiogenic activity on a background of cardiovascular injury. In estrogen-responsive pituitary tumors, vascular channels increased in number and diameter after 20 days of treatment with estrogen, commensurate with LMW FGF2 expression and localization of FGF2 to the mitochondria and euchromatin in the pituitary lactotrophs and gonadotrophs (Mukdsi et al., 2006). The proposed mechanism of FGF2 in this model, including cell communication loops involving Src kinase, Ras-p21, MAPK-p44/42, and cFos that maintain the lactotroph phenotype, may also be involved in communication between endothelial and vascular smooth muscle cells. A recent investigation, in which human melanoma cell lines transfected with vectors encoding only LMW FGF2 or all FGF2 isoforms were used as tumor grafts, showed increased density and smaller diameter of tumor vasculature attributable to LMW FGF2 in one cell line (Fontijn et al., 2007). However, some HMW isoform expression was permitted in the LMW-transfected clones. The differences between the cell lines may a reveal dependence of FGF2 isoform actions on the overall gene expression profile, including levels of other growth factors, which could correlate to regional physiological differences in vascular expression of FGF2 isoforms and their angiogenic effects.
PERSPECTIVES ON FGF2 ISOFORMS IN THE CARDIOVASCULAR SYSTEM
Several investigators have independently produced Fgf2 knockout mice (Zhou, 1997; Dono et al., 1998b; Ortega et al., 1998). These mice have revealed important roles of FGF2 in cardiac hypertrophy (Schultz et al., 1999), I-R injury and myocardial infarction (House et al., 2003; Kardami et al., 2007), neuronal regeneration (Vaccarino et al., 1999), bone development and remodeling (Coffin et al., 1995; Montero et al., 2000), and vascular remodeling and vascular tone control (Zhou et al., 1998). FGF2 has the potential to be a powerful molecular therapeutic tool in the treatment of cardiovascular disease. Although FGF2 has been a candidate for treatment of ischemic heart disease for some time, clinical trials using FGF2 for gene therapy have been disappointing (Simons et al., 2002; Syed et al., 2004). The discovery of multiple isoforms of this growth factor has opened the possibility of therapeutic strategies that take advantage of the individual actions of each isoform to treat various cardiovascular diseases with regional or end-organ effects in mind. Exercise and statin treatments are regularly used in the management of cardiovascular disease, particularly in atherosclerosis. The proposal of the Arnal group (Fontaine et al., 2006), that the estrogen signaling-based induction of FGF2 may apply to other stressors or mitigants of stress, should be explored. Particular attention should be paid to addressing the roles that individual FGF2 isoforms may play in each context. Clearly, a better understanding of isoform-specific function of FGF2 in cardiovascular development and adult physiology will provide useful information that would be helpful in further advancing the field and realizing its clinical benefits.
This review provides evidence that the FGF2 LMW isoform and HMW isoforms have opposing roles in I-R injury and cardioprotection, which may be a reason that several ongoing clinical trials do not observe a sustained improvement in cardiac function (Sellke et al., 1998; Laham et al., 1999, 2000; Udelson et al., 2000; Simons et al., 2002). Most of the studies ignore the presence of the endogenous FGF2 HMW isoforms, which may counteract the beneficial role of the administered FGF2 LMW isoform. Also, a significant limiting factor to clinical trials on the safety and efficacy of FGF2 has been the inability to stably deliver any HMW isoform directly to patients. The development of noncleavable human recombinant HMW FGF2 with isoform specificity and nuclear delivery could provide some insight into the role of this isoform in cardiovascular protection. Additionally, further study of the thrombin-cleaved form of HMW FGF2, ELF-2, could help to delineate the dynamic effects of FGF2 isoforms. The apparent paradox of context-dependent synergy and antagonism between LMW and HMW FGF2 could also be clarified by exploring both acute and chronic changes in gene expression upon introduction of FGF2 isoform overexpression, particularly with regard to the presence and absence of the nuclear FGF2 interacting factor. If a therapy can harness the beneficial effects of both isoforms, it will produce greater cardioprotection. Furthermore, the inhibitor of FGFR (PD173074), which demonstrated that FGFR1 is necessary for LMW-induced postischemic recovery of cardiac function, is currently being evaluated in a clinical trial for cancer treatment (Sessa et al., 2006). However, potential actions of the inhibitor in the cardiovascular system, particularly abrogation of the cardioprotective effects of FGF2, needs to be taken into consideration when considering its usage in cancer treatment. Similarly, it is necessary to consider and account for the cardiovascular effects of both FGF2 LMW and HMW isoforms to develop effective therapies for treating ischemic heart disease.
Finally, development of novel mouse models with targeted ablation and transgenic overexpression of LMW and HMW isoforms of FGF2 have provided new avenues for determining the in vivo developmental and physiological function of these isoforms in the cardiovascular and other systems. Overexpression of human FGF2 isoforms on a murine Fgf2 KO background, as previously proposed (Davis et al., 1997), would likely yield great insight into isoform function without the mitigating factor of endogenous expression. Inducible and/or knockin models may represent direct approaches to delineating and differentiating acute and chronic FGF2 isoform effects.
Acknowledgments
We thank all past and present members of Doetschman and Schultz laboratories for sharing ideas and technical support. T.D. and J.J.S. were funded by the NIH/NHLBI R01 and J.J.S. was funded by the American Heart Association and the Pharmaceutical Research and Manufacturers of America.
