

Abstract
Sialic acids are common terminal carbohydrates on cell surface. Together with internal carbohydrate structures, they play important roles in many physiological and pathological processes. In order to obtain α2–3-sialylated oligosaccharides, a highly efficient one-pot three-enzyme synthetic approach was applied. The P. multocida α2–3-sialyltransferase (PmST1) involved in the synthesis was a multifunctional enzyme with extremely flexible donor and acceptor substrate specificities. Sialyltransferase acceptors, including type 1 structure (Galβ1–3GlcNAcβProN3), type 2 structures (Galβ1–4GlcNAcβProN3 and 6-sulfo-Galβ1–4GlcNAcβProN3), type 4 structure (Galβ1–3GalNAcβProN3), type 3 or core 1 structure (Galβ1–3GalNAcαProN3) and human milk oligosaccharide or lipooligosaccharide lacto-N-tetraose (LNT) (Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3), were chemically synthesized. They were then used in one-pot three-enzyme reactions with sialic acid precursor ManNAc or ManNGc, to synthesize a library of natural occurring α2–3-linked sialosides with different internal sugar units. The sialylated oligosaccharides obtained are valuable probes for their biological studies
Keywords: carbohydrate, chemoenzymatic synthesis, sialic acid, sialylation, sialyltransferase
1 Introduction
In nature, terminal sialic acids are commonly α2–3- orα2–6-linked to galactose (Gal) or N-acetyl-galactosamine (GalNAc) [1]. Sialic acid residues can also be α2–8- or α2–9-linked to each other [2]. Currently, more than 50 different sialic acid forms have been identified in nature. The presentation of different forms is species and tissue specific. They are developmentally regulated and are believed to be closely related to their biological functions [3–5]. In addition to the naturally existing diversity of sialic acids and sialyl linkages, the internal carbohydrates are quite different which provide additional complexity of sialic acid-containing structures. For example, sialic acids can be α2–3- to the terminal galactose residue in glycans containing disaccharides such as Galβ1–3GlcNAcβ-(type 1 glycan), Galβ1–4GlcNAcβ-(type 2 glycan), Galβ1–3GalNAcα-(core 1 or type 3 glycan, TF or T antigen), Galβ1–3GalNAcβ-(type 4 glycan), and Galβ1–4Glcβ-(type 5 glycan). Sialic acids are also commonly seen α2–6-linked to the terminal galactose residue in glycans containing disaccharides such as Galβ1–3GlcNAcβ-(type 1 glycan), Galβ1–4GlcNAcβ-(type 2 glycan), and Galβ1–4Glcβ-(type 5 glycan). α2–6-Sialylated GalNAcα-(Siaα2–6GalNAcα-, STn antigen) has also been found in the O-GalNAc glycans on glycoproteins of certain types of cancer cells and is believed to be a potent cancer marker (Figure 1A) [6–8]. Sialylated lipooligosaccharide (LOS) structures have been found in many bacteria, such as Haemophilus influenzae [9], possibly Pasteurella multocida [10], Escherichia coli K1 and K92 [11]. The core structures for sialylation in LOS are lacto-N-tetraose (LNT) and lacto-N-neotetraose (LNnT). They both can be sialylated and their corresponding sialo-sides are called sialyllacto-N-tetraose (LST) a, b, c and d (Figure 1b) [12, 13]. In addition, sialyl Lewisx and sialyl Lewisa are two of the most important sialosides that have been broadly studied. They both contain a sialic acid α2–3-linked to a trisaccharide composed of galactose, GalNAc and fucose (Figure 1c), with or without sulfation at galatose or N-acetylglucosamine (GlcNAc) [14].
Figure 1.

Common sialic acid-containing structures in nature.
The interaction of sialoside and sialic acid-binding proteins is dependent on the diversity of sialoside structures including sialic acid forms, sialyl linkages, and internal carbohydrate structures [4, 15]. Other than well documented importance of sialic acids and sialyl linkages in host-virus interactions [16–21], the internal carbohydrate structures of sialosides have also been reported to be important for Siglec binding in the immune system [22–25]. For example, among CD-33 related Siglecs, Siglec-8 in human binds strongly only to sialyl Lewisx structures with O-sulfation at C-6 of galactose. Sialyl Lewisx, sialyl LacNAc, and sialyl Lewisx with O-sulfation at C-6 of GlcNAc did not bind to Siglec-8 at all [26–28]. However, Siglec-9 favored sialyl Lewisx structures with O-sulfation at the C-6 of GlcNAc instead of galactose [29]. Another example of the importance of sialic acid form was shown by preferred binding of Siglec-10 to Neu5Gcα2–6LacNAc rather than to Ne5Acα2–6LacNAc [25].
