

Abstract
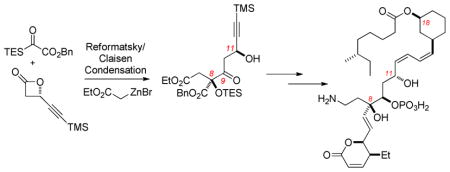
A formal synthesis of leustroducsin B has been completed. The synthesis relies upon a recently developed Reformatsky/Claisen condensation of silyl glyoxylates and enantioenriched β-lactones that establishes two of the molecule’s three core stereocenters and permits further elaboration to an intermediate in Imanishi’s synthesis via reliable chemistry (Prasad reduction, asymmetric pentenylation, Mitsunobu inversion).
The leustroducsin1 and phoslactomycin2 families of natural products were first isolated in 1993 by Kohama et al. from the culture broth of Streptomyces platensis and were later found to exhibit interesting antifungal, antibacterial, and antitumor activities.1b,2b,3ab These natural products boast intriguing molecular architectures: common features include a highly congested and functionalized core flanked by dihydropyrone and cyclohexyl moieties and a central tertiary alcohol with vicinal phosphate and aminoethyl substituents.
The individual members of this natural product family are largely distinguished by the substituent present at C18 of the cyclohexyl ring (Figure 1), a structural feature which has been shown to partially modulate a variety of biological activities attributed to the leustroducsins and phoslactomycins.1a,2a Particularly notable is leustroducsin B (C18: 6-methyloctanoate), which has shown potent in vitro induction of granulocyte-CSF and granulocyte-macrophage-CSF production by KM-102 cells,1a,3c,3d in vivo augmentation of host resistance in E. coli infections,3c and induction of thrombocytosis in mice.3e
Figure 1.

Leustroducsin B and Related Compounds
A number of synthetic studies have been performed on the leustroducsin4–6 and phoslactomycin7 families of molecules, including total syntheses of leustroducsin B by Fukuyama (2003)4 and Imanishi (2006)5 and a formal synthesis by Cossy (2008).6 This Letter details a formal synthesis of leustroducsin B enabled by a tandem Reformatsky/Claisen condensation of silyl glyoxylate 28 and enantioenriched β-lactones9 recently invented for the purpose of preparing the leustroducsin core functionality. The three-component coupling shown in Scheme 1 proceeded reliably on multigram scale to give the derived Claisen products in 61% (1a, SiR3 = TES) or 67% (1b, SiR3 = TBS) yield with excellent diastereoselectivity (>20:1) through the transfer of stereochemical information from the β-lactone. Access to the correct C8-configuration required the use of the illustrated (S)-β-lactone, a circumstance leading to a temporary stereochemical error at C11.10
Scheme 1.

Three-Component Coupling
The 11S alcohol was advantageously deployed to establish the correct configuration of the C9 hydroxyl via a directed Prasad reduction11 of the C9 ketone to the corresponding syn-diol and facilitate the evaluation of a possible late-stage Mitsunobu reaction. Excellent selectivity for the propargylic site was observed (>20:1) in the synthesis of 3, achieving both stereochemical correction at C11 and differentiation of the C9 and C11 hydroxyls. While we encountered difficulties in hydrolysis of the chloroacetate under basic and acidic conditions at this early stage due to competing lactonization and silyl ether deprotection/retro-aldol pathways, we were pleased to observe clean phosphorylation vicinal to the hindered quaternary center (3 → 4, Scheme 2). We therefore chose to delay this sequence until a stage at which the resulting phosphate would survive the remaining transformations in the synthesis and when the corresponding chloroacetate might be more amenable to hydrolysis. Additionally, the TES ether was selected for protection of the C8 hydroxyl group to avoid a tenuous removal of the more robust silyl ether later in the synthesis.
Scheme 2.