Grant sponsor: NIH/NHLBI R01; Grant number: HL070174; Grant number: HL075633; Grant sponsor: American Heart Association; Grant number: SDG 23004N; Grant sponsor: Pharmaceutical Research and Manufacturers of America (Research Starter Grant)
References
- Abraham JA, Whang JL, Tumolo A, Mergia A, Fiddes JC. Human basic fibroblast growth factor: nucleotide sequence, genomic organization, and expression in mammalian cells. Cold Spring Harb Symp Quant Biol. 1986;51(pt 1):657–668. doi: 10.1101/sqb.1986.051.01.078. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- Arese M, Chen Y, Florkiewicz RZ, Gualandris A, Shen B, Rifkin DB. Nuclear activities of basic fibroblast growth factor: potentiation of low-serum growth mediated by natural or chimeric nuclear localization signals. Mol Biol Cell. 1999;10:1429–1444. doi: 10.1091/mbc.10.5.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud E, Touriol C, Boutonnet C, Gensac MC, Vagner S, Prats H, Prats AC. A new 34-kilodalton isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol Cell Biol. 1999;19:505–514. doi: 10.1128/mcb.19.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew DS, Bartholomew C, Ihle JN. Insertional mutagenesis and the transformation of hematopoietic stem cells. Hematol Pathol. 1993;7:1–22. [PubMed] [Google Scholar]
- Association AH 2007. American Heart Association. Dallas TX: American Heart Association; 2007. Heart and stroke statistical update. [Google Scholar]
- Azhar M, Schultz Jel J, Grupp I, Dorn GW, Jr, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffour R, Berman J, Garb JL, Rhee SW, Kaufman J, Friedmann P. Enhanced angiogenesis and growth of collaterals by in vivo administration of recombinant basic fibroblast growth factor in a rabbit model of acute lower limb ischemia: dose-response effect of basic fibroblast growth factor. J Vasc Surg. 1992;16:181–191. [PubMed] [Google Scholar]
- Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- Bouche G, Baldin V, Belenguer P, Prats H, Amalric F. Activation of rDNA transcription by FGF-2: key role of protein kinase CKII. Cell Mol Biol Res. 1994;40:547–554. [PubMed] [Google Scholar]
- Bugler B, Amalric F, Prats H. Alternative initiation of translation determines cytoplasmic or nuclear localization of basic fibroblast growth factor. Mol Cell Biol. 1991;11:573–577. doi: 10.1128/mcb.11.1.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush RL, Pevec WC, Ndoye A, Cheung AT, Sasse J, Pearson DN. Regulation of new blood vessel growth into ischemic skeletal muscle. J Vasc Surg. 1998;28:919–928. doi: 10.1016/s0741-5214(98)70070-9. [DOI] [PubMed] [Google Scholar]
- Butcher JT, Markwald RR. Valvulogenesis: the moving target. Philos Trans R Soc Lond B Biol Sci. 2007;362:1489–1503. doi: 10.1098/rstb.2007.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Bronson RT, Klaman LD, Hampton TG, Wang JF, Green PJ, Magnuson T, Douglas PS, Morgan JP, Neel BG. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet. 2000;24:296–299. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- Cheung WM, Chen SF, Nian GM, Lin TN. Induction of angiogenesis related genes in the contralateral cortex with a rat three-vessel occlusion model. Chin J Physiol. 2000;43:119–124. [PubMed] [Google Scholar]
- Choi J, Ko MK, Kay EP. Subcellular localization of the expressed 18 kDa FGF-2 isoform in corneal endothelial cells. Mol Vis. 2000;6:222–231. [PubMed] [Google Scholar]
- Choy M, Oltjen SL, Otani YS, Armstrong MT, Armstrong PB. Fibroblast growth factor-2 stimulates embryonic cardiac mesenchymal cell proliferation. Dev Dyn. 1996;206:193–200. doi: 10.1002/(SICI)1097-0177(199606)206:2<193::AID-AJA8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Claus P, Doring F, Gringel S, Muller-Ostermeyer F, Fuhlrott J, Kraft T, Grothe C. Differential intranuclear localization of fibroblast growth factor-2 isoforms and specific interaction with the survival of motoneuron protein. J Biol Chem. 2003;278:479–485. doi: 10.1074/jbc.M206056200. [DOI] [PubMed] [Google Scholar]
- Coffin JD, Florkiewicz R, Neumann J, Mort-Hopkins T, Dorn GW, Jr, Lightfoot P, German R, Howles PN, Kier A, O’Toole BA, Doetschman T. Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol Biol Cell. 1995;6:1861–1873. doi: 10.1091/mbc.6.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consigli SA, Joseph-Silverstein J. Immunolocalization of basic fibroblast growth factor during chicken cardiac development. J Cell Physiol. 1991;146:379–385. doi: 10.1002/jcp.1041460307. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Barondes SH. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J Cell Biol. 1990;110:1681–1691. doi: 10.1083/jcb.110.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas P, Barrios V, Gimenez-Gallego G, Martinez-Coso V, Cuevas B, Benavides J, Garcia-Segovia J, Asin-Cardiel E. Serum levels of basic fibroblast growth factor in acute myocardial infarction. Eur J Med Res. 1997a;2:282–284. [PubMed] [Google Scholar]
- Cuevas P, Reimers D, Carceller F, Martinez-Coso V, Redondo-Horcajo M, Saenz de Tejada I, Gimenez-Gallego G. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res. 1997b;2:465–468. [PubMed] [Google Scholar]
- Cuevas P, Carceller F, Martinez-Coso V, Asin-Cardiel E, Gimenez-Gallego G. Fibroblast growth factor cardioprotection against ischemia-reperfusion injury may involve K+ ATP channels. Eur J Med Res. 2000;5:145–149. [PubMed] [Google Scholar]