To better understand the biological significance of sialic acid-containing glycans, homogenous sialosides with different forms of sialic acids and various internal carbohydrates are needed. Chemical sialylation is usually more challenging and more time consuming compared to other chemical glycosylation processes due to the structural complexity of sialic acid [30] including a sterically hindered tertiary anomeric center, lacking of a stereo-directing group adjacent to the anomeric position, and the presence of an electron-withdrawing carboxyl group in sialyl donors. Isolation of sialosides from natural sources is also difficult due to their low abundance, lability, and the complication of diverse sialic acid forms, different linkages, and various internal glycans. To overcome these difficulties, chemoenzymatic methods were developed in our laboratory to obtain homogenous sialosides [31–34].
Herein we report the synthesis of a library of α2–3-sialylated glycans using an efficient one-pot three-enzyme chemoenzymatic approach. Sialyltransferase acceptors, including type 1 (Galβ1–3GlcNAcβProN3), type 2 (Galβ1–4GlcNAcβProN3 and 6-sulfo-Galβ1–4GlcNAcβProN3), type 3 or core 1 (Galβ1–3GalNAcαProN3), and type 4 (Galβ1–3GalNAcβProN3) disaccharides as well as type 1 tetrasaccharide (Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3) were chemically synthesized and used in the one-pot three-enzyme system containing a sialic acid aldolase, a CMP-sialic acid synthetase, and an α2–3-sialyltransferase for the production of a library of natural occurring α2–3-linked sialoside epitopes. Two different sialic acid precursors, N-acetylmannosamine (ManNAc) and N-glycolylmannosamine (ManNGc), were used to produce α2–3-linked sialosides containing N-acetylneuraminic acid (Neu5Ac), the most abundant sialic acid form, and N-glycolylneuraminic acid (Neu5Gc), a common non-human animal sialic acid form, respectively.
2 Experimental
2.1 General experimental section
1H and 13C NMR spectra were recorded on a Varian Mercury-300, a Varian Inova-400, or a Varian Inova-600 spectrometer. Chemical shifts were reported in (ppm) unit using 13C and residual 1H signals from deuterated solvents as references. Assignment of 1H NMR spectra was achieved using 2D methods (COSY) when necessary. Low and high resolution electrospray ionization (ESI) mass spectra were obtained at the Mass Spectrometry Facility at the Ohio State University. Silica gel 60 A (40–63 μm, Sorbent technologies) was used for flash column chromatography. Analytical thin layer chromatography was performed on silica gel plates 60 GF254 (Sorbent technologies). Anisaldehyde stain was used for detection. Gel filtration chromatography was performed on a column (100 cm ×2.5 cm) packed with BioGel P-2 Fine resins (Bio-Rad, Hercules, CA). All reagents were of analytical grade and were used as supplied without further purification unless specified. Solvents used in chemical reactions were distilled under an inert argon atmosphere.
2.2 Chemical synthesis of sialyltransferase acceptors
2.2.1 3-Azidopropyl β-D-galactopyranosyl-(1–3)-2-acetamido-2-deoxy-β-D-gluc opyranoside (Galβ1–3GlcNAcβProN3, 1)
Compound 7 [35] (0.90 g, 1.62 mmol) was dissolve in dry MeOH (20 mL), NaOMe (100 mg) was then added. The reaction mixture was stirred for overnight, neutralized by Dowex-50 (H+) resin, and evaporated to dryness. This dried product was dissolved in 10 mL of anhydrous DMF, fol-lowed by the addition of PhCH(OMe)2 (1 mL, 6.57 mmol). The pH of the solution was adjusted to 2–4 by adding D(+)-10-camphorsulfonic acid and the mixture was heated at 60 °C under reducing pressure at 150 mbar. After 1 hour, the reaction mixture was condensed in vacuo. The product was purified by flash column chromatography (Hexane: EtOAc = 3:2, by volume) to afford a white foam 8 (0.73 g, 87%). 1H NMR (600 MHz, CDCl3) δ 7.83 (dd, 1 H, J = 5.4 Hz and 3.0 Hz), 7.70 (dd, 1 H, J = 6.0 and 3.3 Hz), 7.50–7.49 (m, 2 H), 7.37–7.36 (m, 3 H), 5.56 (s, 1 H), 5.23 (d, 1 H, J = 8.4 Hz), 4.64 (t, 1 H, J = 9.6 Hz), 4.37 (dd, 1 H, J = 10.5 and 4.5 Hz), 4.21 (dd, 1 H, J = 10.8 and 2.4 Hz), 3.94–3.91 (m, 1 H), 3.81 (t, 1 H, J = 10.5 Hz), 3.64–3.56 (m, 3 H), 3.28–3.19 (m, 2 H), 1.94–1.90 (m, 1 H), 1.82–1.76 (m, 1 H). 13C NMR (75 MHz, CDCl3) δ 137.21, 134.43, 131.85, 129.60, 128.62, 126.57, 102.14, 99.33, 83.38, 68.88, 68.67, 67.48, 66.36, 56.82, 41.48, 32.25.