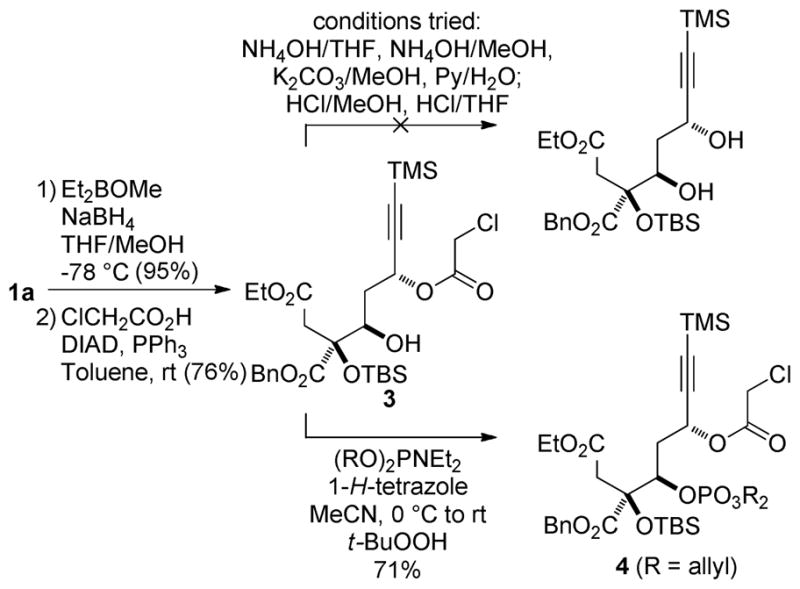
Evaluation of Late-Stage Strategy
The syn-diol was protected as the acetonide (Scheme 3), and we sought to convert the resulting diester 5 to a suitable precursor for introduction of the dihydropyrone, amine, and diene moieties. Reduction of the diester functionality to the corresponding diol 6 required the use of lithium triethylborohydride at reduced temperatures in CH2Cl2 to suppress migration of the triethylsilyl group to the vicinal primary hydroxyl. This migration was exacerbated at higher temperatures, in toluene or Et2O, and when employing standard basic workup conditions (NaOH/H2O2).10
Scheme 3.

Synthesis of Dihydropyrone Precursor
Selective TBS protection of the more accessible primary hydroxyl group and subsequent oxidation with Dess-Martin’s periodinane afforded aldehyde 7 in 75% yield over two steps. Two-carbon homologation to enal 8 was achieved in an excellent yield (88%, 2 steps) through a Horner-Wadsworth-Emmons/DIBAL-H reduction sequence (Scheme 3).
Although several strategies were investigated, a Brown-type asymmetric pentenylation6,12 proved to be the optimal method for introduction of the dihydropyrone from enal 8, setting the stereocenters at C4 and C5 and introducing the functionality required for elaboration of the dihydropyrone. Deprotection of the TMS-protected alkyne (K2CO3/MeOH) and acryloylation proceeded uneventfully (Scheme 4), but acrylate 9 was found to be inert under all ring-closing metathesis conditions screened.10 We conjectured that the presence of the alkyne might be impeding reactivity, as coordination of ruthenium to free alkynes has been shown to hinder RCM reactions.13 The TMS-protected alkyne was similarly unreactive.14
Scheme 4.

Dihydropyrone Installation
Protection of the terminal alkyne as the dicobalt hexacarbonyl complex15 allowed alkene metathesis to proceed at room temperature and afforded dihydropyrone 11 after deprotection with ceric ammonium nitrate. Few examples of ring-closing metatheses in the presence of terminal alkyne functionalities exist; only recently has the use of dicobalt hexacarbonyl alkyne protection been employed to this end.13b,c While the use of an alkyne protecting group adds two additional steps to the synthesis, its near quantitative introduction and removal consequently result in minimal loss of material.
Having accomplished the introduction of the dihydropyrone moiety, we focused on the remaining challenges in the synthesis: (1) introduction of the amine functionality; (2) stereochemical correction at C11 and subsequent phosphorylation at C9; and (3) conversion of the terminal alkyne to the requisite Z,Z-diene. At this stage, deprotection of the acetonide to reveal the C9/C11 diol was problematic, even when the primary silyloxy substituent was first converted to a protected amine functionality (via deprotection and Mitsunobu reaction with a biscarbamate16a,b). The increased stability afforded by a TBS group at C8 allowed for facile and selective deprotection with propanedithiol and BF3•OEt2 in a model system,17 yet in the presence of the more labile TES group, only decomposition was observed under identical conditions.