- Davis MG, Zhou M, Ali S, Coffin JD, Doetschman T, Dorn GW., Jr Intracrine and autocrine effects of basic fibroblast growth factor in vascular smooth muscle cells. J Mol Cell Cardiol. 1997;29:1061–1072. doi: 10.1006/jmcc.1997.0383. [DOI] [PubMed] [Google Scholar]
- Delrieu I, Arnaud E, Ferjoux G, Bayard F, Faye JC. Overexpression of the FGF-2 24-kDa isoform up-regulates IL-6 transcription in NIH-3T3 cells. FEBS Lett. 1998;436:17–22. doi: 10.1016/s0014-5793(98)01086-2. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res. 2000;47:446–456. doi: 10.1016/s0008-6363(00)00078-x. [DOI] [PubMed] [Google Scholar]
- Doble BW, Fandrich RR, Liu L, Padua RR, Kardami E. Calcium protects pituitary basic fibroblast growth factors from limited proteolysis by co-purifying proteases. Biochem Biophys Res Commun. 1990;173:1116–1122. doi: 10.1016/s0006-291x(05)80901-5. [DOI] [PubMed] [Google Scholar]
- Doble BW, Chen Y, Bosc DG, Litchfield DW, Kardami E. Fibroblast growth factor-2 decreases metabolic coupling and stimulates phosphorylation as well as masking of connexin43 epitopes in cardiac myocytes. Circ Res. 1996;79:647–658. doi: 10.1161/01.res.79.4.647. [DOI] [PubMed] [Google Scholar]
- Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- Dono R, Zeller R. Cell-type-specific nuclear translocation of fibroblast growth factor-2 isoforms during chicken kidney and limb morphogenesis. Dev Biol. 1994;163:316–330. doi: 10.1006/dbio.1994.1151. [DOI] [PubMed] [Google Scholar]
- Dono R, James D, Zeller R. A GRmotif functions in nuclear accumulation of the large FGF-2 isoforms and interferes with mitogenic signalling. Oncogene. 1998a;16:2151–2158. doi: 10.1038/sj.onc.1201746. [DOI] [PubMed] [Google Scholar]
- Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998b;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson AE, Cousens LS, Weaver LH, Matthews BW. Three-dimensional structure of human basic fibroblast growth factor. Proc Natl Acad Sci U S A. 1991;88:3441–3445. doi: 10.1073/pnas.88.8.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige JJ, Baird A. Basic fibroblast growth factor is a substrate for protein phosphorylation and is phosphorylated by capillary endothelial cells in culture. Proc Natl Acad Sci U S A. 1989;86:3174–3178. doi: 10.1073/pnas.86.9.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkiewicz RZ, Anchin J, Baird A. The inhibition of fibroblast growth factor-2 export by cardenolides implies a novel function for the catalytic subunit of Na+,K+-ATPase. J Biol Chem. 1998;273:544–551. doi: 10.1074/jbc.273.1.544. [DOI] [PubMed] [Google Scholar]
- Foletti A, Vuadens F, Beermann F. Nuclear localization of mouse fibroblast growth factor 2 requires N-terminal and C-terminal sequences. Cell Mol Life Sci. 2003;60:2254–2265. doi: 10.1007/s00018-003-3258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavsky I. A heparin-binding angiogenic protein—basic fibroblast growth factor—is stored within basement membrane. Am J Pathol. 1988;130:393–400. [PMC free article] [PubMed] [Google Scholar]
- Fontaine V, Filipe C, Werner N, Gourdy P, Billon A, Garmy-Susini B, Brouchet L, Bayard F, Prats H, Doetschman T, Nickenig G, Arnal JF. Essential role of bone marrow fibroblast growth factor-2 in the effect of estradiol on reendothelialization and endothelial progenitor cell mobilization. Am J Pathol. 2006;169:1855–1862. doi: 10.2353/ajpath.2006.060260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontijn D, Duyndam MC, Belien JA, Gallegoz Ruiz MI, Pinedo HM, Boven E. The 18 kDa isoform of basic fibroblast growth factor is sufficient to stimulate human melanoma growth and angiogenesis. Melanoma Res. 2007;17:155–168. doi: 10.1097/CMR.0b013e328184451e. [DOI] [PubMed] [Google Scholar]
- Franciosi JP, Bolender DL, Lough J, Kolesari GL. FGF-2-induced imbalance in early embryonic heart cell proliferation: a potential cause of late cardiovascular anomalies. Teratology. 2000;62:189–194. doi: 10.1002/1096-9926(200010)62:4<189::AID-TERA4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Freed DH, Moon MC, Borowiec AM, Jones SC, Zahradka P, Dixon IM. Cardiotrophin-1: expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol Cell Biochem. 2003;254:247–256. doi: 10.1023/a:1027332504861. [DOI] [PubMed] [Google Scholar]
- Frelin C, Ladoux A, D’Angelo G. Vascular endothelial growth factors and angiogenesis. Ann Endocrinol (Paris) 2000;61:70–74. [PubMed] [Google Scholar]
- Friesel RE, Maciag T. Molecular mechanisms of angiogenesis: fibroblast growth factor signal transduction. FASEB J. 1995;9:919–925. doi: 10.1096/fasebj.9.10.7542215. [DOI] [PubMed] [Google Scholar]
- Fu XB, Yang YH, Sun TZ, Chen W, Li JY, Sheng ZY. Rapid mitogen-activated protein kinase by basic fibroblast growth factor in rat intestine after ischemia/reperfusion injury. World J Gastroenterol. 2003;9:1312–1317. doi: 10.3748/wjg.v9.i6.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Ikemoto M, Kishishita M, Otani H, Nohara R, Tanaka T, Tamaki S, Yamazato A, Sasayama S. Elevated basic fibroblast growth factor in pericardial fluid of patients with unstable angina. Circulation. 1996;94:610–613. doi: 10.1161/01.cir.94.4.610. [DOI] [PubMed] [Google Scholar]