A solution of compound 8 (0.69 g, 1.32 mmol) and galactosyl trichloroacetimidate 9 [36] (0.79 g, 1.60 mmol) in anhydrous CH2Cl2 (10 mL) was stirred with activated 4 A molecular sieves (1.20 g) under argon atmosphere for 30 min. The reaction mixture was cooled to −20 °C and TMSOTf (20μL) was added drop wisely. The reaction mixture was quenched with Et3N (0.5 mL) after 2 hrs and was filtered through Celite. The filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (Toluene:EtOAc = 3:1, by volume) to give the glycosylation product 10 (0.60 g, 53%). 1H NMR (600 MHz, CDCl3) δ 7.86 (dd, 2 H, J = 5.4 Hz and 3.0 Hz), 7.75 (dd, 2 H, J = 5.4 Hz and 3.0 Hz), 7.48–7.46 (m, 2 H), 7.36–7.35 (m, 3 H), 5.56 (m, 1 H), 5.17 (dd, 1 H, J = 3.3 and 0.9 Hz), 5.13 (d, 1 H, J = 9.0 Hz), 4.97 (dd, 1 H, J = 10.2 and 7.8 Hz), 4.77–4.72 (m, 2 H), 4.53 (d, 1 H, J = 8.4 Hz), 4.36 (dd, 1 H, J = 10.2 Hz and 4.8 Hz), 4.27 (dd, 1 H, J = 10.2 and 8.4 Hz), 4.01 (dd, 1 H, J = 10.8 and 7.8 Hz), 3.91–3.88 (m, 1 H), 3.84 (t, 1 H, J = 10.2 Hz), 3.81–3.77 (m, 2 H), 3.65–3.62 (m, 1 H), 3.56–3.53 (m, 1 H), 3.49–3.46 (m, 1 H), 3.23–3.20 (m, 1 H), 3.17–3.13 (m, 1 H), 1.94–1.90 (m, 1 H), 1.89 (s, 3 H), 1.82 (s, 3 H), 1.81–1.75 (m, 1 H), 1.54 (s, 3 H). 13C NMR (150 MHz, CDCl3) δ 170.47, 170.26, 170.24, 169.09, 137.23, 134.54, 131.86, 129.50, 128.58, 126.24, 101.68, 100.73, 99.17, 81.16, 75.80, 71.20, 70.51, 69.38, 68.90, 67.35, 66.85, 66.51, 60.99, 55.54, 41.48, 32.25, 20.82, 20.76, 20.65, 20.31.
The protected disaccharide 10 (600 mg, 0.70 mmol) was dissolved in 10 mL of methanol and 100 mg Pd/C was added. The mixture was shaken under H2 (4 Bar) for overnight, filtered, and dried. The dried product, TBAI (100 mg, 0.27 mmol), and sodium azide (228 mg, 3.5 mmol) were dissolved in 10 mL of anhydrous DMF. After stirred at 60 °C for 3 hours, the reaction mixture was poured into water (50 mL) and extracted by ethyl acetate (30 mL). The organic layer was dried to afford product 11 (485 mg, 95%), which was dissolved in ethanol (5 mL), followed by the addition of hydrazine hydrate (64% hydrazine, 10 mL). After being refluxed at 100°C for 3 hours, the reaction mixture was directly dried in vacuo. To the dried white powder was added pyridine (10 mL) and acetic anhydride (2 mL). After overnight stirring, the mixture was concentrated and purified by flash column chromatography (Hexane:Acetone = 1:1, by volume). The purified white foam product was dissolved in dry MeOH (10 mL), and NaOMe (50 mg) was added. The reaction was stirred for overnight, neutralized by Dowex-50 (H+) resin, and evaporated to dryness to afford Galβ1–3GlcNAcβProN3 (1) (262 mg, 84%). 1H NMR (600 MHz, D2O) δ 4.51 (d, 1 H, J = 8.4 Hz), 4.39 (d, 1 H, J = 7.8 Hz), 3.96–3.92 (m, 1 H), 3.90–3.87 (m, 2 H), 3.80–3.59 (m, 8 H), 3.51–3.44 (m, 3 H), 3.35–3.32 (m, 2 H), 2.00 (s, 3 H), 1.80 (pentalet, 2 H, J = 6.6 Hz). 13C NMR (75 MHz, D2O)δ 174.73, 103.64, 101.04, 82.48, 75.44, 75.38, 72.56, 70.77, 68.81, 68.61, 67.26, 61.13, 60.80, 54.66, 47.88, 28.21, 22.32. HRMS (ESI) m/z calcd for C17H30N4O11 (M-H) 465.1828, found 465.1833.