After evaluating the stepwise introduction of functionalities at this stage, we recognized the need to access a more easily modified intermediate. In light of the remaining transformations needed to complete the synthesis, dioxane 11 was converted to dioxolane 13 via the intermediate tetraol 12; the latter was generated by global deprotection with CSA/MeOH (Scheme 5). Careful purification of the sensitive tetraol 12 through a short plug of deactivated silica gel was necessary to avoid significant decomposition. Selective silylation of the primary and propargylic hydroxyl groups could be achieved at low temperature, and protection of the remaining diol as the acetonide afforded 13 in 73% yield over three steps.
Scheme 5.
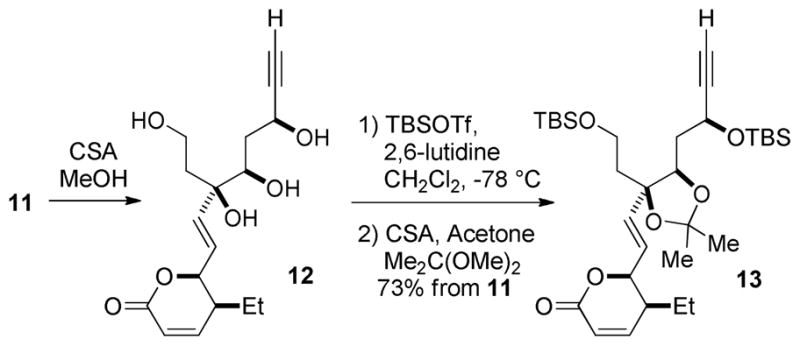
Dioxane to Dioxolane Conversion
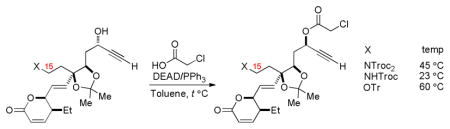
Dioxolane 13 proved far more amenable to selective functionalization and offered relief to what had appeared to be a synthetic impasse. Protecting group exchange afforded propargyl alcohol 14, which underwent clean Mitsunobu inversion with chloroacetic acid16 at 60 °C. The temperature required for this stereochemical correction was found through various abandoned routes to be dependent on the substitutent at C15 (Scheme 6), likely indicative of the spatial proximity of the alkyl side chain to the C11 stereocenter.18 Saponification of the chloroacetate and TBS protection afforded 15 in high yield, which proved a viable precursor to the vinyl iodide through iodination with NIS/AgNO3 and diimide reduction19 (84%, 2 steps). Selective deprotection of the trityl ether with BCl3 afforded 16, an intermediate present in Imanishi’s synthesis of leustroducsin B;5 its interception thus constitutes a formal synthesis of the natural product.
Scheme 6.

Completion of Formal Synthesis
In conclusion, we have completed a formal synthesis of leustroducsin B by preparing vinyl iodide 16 in 24 steps and an overall 4% yield from hydroxyketone 1. Rapid access to the core of the molecule was achieved via a tandem Reformatsky/Claisen condensation of a silyl glyoxylate and an enantioenriched β-lactone, a method relying on unusual 1,4-induced stereotransmission from the electrophile. This strategy allowed for the use of well-precedented, synthetically attractive chemistry to establish the molecule’s remaining stereocenters (Prasad reduction, Brown pentenylation, Mitsunobu reaction). A late-stage shift in protection strategy (dioxane to dioxolane) permitted the necessary selective functionalization of polyhydroxylated intermediates and allowed for completion of the formal synthesis upon reaching an advanced intermediate (16) in Imanishi’s total synthesis.
Supplementary Material
Acknowledgments
The project described was supported by Award R01 GM084927 from the National Institute of General Medical Sciences. Additional support from Novartis is gratefully acknowledged. The authors acknowledge Adam Gray for his assistance in the early stages of the synthesis.