- Gajdusek CM, Carbon S. Injury-induced release of basic fibroblast growth factor from bovine aortic endothelium. J Cell Physiol. 1989;139:570–579. doi: 10.1002/jcp.1041390317. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M, Inserte J, Agullo L, Cabestrero A. The end-effectors of preconditioning protection against myocardial cell death secondary to ischemiareperfusion. Cardiovasc Res. 2006;70:274–285. doi: 10.1016/j.cardiores.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Garmy-Susini B, Delmas E, Gourdy P, Zhou M, Bossard C, Bugler B, Bayard F, Krust A, Prats AC, Doetschman T, Prats H, Arnal JF. Role of fibroblast growth factor-2 isoforms in the effect of estradiol on endothelial cell migration and proliferation. Circ Res. 2004;94:1301–1309. doi: 10.1161/01.RES.0000127719.13255.81. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Auchampach JA. Reperfusion injury: does it exist? J Mol Cell Cardiol. 2007;42:12–18. doi: 10.1016/j.yjmcc.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasdai D, Barak V, Leibovitz E, Herz I, Sclarovsky S, Eldar M, Scheinowitz M. Serum basic fibroblast growth factor levels in patients with ischemic heart disease. Int J Cardiol. 1997;59:133–138. doi: 10.1016/s0167-5273(97)02921-5. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- He ZY, Feng B, Yang SL, Luo HL. Intracardiac basic fibroblast growth factor and transforming growth factor-beta 1 mRNA and their proteins expression level in patients with pressure or volume-overload right or left ventricular hypertrophy. Acta Cardiol. 2005;60:21–25. doi: 10.2143/AC.60.1.2005044. [DOI] [PubMed] [Google Scholar]
- Hellman U, Hellstrom M, Morner S, Engstrom-Laurent A, Aberg AM, Oliviero P, Samuel JL, Waldenstrom A. Parallel up-regulation of FGF-2 and hyaluronan during development of cardiac hypertrophy in rat. Cell Tissue Res. 2008;332:49–56. doi: 10.1007/s00441-007-0562-8. [DOI] [PubMed] [Google Scholar]
- Horrigan MC, Malycky JL, Ellis SG, Topol EJ, Nicolini FA. Reduction in myocardial infarct size by basic fibroblast growth factor following coronary occlusion in a canine model. Int J Cardiol. 1999;68(suppl 1):S85–S91. doi: 10.1016/s0167-5273(98)00296-4. [DOI] [PubMed] [Google Scholar]
- House SL. Pharmacology and cell biophysics. Cincinnati: University of Cincinnati; 2005. Role of fibroblast growth factor 2 in cardiac ischemia-reperfusion injury and cardiac hypertrophy. [Google Scholar]
- House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz Jel J. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation. 2003;108:3140–3148. doi: 10.1161/01.CIR.0000105723.91637.1C. [DOI] [PubMed] [Google Scholar]
- House SL, Branch K, Newman G, Doetschman T, Schultz Jel J. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am J Physiol Heart Circ Physiol. 2005;289:H2167–H2175. doi: 10.1152/ajpheart.00392.2005. [DOI] [PubMed] [Google Scholar]
- House SL, Melhorn SJ, Newman G, Doetschman T, Schultz Jel J. The protein kinase C pathway mediates cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2. Am J Physiol Heart Circ Physiol. 2007;293:H354–H365. doi: 10.1152/ajpheart.00804.2006. [DOI] [PubMed] [Google Scholar]
- Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem. 1997;45:1005–1019. doi: 10.1177/002215549704500710. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. 2008;237:18–27. doi: 10.1002/dvdy.21388. [DOI] [PubMed] [Google Scholar]
- Jaye M, Howk R, Burgess W, Ricca GA, Chiu IM, Ravera MW, O’Brien SJ, Modi WS, Maciag T, Drohan WN. Human endothelial cell growth factor: cloning, nucleotide sequence, and chromosome localization. Science. 1986;233:541–545. doi: 10.1126/science.3523756. [DOI] [PubMed] [Google Scholar]
- Jiang N, Finklestein SP, Do T, Caday CG, Charette M, Chopp M. Delayed intravenous administration of basic fibroblast growth factor (bFGF) reduces infarct volume in a model of focal cerebral ischemia/reperfusion in the rat. J Neurol Sci. 1996;139:173–179. [PubMed] [Google Scholar]
- Jiang ZS, Padua RR, Ju H, Doble BW, Jin Y, Hao J, Cattini PA, Dixon IM, Kardami E. Acute protection of ischemic heart by FGF-2: involvement of FGF-2 receptors and protein kinase C. Am J Physiol Heart Circ Physiol. 2002;282:H1071–H1080. doi: 10.1152/ajpheart.00290.2001. [DOI] [PubMed] [Google Scholar]
- Jiang ZS, Srisakuldee W, Soulet F, Bouche G, Kardami E. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc Res. 2004;62:154–166. doi: 10.1016/j.cardiores.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Jiang ZS, Jeyaraman M, Wen GB, Fandrich RR, Dixon IM, Cattini PA, Kardami E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J Mol Cell Cardiol. 2007;42:222–233. doi: 10.1016/j.yjmcc.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Kardami E, Fandrich RR. Basic fibroblast growth factor in atria and ventricles of the vertebrate heart. J Cell Biol. 1989;109:1865–1875. doi: 10.1083/jcb.109.4.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardami E, Liu L, Doble BW. Basic fibroblast growth factor in cultured cardiac myocytes. Ann N Y Acad Sci. 1991a;638:244–255. doi: 10.1111/j.1749-6632.1991.tb49035.x. [DOI] [PubMed] [Google Scholar]
- Kardami E, Stoski RM, Doble BW, Yamamoto T, Hertzberg EL, Nagy JI. Biochemical and ultrastructural evidence for the association of basic fibroblast growth factor with cardiac gap junctions. J Biol Chem. 1991b;266:19551–19557. [PubMed] [Google Scholar]
- Kardami E, Jiang ZS, Jimenez SK, Hirst CJ, Sheikh F, Zahradka P, Cattini PA. Fibroblast growth factor 2 isoforms and cardiac hypertrophy. Cardiovasc Res. 2004;63:458–466. doi: 10.1016/j.cardiores.2004.04.024. [DOI] [PubMed] [Google Scholar]