2.2.2 2.3-Azidopropyl β-D-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-β-D-glucopyranoside (Galβ1–4GlcNAcβProN3, 2)
To a solution of compound 7 (2.0 g, 3.91 mmol) in methanol (30 mL) was added NaOMe (100 mg). After stirring at RT for 2 hrs, TLC (Hexane:Acetone = 1:2, by volume) showed the completion of the reaction. The reaction mixture was neutralized by Dowex-50 (H+) resin, filtered, and evaporated to dryness. The dried product was then dissolved in anhydrous CH2Cl2, followed by the addition of imidazole (531 mg, 7.82 mmol) and TBSCl (718 mg, 4.69 mmol). After overnight stirring, the mixture was condensed and purified by flash column chromatography (Hexane:EtOAc = 2:1, by volume) to afford a white foam 12 (1.64 g, 84%). 1H NMR (600 MHz, CDCl3) δ 7.83 (dd, 2 H, J = 5.4 Hz and 3.0 Hz), 7.71 (dd, 2 H, J = 5.4 Hz and 3.0 Hz), 5.20 (d, 1 H, J = 8.4 Hz), 4.36 (dd, 1 H, J = 11.4 Hz and 9.0 Hz), 4.09 (dd, 1 H, J = 10.8 Hz and 4.8 Hz), 3.98 (dd, 1 H, J = 10.8 Hz and 4.8 Hz), 3.91–3.86 (m, 2 H), 3.63–3.56 (m, 2 H), 3.54–3.51 (m, 1 H), 3.40–3.34 (m, 2 H), 1.96–1.90 (m, 1 H), 1.85–1.80 (m, 1 H), 0.91 (s, 9 H), 0.12 (s, 3 H), 0.11 (s, 3 H).
A solution of acceptor 12 (0.75 g, 1.50 mmol) and trichloroacetimidate donor 9 (0.89 g, 1.80 mmol) in anhydrous CH2Cl2 (20 mL) was stirred with activated 4 A molecular sieves (2 g) under argon atmosphere for 30 min. The reaction mixture was cooled to −40 °C and TMSOTf (15 μL) was added drop wisely. The reaction mixture was quenched with Et3N (0.5 mL) after 10 hours, and filtered through celite. The filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (Hexane:EtOAc = 2:1, by volume) to afford the coupled product 13 (0.98 g, 79%). 1H NMR (600 MHz, CDCl3) δ 7.75–7.73 (m, 2 H), 7.64–7.62 (m, 2 H), 5.27 (d, 1 H, J = 3.0 Hz), 5.14 (dd, 1 H, J = 10.2 Hz and 8.4 Hz), 5.11 (d, 1 H, J = 8.4 Hz), 4.90 (dd, 1 H, J = 10.5 Hz and 3.3 Hz), 4.53 (d, 1 H, J = 7.8 Hz), 4.31 (dd, 1 H, J = 10.8 Hz and 8.4 Hz), 4.02–3.94 (m, 4 H), 3.92–3.89 (m, 1 H), 3.82–3.77 (m, 2 H), 3.68 (dd, 1 H, J = 11.4 Hz and 3.6 Hz), 3.56 (t, 1 H, J = 9.0 Hz), 3.51–3.47 (m, 1 H), 3.42 (dd, 1 H, J = 7.8 Hz and 2.4 Hz), 3.35–3.28 (m, 2 H), 2.03 (s, 3 H), 2.00 (s, 3 H), 1.88 (s, 3 H), 1.80 (s, 3 H), 1.88–1.70 (m, 2 H), 0.83 (s, 9 H), 0.03 (s, 3 H), 0.00 (s, 3 H). 13C NMR (150 MHz, CDCl3) δ 170.59, 170.18, 169.43, 134.27, 131.89, 101.92, 98.19, 82.12, 74.90, 71.35, 71.08, 69.81, 68.91, 66.99, 65.91, 61.75, 61.70, 56.23, 41.61, 32.52, 26.08, 20.90, 20.76, 20.70, 20.45, 18.48, −4.69, −5.00.