Footnotes
Supporting Information Available: Experimental details and spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Kohama T, Enokita R, Okazaki T, Miyaoka H, Torikata A, Inukai M, Kaneko I, Kagasaki T, Sakaida Y, Satoh A, Shirashi A. J Antibiot. 1993;46:1503. doi: 10.7164/antibiotics.46.1503. [DOI] [PubMed] [Google Scholar]; (b) Kohama T, Nakamura T, Kinoshita T, Kaneko I, Shiraishi A. J Antibiot. 1993;46:1512. doi: 10.7164/antibiotics.46.1512. [DOI] [PubMed] [Google Scholar]; (c) Matsuhashi H, Shimada K. Tetrahedron. 2002;58:5619. [Google Scholar]
- 2.(a) Fushimi S, Nishikawa S, Shimazo A, Seto H. J Antibiot. 1989;42:1019. doi: 10.7164/antibiotics.42.1019. [DOI] [PubMed] [Google Scholar]; (b) Fushimi S, Furihata K, Seto H. J Antibiot. 1989;42:1026. doi: 10.7164/antibiotics.42.1026. [DOI] [PubMed] [Google Scholar]; (c) Ozasa T, Suzuki K, Sasamata M, Tanaka K, Koburi M, Kadota S, Nagai K, Saito T, Watanabe S, Iwanami M. J Antibiot. 1989;42:1331. doi: 10.7164/antibiotics.42.1331. [DOI] [PubMed] [Google Scholar]; (d) Ozasa T, Tanaka K, Sasamata M, Kaniwa H, Shimizu M, Matsumoto H, Iwanami M. J Antibiot. 1989;42:1339. doi: 10.7164/antibiotics.42.1339. [DOI] [PubMed] [Google Scholar]; (e) Shibata T, Kurihara S, Yoda K, Haruyama H. Tetrahedron. 1995;51:11999. [Google Scholar]; (f) Sekiyama Y, Palaniappan N, Reynolds KA, Osada H. Tetrahedron. 2003;59:7465. doi: 10.1074/jbc.M305082200. [DOI] [PubMed] [Google Scholar]; (g) Choudhuri SD, Ayers S, Soine WH, Reynolds KA. J Antibiot. 2005;58:573. doi: 10.1038/ja.2005.78. [DOI] [PubMed] [Google Scholar]; (h) Mizuhara N, Usuki Y, Ogita M, Fujita K, Kuroda M, Doe M, Lio H, Tanaka T. J Antibiot. 2007;60:762. doi: 10.1038/ja.2007.101. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lewy DS, Gauss CM, Soenen DR, Boger DL. Curr Med Chem. 2002;9:2005. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]; (b) Uramoto M, Shen YC, Takizawa N, Kusakabe H, Isono K. J Antibiot. 1985;38:665. doi: 10.7164/antibiotics.38.665. [DOI] [PubMed] [Google Scholar]; (c) Kohama T, Katayama T, Kinoshita T, Kaneko I, Shiraishi A. Microbiol Immunol. 1994;38:741. doi: 10.1111/j.1348-0421.1994.tb01850.x. [DOI] [PubMed] [Google Scholar]; (d) Koishi R, Serizawa N, Kohama T. J Interferon Cytokine Res. 1998;18:863. doi: 10.1089/jir.1998.18.863. [DOI] [PubMed] [Google Scholar]; (e) Kohama T, Maeda H, Imada-Sakai J, Shiraishi A, Yamashita K. J Antibiot. 1996;49:91. doi: 10.7164/antibiotics.49.91. [DOI] [PubMed] [Google Scholar]
- 4.Shimada K, Kaburagi Y, Fukuyama T. J Am Chem Soc. 2003;125:4048. doi: 10.1021/ja0340679. [DOI] [PubMed] [Google Scholar]
- 5.(a) Miyashita K, Tsunemi T, Hosokawa T, Ikejiri M, Imanishi T. Tetrahedron Lett. 2007;48:3829. [Google Scholar]; (b) Miyashita K, Tsunemi T, Hosokawa T, Ikejiri M, Imanishi T. J Org Chem. 2008;73:5360. doi: 10.1021/jo8005599. [DOI] [PubMed] [Google Scholar]
- 6.Moise J, Sonowane RP, Corsi C, Wendeborn SV, Arseniyadis S, Cossy J. Synlett. 2008:2617. [Google Scholar]