- Kardami E, Detillieux K, Ma X, Jiang Z, Santiago JJ, Jimenez SK, Cattini PA. Fibroblast growth factor-2 and cardioprotection. Heart Fail Rev. 2007;12:267–277. doi: 10.1007/s10741-007-9027-0. [DOI] [PubMed] [Google Scholar]
- Kaye D, Pimental D, Prasad S, Maki T, Berger HJ, McNeil PL, Smith TW, Kelly RA. Role of transiently altered sarcolemmal membrane permeability and basic fibroblast growth factor release in the hypertrophic response of adult rat ventricular myocytes to increased mechanical activity in vitro. J Clin Invest. 1996;97:281–291. doi: 10.1172/JCI118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler DA, Langer RS, Pless NA, Folkman J. Mast cells and tumor angiogenesis. Int J Cancer. 1976;18:703–709. doi: 10.1002/ijc.2910180520. [DOI] [PubMed] [Google Scholar]
- Kim-Schulze S, Lowe WL Jr, Schnaper HW. Estrogen stimulates delayed mitogen-activated protein kinase activity in human endothelial cells via an autocrine loop that involves basic fibroblast growth factor. Circulation. 1998;98:413–421. doi: 10.1161/01.cir.98.5.413. [DOI] [PubMed] [Google Scholar]
- Kruithof BP, van Wijk B, Somi S, Kruithof-de Julio M, Perez Pomares JM, Weesie F, Wessels A, Moorman AF, van den Hoff MJ. BMP and FGF regulate the differentiation of multipotential pericardial mesoderm into the myocardial or epicardial lineage. Dev Biol. 2006;295:507–522. doi: 10.1016/j.ydbio.2006.03.033. [DOI] [PubMed] [Google Scholar]
- Laham RJ, Sellke FW, Edelman ER, Pearlman JD, Ware JA, Brown DL, Gold JP, Simons M. Local perivascular delivery of basic fibroblast growth factor in patients undergoing coronary bypass surgery: results of a phase I randomized, double-blind, placebo-controlled trial. Circulation. 1999;100:1865–1871. doi: 10.1161/01.cir.100.18.1865. [DOI] [PubMed] [Google Scholar]
- Laham RJ, Post M, Sellke FW, Simons M. Therapeutic Angiogenesis Using Local Perivascular and Pericardial Delivery. Curr Interv Cardiol Rep. 2000;2:213–217. [PubMed] [Google Scholar]
- Landriscina M, Soldi R, Bagala C, Micucci I, Bellum S, Tarantini F, Prudovsky I, Maciag T. S100A13 participates in the release of fibroblast growth factor 1 in response to heat shock in vitro. J Biol Chem. 2001;276:22544–22552. doi: 10.1074/jbc.M100546200. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Ornitz DM. Fibroblast growth factors and Hedgehogs: at the heart of the epicardial signaling center. Trends Genet. 2008;24:33–40. doi: 10.1016/j.tig.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Levin EG, Sikora L, Ding L, Rao SP, Sriramarao P. Suppression of tumor growth and angiogenesis in vivo by a truncated form of 24-kd fibroblast growth factor (FGF)-2. Am J Pathol. 2004;164:1183–1190. doi: 10.1016/S0002-9440(10)63206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Newman G, Doetschman T, Schultz Jel J. Role of high molecular weight isoforms of fibroblast growth factor-2 (FGF-2) in cardiac ischemia-reperfusion injury (I/R) (abstract) J Mol Cell Cardiol. 2007a;42:S187. [Google Scholar]
- Liao S, Porter D, Scott A, Newman G, Doetschman T, Schultz Jel J. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: the role of JNK signaling. J Mol Cell Cardiol. 2007b;42:106–120. doi: 10.1016/j.yjmcc.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, Hoeben RC, deRuiter MC, Poelmann RE, Gittenberger-de Groot AC. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorld-Journal. 2007;7:1777–1798. doi: 10.1100/tsw.2007.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Dang X, Claus P, Hirst C, Fandrich RR, Jin Y, Grothe C, Kirshenbaum LA, Cattini PA, Kardami E. Chromatin compaction and cell death by high molecular weight FGF-2 depend on its nuclear localization, intracrine ERK activation, and engagement of mitochondria. J Cell Physiol. 2007;213:690–698. doi: 10.1002/jcp.21139. [DOI] [PubMed] [Google Scholar]
- McNeil PL, Ito S. Gastrointestinal cell plasma membrane wounding and resealing in vivo. Gastroenterology. 1989;96:1238–1248. doi: 10.1016/s0016-5085(89)80010-1. [DOI] [PubMed] [Google Scholar]
- Mellman I, Warren G. The road taken: past and future foundations of membrane traffic. Cell. 2000;100:99–112. doi: 10.1016/s0092-8674(00)81687-6. [DOI] [PubMed] [Google Scholar]
- Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, Burch J, Kubalak SW, Kaliman P, Belmonte JC, Chien KR, Ruiz-Lozano P. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci U S A. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignatti P, Morimoto T, Rifkin DB. Basic fibroblast growth factor, a protein devoid of secretory signal sequence, is released by cells via a pathway independent of the endoplasmic reticulum-Golgi complex. J Cell Physiol. 1992;151:81–93. doi: 10.1002/jcp.1041510113. [DOI] [PubMed] [Google Scholar]
- Molin D, Post MJ. Therapeutic angiogenesis in the heart: protect and serve. Curr Opin Pharmacol. 2007;7:158–163. doi: 10.1016/j.coph.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Dorn IG., Jr Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, Doetschman T, Coffin JD, Hurley MM. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093. doi: 10.1172/JCI8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano R, Vassalli JD, Baird A, Guillemin R, Orci L. Basic fibroblast growth factor induces angiogenesis in vitro. Proc Natl Acad Sci U S A. 1986;83:7297–7301. doi: 10.1073/pnas.83.19.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morabito CJ, Dettman RW, Kattan J, Collier JM, Bristow J. Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev Biol. 2001;234:204–215. doi: 10.1006/dbio.2001.0254. [DOI] [PubMed] [Google Scholar]