Compound 13 (0.98 g, 1.18 mmol) was treated by TBAF (4 mL, 1 M in THF) in THF (20 mL) for 2 hours. The TLC (Hexane:EtOAc = 1:2, by volume) showed the TBS group was removed. After flash column chromatography purification, the product was dissolved in DMF (10 mL). Sodium azide (325 mg, 5 mmol) and TBAI (50 mg, 0.14 mmol) were added. The mixture was heated at 70 °C for 4 hrs, condensed in vacuo, and purified by flash column chromatography (Hexane:EtOAc = 1:2, by volume) to afford compound 14 (647 mg, 76%). Deprotection and N-acetylation of compound 14 (647 mg, 0.89 mmol) were achieved by using a similar approach as described for compound 11 to give Galβ1–4GlcNAcβProN3 (2) (343 mg, 82%) as a white foam. 1H NMR (600 MHz, D2O) δ 4.51 (d, 1 H, J = 7.8 Hz), 4.45 (d, 1 H, J = 7.8 Hz), 3.98–3.95 (m, 2 H), 3.90 (d, 1 H, J = 3.6 Hz), 3.81 (dd, 1 H, J = 12.3 Hz and 5.1 Hz), 3.76–3.67 (m, 7 H), 3.65 (dd, 1 H, J = 9.6 Hz and 3.6 Hz), 3.59–3.56 (m, 1 H), 3.52 (dd, 1 H, J = 9.6 Hz and 7.8 Hz), 3.37–3.34 (m, 2 H), 2.03 (s, 3 H), 1.82 (pentalet, 2 H, J = 6.6 Hz). 13C NMR (150 MHz, D2O) δ 174.62, 103.01, 101.24, 78.56, 75.48, 74.88, 72.63, 72.50, 71.09, 68.68, 67.26, 61.16, 60.18, 55.22, 47.90, 28.24, 22.31. HRMS (ESI) m/z calcd for C17H30N4O11 (M-H) 465.1828, found 465.1840.
2.2.3 3-Azidopropyl β-D-galactopyranosyl-(1–4)-2-acetamido-2-deoxy-6-O-sulfo-β-D-glucopyranoside (Galβ1–4GlcNAc6SβProN3, 3)
To a solution of compound 13 (500 mg, 0.60 mmol) in pyridine (10 mL) was added acetic anhydride (1 mL). After overnight stirring, the reaction mixture was diluted by EtOAc (100 mL) and washed sequentially with 2N HCl (100 mL), saturated NaHCO3 (100 mL), and brine (100 mL). The organic layer was dried by MgSO4 and evaporated to produce a yellow syrup, which was treated by TBAF and sodium azide (with TBAI) sequentially to give compound 15 (434 mg, 85%) with a free hydroxyl group at the C-6 of GlcNAc. 1H NMR (600 MHz, CDCl3) δ 7.86–7.81 (m, 2 H), 7.74–7.71 (m, 2 H), 5.72 (dd, 1 H, J = 10.8 Hz and 9.0 Hz), 5.39 (d, 1 H, J = 8.4 Hz), 5.32 (dd, 1 H, J = 3.3 and 0.9 Hz), 5.11 (dd, 1 H, J = 10.8 and 7.8 Hz), 4.98 (dd, 1 H, J = 10.2 and 3.6 Hz), 4.65 (d, 1 H, J = 7.8 Hz), 4.15 (dd, 1 H, J = 10.8 and 8.4 Hz), 4.09–4.00 (m, 3 H), 3.94–3.92 (m, 1 H), 3.89–3.86 (m, 2 H), 3.80–3.76 (m, 1 H), 3.59–3.57 (m, 1 H), 3.54–3.51 (m, 1 H), 3.23–3.19 (m, 1 H), 3.16–3.12 (m, 1 H), 2.12 (s, 3 H), 2.02 (s, 3 H), 1.95 (s, 3 H), 1.89 (s, 3 H), 1.70–1.67 (m, 2 H). 13C NMR (75 MHz, CDCl3) δ 170.61, 170.45, 170.36, 170.11, 169.46, 134.66, 134.47, 123.78, 101.23, 98.16, 75.79, 75.03, 71.56, 71.18, 70.70, 69.40, 66.91, 66.47, 61.00, 60.67, 55.15, 48.08, 29.03, 20.95, 20.91, 20.87, 20.81.