- 7.For phoslactomycin B syntheses, see: Wang YG, Takeyama T, Kobayashi Y. Angew Chem Int Ed. 2006;45:3320. doi: 10.1002/anie.200600458.Nonaka H, Maeda N, Kobayashi Y. Tetrahedron Lett. 2007;48:5601.Shibahara S, Fujina M, Tashiro Y, Takahashi K, Ishihara J, Hatakeyama S. Org Lett. 2008;10:2139. doi: 10.1021/ol8004672.Druais V, Hall MJ, Corsi C, Wendeborn SV, Meyer C, Cossy J. Org Lett. 2009;11:935. doi: 10.1021/ol8029142.Konig CM, Gebhardt B, Schleth C, Dauber M, Koert U. Org Lett. 2009;11:2728. doi: 10.1021/ol900757k.Shibahara S, Fujino M, Tashiro Y, Nanako O, Esumi T, Takahashi K, Ishihara J, Hatakeyama S. Synthesis. 2009:2935.Gebhardt B, König CM, Schleth C, Dauber M, Koert U. Chem Eur J. 2010;16:5934–5941. doi: 10.1002/chem.201000104.
- 8.Greszler SN, Malinowski JT, Johnson JS. J Am Chem Soc. 2010;132:17393. doi: 10.1021/ja108848d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Nelson SG, Peelen TJ, Wan Z. J Am Chem Soc. 1999;121:9742. [Google Scholar]; (b) Nelson SG, Zhu C, Shen X. J Am Chem Soc. 2004;126:14. doi: 10.1021/ja0391208. [DOI] [PubMed] [Google Scholar]; (c) Nelson SG, Zhu C, Shen X. J Am Chem Soc. 2004;126:5352. doi: 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]
- 10.See the Supporting Information for additional details.
- 11.Chen KM, Gunderson KG, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Chem Lett. 1987:1923–1926. [Google Scholar]
- 12.Brown HC, Bhat KS. J Am Chem Soc. 1986;108:293. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 13.(a) Bressy C, Vors JP, Hillebrand S, Arseniyadis S, Cossy J. Angew Chem Int Ed. 2008;47:10137. doi: 10.1002/anie.200802423. [DOI] [PubMed] [Google Scholar]; (b) Ono K, Nagata T, Nishida A. Synlett. 2003:1207. [Google Scholar]; (c) Ono K, Nakagawa M, Nishida A. Angew Chem Int Ed. 2004;43:2020. doi: 10.1002/anie.200453673. [DOI] [PubMed] [Google Scholar]
- 14.A recent synthesis of phoslactomycin B was able to utilize a similarly unreactive trimethylsilyl-protected alkyne by employing a relay ring-closing metathesis reaction: Druais V, Hall MJ, Corsi C, Wendeborn SV, Meyer C, Cossy J. Tetrahedron. 2010;66:6358.
- 15.(a) Nicholas KM, Pettit R. Tetrahedron Lett. 1971;37:3475. [Google Scholar]; (b) Seyferth D, Nestle MO, Wehman AT. J Am Chem Soc. 1975;97:7417. [Google Scholar]; (c) Greenfield H, Sternberg HW, Friedel RA, Wotiz JH, Markby R, Wender I. J Am Chem Soc. 1956;78:120. [Google Scholar]
- 16.(a) Mitsunobu O, Yamada Y. Bull Chem Soc Japan. 1967;40:2380. [Google Scholar]; (b) Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem Rev. 2010;109:2551. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]; (c) Saiah M, Bessodes M, Antonakis K. Tetrahedron Lett. 1992;33:4317. [Google Scholar]
-
17.Successful dioxane deprotection (C8 –OTBS):
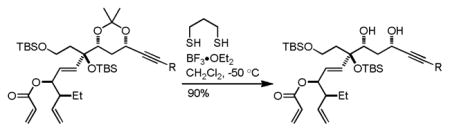
-
18.Intermediates screened in Mitsunobu reactions:
- 19.Myers AG, Zheng B, Movassaghi M. J Org Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.