- Moy FJ, Seddon AP, Bohlen P, Powers R. High-resolution solution structure of basic fibroblast growth factor determined by multidimensional heteronuclear magnetic resonance spectroscopy. Biochemistry. 1996;35:13552–13561. doi: 10.1021/bi961260p. [DOI] [PubMed] [Google Scholar]
- Mukdsi JH, De Paul AL, Petiti JP, Gutierrez S, Aoki A, Torres AI. Pattern of FGF-2 isoform expression correlated with its biological action in experimental prolactinomas. Acta Neuropathol. 2006;112:491–501. doi: 10.1007/s00401-006-0101-9. [DOI] [PubMed] [Google Scholar]
- Narine K, De Wever O, Van Valckenborgh D, Francois K, Bracke M, DeSmet S, Mareel M, Van Nooten G. Growth factor modulation of fibroblast proliferation, differentiation, and invasion: implications for tissue valve engineering. Tissue Eng. 2006;12:2707–2716. doi: 10.1089/ten.2006.12.2707. [DOI] [PubMed] [Google Scholar]
- Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171:53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, Compton LA, Barnett JV. Coronary vessel development: the epicardium delivers. Trends Cardiovasc Med. 2004;14:247–251. doi: 10.1016/j.tcm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padua RR, Kardami E. Increased basic fibroblast growth factor (bFGF) accumulation and distinct patterns of localization in isoproterenol-induced cardiomyocyte injury. Growth Factors. 1993;8:291–306. doi: 10.3109/08977199308991574. [DOI] [PubMed] [Google Scholar]
- Padua RR, Sethi R, Dhalla NS, Kardami E. Basic fibroblast growth factor is cardioprotective in ischemia-reperfusion injury. Mol Cell Biochem. 1995;143:129–135. doi: 10.1007/BF01816946. [DOI] [PubMed] [Google Scholar]
- Padua RR, Merle PL, Doble BW, Yu CH, Zahradka P, Pierce GN, Panagia V, Kardami E. FGF-2-induced negative inotropism and cardioprotection are inhibited by chelerythrine: involvement of sarcolemmal calcium-independent protein kinase C. J Mol Cell Cardiol. 1998;30:2695–2709. doi: 10.1006/jmcc.1998.0832. [DOI] [PubMed] [Google Scholar]
- Parker TG, Packer SE, Schneider MD. Peptide growth factors can provoke “fetal” contractile protein gene expression in rat cardiac myocytes. J Clin Invest. 1990;85:507–514. doi: 10.1172/JCI114466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasumarthi KB, Kardami E, Cattini PA. High and low molecular weight fibroblast growth factor-2 increase proliferation of neonatal rat cardiac myocytes but have differential effects on binucleation and nuclear morphology. Evidence for both paracrine and intracrine actions of fibroblast growth factor-2. Circ Res. 1996;78:126–136. doi: 10.1161/01.res.78.1.126. [DOI] [PubMed] [Google Scholar]
- Pellieux C, Foletti A, Peduto G, Aubert JF, Nussberger J, Beermann F, Brunner HR, Pedrazzini T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J Clin Invest. 2001;108:1843–1851. doi: 10.1172/JCI13627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi DJ, Ballard VL, Mikawa T. Epicardium is required for the full rate of myocyte proliferation and levels of expression of myocyte mitogenic factors FGF2 and its receptor, FGFR-1, but not for transmural myocardial patterning in the embryonic chick heart. Dev Dyn. 2003;228:161–172. doi: 10.1002/dvdy.10360. [DOI] [PubMed] [Google Scholar]
- Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- Peters NS, Wit AL. Gap junction remodeling in infarction: does it play a role in arrhythmogenesis? J Cardiovasc Electrophysiol. 2000;11:488–490. doi: 10.1111/j.1540-8167.2000.tb00348.x. [DOI] [PubMed] [Google Scholar]
- Pintucci G, Yu PJ, Saponara F, Kadian-Dodov DL, Galloway AC, Mignatti P. PDGF-BB induces vascular smooth muscle cell expression of high molecular weight FGF-2, which accumulates in the nucleus. J Cell Biochem. 2005;95:1292–1300. doi: 10.1002/jcb.20505. [DOI] [PubMed] [Google Scholar]
- Piotrowicz RS, Ding L, Maher P, Levin EG. Inhibition of cell migration by 24-kDa fibroblast growth factor-2 is dependent upon the estrogen receptor. J Biol Chem. 2001;276:3963–3970. doi: 10.1074/jbc.M004868200. [DOI] [PubMed] [Google Scholar]
- Plouet J, Mascarelli F, Loret MD, Faure JP, Courtois Y. Regulation of eye derived growth factor binding to membranes by light, ATP or GTP in photoreceptor outer segments. EMBO J. 1988;7:373–376. doi: 10.1002/j.1460-2075.1988.tb02823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post MJ, Laham R, Sellke FW, Simons M. Therapeutic angiogenesis in cardiology using protein formulations. Cardiovasc Res. 2001;49:522–531. doi: 10.1016/s0008-6363(00)00216-9. [DOI] [PubMed] [Google Scholar]
- Powell PP, Klagsbrun M. Three forms of rat basic fibroblast growth factor are made from a single mRNA and localize to the nucleus. J Cell Physiol. 1991;148:202–210. doi: 10.1002/jcp.1041480204. [DOI] [PubMed] [Google Scholar]
- Presta M, Rusnati M, Urbinati C, Sommer A, Ragnotti G. Biologically active synthetic fragments of human basic fibroblast growth factor (bFGF): identification of two Asp-Gly-Arg-containing domains involved in the mitogenic activity of bFGF in endothelial cells. J Cell Physiol. 1991;149:512–524. doi: 10.1002/jcp.1041490322. [DOI] [PubMed] [Google Scholar]