Compound 15 (434 mg, 0.51 mmol) and pyridine sulfur trioxide (182 mg, 1.02 mmol) were dissolved in pyridine (10 mL) and stirred at room temperature. The reaction was completed in 1.5 h. Purification by flash column chromatography (Hexane:EtOAc = 1:1, by volume) afforded the sulfated product. After this sulfated product was treated similarly as described for compound 11, the sulfated disaccharide Galβ1–4GlcNAc6SβProN3 (3) was obtained as a white foam (184 mg, 66%). 1H NMR (600 MHz, D2O) δ 4.54 (d, 1 H, J = 8.4 Hz), 4.52 (d, 1 H, J = 7.8 Hz), 4.38 (dd, 1 H, J = 11.1 and 1.5 Hz), 4.31 (dd, 1 H, J = 11.1 and 4.5 Hz), 3.97–3.94 (m, 1 H), 3.91 (d, 1 H, J = 3.6 Hz), 3.81–3.64 (m, 9 H), 3.51 (dd, 1 H, J = 7.8 and 1.8 Hz), 3.37–3.34 (m, 2 H), 2.04 (s, 3 H), 1.84–1.81 (m, 2 H). 13C NMR (75 MHz, D2O)δ 174.64, 102.65, 101.32, 77.48, 75.49, 72.67, 72.61, 72.40, 71.13, 68.75, 67.41, 66.39, 61.20, 55.22, 47.92, 28.26, 22.32. HRMS (ESI) m/z calcd for C17H29N4O14SNa (M-Na) 545.1407, found 545.1398
2.2.4 3.3-Azidopropyl β-D-galactopyranosyl-(1–3)-2-acetamido-2-deoxy-α-D-galactopyranoside (Galβ1–3GalNAcαProN3, 4)
To a solution of compound 16 [37] (2.5 g, 5.74 mmol) in 3-chloro-1-propanol (15 mL) was added acetyl chloride (1 mL). The solution was heated to 90 °C. Four hours later, TLC showed the disappearance of the starting material. The mixture was cooled to room temperature and transferred directly to silica gel column for purification (Hexanes:EtOAc = 1:1 followed by EtOAc, and then Hexanes:Acetone = 1:1, by volume). According to the proton NMR spectroscopy, the purified product was an α/β mixture without any O-acetylation. The product was then dissolved in DMF (20 mL), followed by the addition of PhCH(OMe)2 (2 mL). D(+)-10-Camphorsulfonic acid was added to adjust the pH to 2–4. The reaction mixture was heated at 50 °C at 150 mBar for 30 minutes, condensed, and purified by flash column chromatography (Hexane:EtOAc = 3:2, by volume) to afford two products, compound 17 (0.57 g, 21%) and 18 (1.55 g, 57%). For compound 17, 1H NMR (600 MHz, CDCl3) δ 7.79 (dd, 2 H, J = 5.4 Hz and 3.0 Hz), 7.60 (dd, 2 H, J = 5.0 Hz and 3.0 Hz), 7.54 (dd, 2 H, J = 7.2 Hz and 1.5 Hz), 7.39–7.35 (m, 3 H), 5.61 (s, 1 H), 5.44 (dd, 1 H, J = 11.4 Hz and 3.6 Hz), 5.04 (d, 1 H, J = 3.0 Hz), 4.65 (dd, 1 H, J = 11.4 Hz and 3.6 Hz), 4.37 (d, 1 H, J = 4.2 Hz), 4.29 (dd, 1 H, J = 12.6 Hz and 1.2 Hz), 4.11 (dd, 1 H, J = 12.6 Hz and 1.2 Hz), 3.88–3.84 (m, 2 H), 3.42–3.39 (m, 1 H), 1.94–1.88 (m, 2 H). 13C NMR (150 MHz, CDCl3) δ 168.85, 137.70, 134.28, 131.92, 129.50, 128.53, 126.65, 123.45, 101.59, 98.84, 75.92, 69.72, 64.40, 63.34, 54.27, 41.90, 31.93. For compound 18, 1H NMR (600 MHz, CDCl3) δ 7.83–7.80 (m, 2 H), 7.71–7.69 (m, 2 H), 7.55–7.52 (m, 2 H), 7.40–7.36 (m, 3 H), 5.60 (s, 1 H), 5.25 (d, 1 H, J = 8.4 Hz), 4.52 (dd, 1 H, J = 11.1 Hz and 3.9 Hz), 4.42–4.36 (m, 2 H), 4.28 (d, 1 H, J = 3.6 Hz), 4.12 (dd, 1 H, J = 12.0 Hz and 1.8 Hz), 3.98–3.95 (m, 1 H), 3.64–3.59 (m, 2 H), 3.42–3.34 (m, 2 H), 1.98–1.93 (m, 1 H), 1.85–1.81 (m, 1 H). 13C NMR (150 MHz, CDCl3) δ 168.85, 137.57, 134.33, 129.57, 128.54, 126.71, 101.73, 98.74, 75.34, 69.45, 68.05, 67.00, 66.14, 54.94, 41.70, 32.37.