- Prudovsky I, Bagala C, Tarantini F, Mandinova A, Soldi R, Bellum S, Maciag T. The intracellular translocation of the components of the fibroblast growth factor 1 release complex precedes their assembly prior to export. J Cell Biol. 2002;158:201–208. doi: 10.1083/jcb.200203084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudovsky I, Mandinova A, Soldi R, Bagala C, Graziani I, Landriscina M, Tarantini F, Duarte M, Bellum S, Doherty H, Maciag T. The non-classical export routes: FGF1 and IL-1alpha point the way. J Cell Sci. 2003;116:4871–4881. doi: 10.1242/jcs.00872. [DOI] [PubMed] [Google Scholar]
- Quarto N, Finger FP, Rifkin DB. The NH2-terminal extension of high molecular weight bFGF is a nuclear targeting signal. J Cell Physiol. 1991;147:311–318. doi: 10.1002/jcp.1041470217. [DOI] [PubMed] [Google Scholar]
- Quarto N, Fong KD, Longaker MT. Gene profiling of cells expressing different FGF-2 forms. Gene. 2005;356:49–68. doi: 10.1016/j.gene.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Rajanayagam MA, Shou M, Thirumurti V, Lazarous DF, Quyyumi AA, Goncalves L, Stiber J, Epstein SE, Unger EF. Intracoronary basic fibroblast growth factor enhances myocardial collateral perfusion in dogs. J Am Coll Cardiol. 2000;35:519–526. doi: 10.1016/s0735-1097(99)00550-1. [DOI] [PubMed] [Google Scholar]
- Renko M, Quarto N, Morimoto T, Rifkin DB. Nuclear and cytoplasmic localization of different basic fibroblast growth factor species. J Cell Physiol. 1990;144:108–114. doi: 10.1002/jcp.1041440114. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Schafer T, Zentgraf H, Zehe C, Brugger B, Bernhagen J, Nickel W. Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J Biol Chem. 2004;279:6244–6251. doi: 10.1074/jbc.M310500200. [DOI] [PubMed] [Google Scholar]
- Schultz JE, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest. 1999;104:709–719. doi: 10.1172/JCI7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109:787–796. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellke FW, Laham RJ, Edelman ER, Pearlman JD, Simons M. Therapeutic angiogenesis with basic fibroblast growth factor: technique and early results. Ann Thorac Surg. 1998;65:1540–1544. doi: 10.1016/s0003-4975(98)00340-3. [DOI] [PubMed] [Google Scholar]
- Sessa C, Vigano L, Grasselli G, Trigo J, Marimon I, Llado A, Locatelli A, Ielmini N, Marsoni S, Gianni L. Phase I clinical and pharmacological evaluation of the multi-tyrosine kinase inhibitor SU006668 by chronic oral dosing. Eur J Cancer. 2006;42:171–178. doi: 10.1016/j.ejca.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Sheikh F, Sontag DP, Fandrich RR, Kardami E, Cattini PA. Overexpression of FGF-2 increases cardiac myocyte viability after injury in isolated mouse hearts. Am J Physiol Heart Circ Physiol. 2001;280:H1039–H1050. doi: 10.1152/ajpheart.2001.280.3.H1039. [DOI] [PubMed] [Google Scholar]
- Sheng Z, Lewis JA, Chirico WJ. Nuclear and nucleolar localization of 18-kDa fibroblast growth factor-2 is controlled by C-terminal signals. J Biol Chem. 2004;279:40153–40160. doi: 10.1074/jbc.M400123200. [DOI] [PubMed] [Google Scholar]
- Simons M, Annex BH, Laham RJ, Kleiman N, Henry T, Dauerman H, Udelson JE, Gervino EV, Pike M, Whitehouse MJ, Moon T, Chronos NA. Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation. 2002;105:788–793. doi: 10.1161/hc0802.104407. [DOI] [PubMed] [Google Scholar]
- Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- Sommer A, Brewer MT, Thompson RC, Moscatelli D, Presta M, Rifkin DB. A form of human basic fibroblast growth factor with an extended amino terminus. Biochem Biophys Res Commun. 1987;144:543–550. doi: 10.1016/s0006-291x(87)80001-3. [DOI] [PubMed] [Google Scholar]
- Sorensen V, Nilsen T, Wiedlocha A. Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays. 2006;28:504–514. doi: 10.1002/bies.20405. [DOI] [PubMed] [Google Scholar]
- Speir E, Tanner V, Gonzalez AM, Farris J, Baird A, Casscells W. Acidic and basic fibroblast growth factors in adult rat heart myocytes. Localization, regulation in culture, and effects on DNA synthesis. Circ Res. 1992;71:251–259. doi: 10.1161/01.res.71.2.251. [DOI] [PubMed] [Google Scholar]
- Spirito P, Fu YM, Yu ZX, Epstein SE, Casscells W. Immunohistochemical localization of basic and acidic fibroblast growth factors in the developing rat heart. Circulation. 1991;84:322–332. doi: 10.1161/01.cir.84.1.322. [DOI] [PubMed] [Google Scholar]
- Spruill LS, Baicu CF, Zile MR, McDermott PJ. Selective translation of mRNAs in the left ventricular myocardium of the mouse in response to acute pressure overload. J Mol Cell Cardiol. 2008;44:69–75. doi: 10.1016/j.yjmcc.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisakuldee W, Nickel BE, Fandrich RR, Jiang ZS, Kardami E. Administration of FGF-2 to the heart stimulates connexin-43 phosphorylation at protein kinase C target sites. Cell Commun Adhes. 2006;13:13–19. doi: 10.1080/15419060600631326. [DOI] [PubMed] [Google Scholar]
- Sugi Y, Sasse J, Barron M, Lough J. Developmental expression of fibroblast growth factor receptor-1 (cek-1; flg) during heart development. Dev Dyn. 1995;202:115–125. doi: 10.1002/aja.1002020203. [DOI] [PubMed] [Google Scholar]
- Sun G, Doble BW, Sun JM, Fandrich RR, Florkiewicz R, Kirshenbaum L, Davie JR, Cattini PA, Kardami E. CUG-initiated FGF-2 induces chromatin compaction in cultured cardiac myocytes and in vitro. J Cell Physiol. 2001;186:457–467. doi: 10.1002/1097-4652(2000)9999:999<000::AID-JCP1044>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Syed IS, Sanborn TA, Rosengart TK. Therapeutic angiogenesis: a biologic bypass. Cardiology. 2004;101:131–143. doi: 10.1159/000075994. [DOI] [PubMed] [Google Scholar]
- Taverna S, Ghersi G, Ginestra A, Rigogliuso S, Pecorella S, Alaimo G, Saladino F, Dolo V, Dell’Era P, Pavan A, Pizzolanti G, Mignatti P, Presta M, Vittorelli ML. Shedding of membrane vesicles mediates fibroblast growth factor-2 release from cells. J Biol Chem. 2003;278:51911–51919. doi: 10.1074/jbc.M304192200. [DOI] [PubMed] [Google Scholar]
- Thompson SA. The disulfide structure of bovine pituitary basic fibroblast growth factor. J Biol Chem. 1992;267:2269–2273. [PubMed] [Google Scholar]
- Thompson RW, Whalen GF, Saunders KB, Hores T, D’Amore PA. Heparinmediated release of fibroblast growth factor-like activity into the circulation of rabbits. Growth Factors. 1990;3:221–229. doi: 10.3109/08977199009043906. [DOI] [PubMed] [Google Scholar]
- Udelson JE, Dilsizian V, Laham RJ, Chronos N, Vansant J, Blais M, Galt JR, Pike M, Yoshizawa C, Simons M. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 improves stress and rest myocardial perfusion abnormalities in patients with severe symptomatic chronic coronary artery disease. Circulation. 2000;102:1605–1610. doi: 10.1161/01.cir.102.14.1605. [DOI] [PubMed] [Google Scholar]
- Uhlen P, Burch PM, Zito CI, Estrada M, Ehrlich BE, Bennett AM. Gain-offunction/Noonan syndrome SHP-2/Ptpn11 mutants enhance calcium oscillations and impair NFAT signaling. Proc Natl Acad Sci U S A. 2006;103:2160–2165. doi: 10.1073/pnas.0510876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger EF, Banai S, Shou M, Lazarous DF, Jaklitsch MT, Scheinowitz M, Correa R, Klingbeil C, Epstein SE. Basic fibroblast growth factor enhances myocardial collateral flow in a canine model. Am J Physiol. 1994;266:H1588–H1595. doi: 10.1152/ajpheart.1994.266.4.H1588. [DOI] [PubMed] [Google Scholar]
- Unger EF, Goncalves L, Epstein SE, Chew EY, Trapnell CB, Cannon RO, III, Quyyumi AA. Effects of a single intracoronary injection of basic fibroblast growth factor in stable angina pectoris. Am J Cardiol. 2000;85:1414–1419. doi: 10.1016/s0002-9149(00)00787-6. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:848. doi: 10.1038/12226. [DOI] [PubMed] [Google Scholar]
- Villanueva S, Cespedes C, Gonzalez A, Vio CP. bFGF induces an earlier expression of nephrogenic proteins after ischemic acute renal failure. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1677–R1687. doi: 10.1152/ajpregu.00023.2006. [DOI] [PubMed] [Google Scholar]
- Walgenbach KJ, Gratas C, Shestak KC, Becker D. Ischaemia-induced expression of bFGF in normal skeletal muscle: a potential paracrine mechanism for mediating angiogenesis in ischaemic skeletal muscle. Nat Med. 1995;1:453–459. doi: 10.1038/nm0595-453. [DOI] [PubMed] [Google Scholar]
- Walter P, Gilmore R, Blobel G. Protein translocation across the endoplasmic reticulum. Cell. 1984;38:5–8. doi: 10.1016/0092-8674(84)90520-8. [DOI] [PubMed] [Google Scholar]
- Wang JM, Hayashi T, Zhang WR, Sakai K, Shiro Y, Abe K. Reduction of ischemic brain injury by topical application of insulin-like growth factor-I after transient middle cerebral artery occlusion in rats. Brain Res. 2000;859:381–385. doi: 10.1016/s0006-8993(00)02008-4. [DOI] [PubMed] [Google Scholar]
- Witte L, Fuks Z, Haimovitz-Friedman A, Vlodavsky I, Goodman DS, Eldor A. Effects of irradiation on the release of growth factors from cultured bovine, porcine, and human endothelial cells. Cancer Res. 1989;49:5066–5072. [PubMed] [Google Scholar]
- Yanagisawa-Miwa A, Uchida Y, Nakamura F, Tomaru T, Kido H, Kamijo T, Sugimoto T, Kaji K, Utsuyama M, Kurashima C, et al. Salvage of infarcted myocardium by angiogenic action of basic fibroblast growth factor. Science. 1992;257:1401–1403. doi: 10.1126/science.1382313. [DOI] [PubMed] [Google Scholar]
- Yu PJ, Ferrari G, Pirelli L, Galloway AC, Mignatti P, Pintucci G. Thrombin cleaves the high molecular weight forms of basic fibroblast growth factor (FGF-2): a novel mechanism for the control of FGF-2 and thrombin activity. Oncogene. 2008;27:2594–2601. doi: 10.1038/sj.onc.1210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehe C, Engling A, Wegehingel S, Schafer T, Nickel W. Cell-surface heparan sulfate proteoglycans are essential components of the unconventional export machinery of FGF-2. Proc Natl Acad Sci U S A. 2006;103:15479–15484. doi: 10.1073/pnas.0605997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Ma YF, Gan JX, Jiang GY, Xu SX, Tao XL, Hong A, Li JK. Basic fibroblast growth factor alleviates brain injury following global ischemia reperfusion in rabbits. J Zhejiang Univ Sci B. 2005;6:637–643. doi: 10.1631/jzus.2005.B0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Rivkees SA. Programmed cell death in the developing heart: regulation by BMP4 and FGF2. Dev Dyn. 2000;217:388–400. doi: 10.1002/(SICI)1097-0177(200004)217:4<388::AID-DVDY6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Zhou M. Molecular genetics. Cincinnati: University of Cincinnati; 1997. Role of fibroblast growth factor 2 in cardiovascular development and function. [Google Scholar]
- Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med. 1998;4:201–207. doi: 10.1038/nm0298-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Komiya H, Chirino A, Faham S, Fox GM, Arakawa T, Hsu BT, Rees DC. Three-dimensional structures of acidic and basic fibroblast growth factors. Science. 1991;251:90–93. doi: 10.1126/science.1702556. [DOI] [PubMed] [Google Scholar]