Compound 17 (570 mg, 1.20 mmol) was coupled to trichloroacetimidate 9 (710 mg, 1.44 mmol) using a similar approach as described for compound 10. Purification by flash column chromatography (Hexane:EtOAc = 1:1, by volume) gave product 19 (461 mg, 62%) as a white foam. 1H NMR (600 MHz, CDCl3) δ 7.85–7.81 (m, 2 H), 7.72–7.68 (m, 2 H), 7.59 (d, 2 H, J = 7.2 Hz), 7.39–7.33 (m, 3 H), 5.60 (s, 1 H), 5.36 (dd, 1 H, J = 11.7 Hz and 3.3 Hz), 5.32 (d, 1 H, J = 3.6 Hz), 5.06 (dd, 1 H, J = 10.5 Hz and 2.1 Hz), 5.00 (d, 1 H, J = 3.6 Hz), 4.93–4.89 (m, 2 H), 4.81 (d, 1 H, J = 8.4 Hz), 4.48 (d, 1 H, J = 3.0 Hz), 4.28 (d, 1 H, J = 12.6 Hz), 4.19 (dd, 1 H, J = 11.7 Hz and 5.7 Hz), 4.10–4.06 (m, 2 H), 3.92 (t, 1 H, J = 6.6 Hz), 3.88–3.85 (m, 1 H), 3.82 (s, 1 H), 3.60–3.53 (m, 2 H), 3.43–3.39 (m, 1 H), 2.10 (s, 3 H), 2.04 (s, 3 H), 1.95–1.88 (m, 2 H), 1.86 (s, 3 H), 1.15 (s, 3 H). 13C NMR (150 MHz, CDCl3) δ 170.62, 170.55, 168.88, 168.48, 137.98, 134.50, 134.36, 132.57, 131.20, 129.09, 128.41, 126.54, 123.58, 123.53, 101.82, 100.98, 98.96, 76.50, 72.51, 71.37, 70.87, 69.69, 68.98, 67.26, 64.44, 63.57, 61.63, 51.31, 41.83, 31.88, 21.00, 20.95, 20.73, 19.64.
Compound 19 (461 mg, 0.57 mmol) was dissolved in DMF (10 mL). Sodium azide (195 mg, 3.0 mmol) and TBAI (30 mg, 0.08 mmol) were added. The mixture was heated at 70 °C for 4 hrs, condensed in vacuo, diluted by water, and extracted by CH2Cl2. The organic layer was evaporated to dryness and 80% HOAc (15 mL) was added to dissolve it. The solution was then heated at 60 °C for 3 hrs to remove the benzylidene protecting group. After removal of the solvent, compound 21 (387 mg, 94%) was obtained without purification. Deprotection and N-acetylation of compound 21 (387 mg, 0.54 mmol) were achieved using a similar approach as described for compound 11 to give Galβ1–4GalNAcαProN3 (4) (214 mg, 85%) as the final product. 1H NMR (600 MHz, D2O) δ 4.90 (d, 1 H, J = 3.6 Hz), 4.48 (d, 1 H, J = 7.8 Hz), 4.35 (dd, 1 H, J = 10.8 Hz and 3.6 Hz), 4.26 (d, 1 H, J = 2.4 Hz), 4.04 (dd, 1 H, J = 10.8 Hz and 3.0 Hz), 4.00 (t, 1 H, J = 6.0 Hz), 3.92 (d, 1 H, J = 3.0 Hz), 3.83–3.74 (m, 5 H), 3.68–3.63 (m, 2 H), 3.58–3.45 (m, 4 H), 2.04 (s, 3 H), 1.92 (quintet, 2 H, J = 6.6 Hz). 13C NMR (150 MHz, D2O) δ 174.70, 104.89, 97.41, 77.40, 75.18, 72.71, 70.81, 68.92, 68.79, 65.13, 61.39, 61.19, 48.87, 48.41, 28.17, 22.22. HRMS (ESI) m/z calcd for C17H30N4O11 (M-H) 465.1828, found 465.1829.
2.2.5 3-Azidopropylβ-D-galactopyranosyl-(1–3)-2-acetamido-2-deoxy-β-D-galactopyranoside (Galβ1–3GalNAcβProN3, 5)
Compound 18 (1.0 g, 2.11 mmol) was coupled to trichloroacetimidate 9 (1.25 g, 2.53 mmol) using a similar approach as described for compound 10. Purification by flash column chromatography (Hexane:EtOAc = 1:1, by volume) gave product 20 (812 mg, 48%) as a white foam. 1H NMR (600 MHz, CDCl3) δ 7.84–7.82 (m, 2 H), 7.73–7.71 (m, 2 H), 7.57 (d, 2 H, J = 7.8 Hz), 7.38–7.31 (m, 3 H), 5.57 (s, 1 H), 5.27 (d, 1 H, J = 3.6 Hz), 5.08–5.05 (m, 2 H), 4.80 (dd, 1 H, J = 10.5 Hz and 3.3 Hz), 4.75 (dd, 1 H, J = 11.4 Hz and 3.0 Hz), 4.68 (dd, 1 H, J = 11.4 Hz and 8.4 Hz), 4.54 (d, 1 H, J = 8.4 Hz), 4.36 (d, 1 H, J = 3.0 Hz), 4.33 (d, 1 H, J = 12.6 Hz), 4.10–4.02 (m, 3 H), 3.95–3.92 (m, 1 H), 3.80 (t, 1 H, J = 6.6 Hz), 3.57 (s, 1 H), 3.56–3.52 (m, 1 H), 3.35–3.32 (m, 1 H), 3.30–3.26 (m, 1 H), 2.09 (s, 3 H), 2.00 (s, 3 H), 1.97–1.92 (m, 1 H), 1.87 (s, 3 H), 1.81–1.72 (m, 1 H), 1.39 (s, 3 H). 13C NMR (150 MHz, CDCl3) δ 170.48, 170.31, 169.40, 168.96, 167.53, 137.93, 134.60, 134.49, 131.79, 129.10, 128.38, 126.59, 123.82, 123.53, 101.68, 101.08, 98.94, 75.77, 75.04, 71.18, 71.00, 69.44, 68.81, 67.09, 66.96, 65.81, 61.51, 51.93, 41.66, 32.13, 20.95, 20.91, 20.70, 19.99.
Compound 22 (664 mg, 91%) was obtained from compound 20 (812 mg, 1.01 mmol) using a similar approach as described for compound 21. Deprotection and N-acetylation of compound 22 (664 mg, 0.92 mmol) were achieved using a similar approach as described for compound 11 to give Galβ1–4GalNAcβProN3 (5) (326 mg, 76%) as the final product. 1H NMR (600 MHz, D2O) δ 4.51 (d, 1 H, J = 8.4 Hz), 4.45 (d, 1 H, J = 7.8 Hz), 4.19 (d, 1 H, J = 3.0 Hz), 4.02–3.97 (m, 2 H), 3.92 (d, 1 H, J = 3.6 Hz), 3.88 (dd, 1 H, J = 10.8 Hz and 3.0 Hz), 3.83–3.65 (m, 7 H), 3.62 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 3.53 (dd, 1 H, J = 10.2 Hz and 7.8 Hz), 3.41–3.38 (m, 2 H), 2.04 (s, 3 H), 1.86 (pentalet, 2 H, J = 6.6 Hz). 13C NMR (150 MHz, D2O) δ 174.88, 105.00, 101.57, 80.05, 75.15, 74.91, 72.65, 70.77, 68.76, 68.17, 67.19, 61.18, 61.11, 51.47, 47.99, 28.31, 22.45. HRMS (ESI) m/z calcd for C17H30N4O11 (M-H) 465.1828, found 465.1835.
2.2.6 3-Azidopropyl β-D-galactopyranosyl-(1–3)-2-acetamido-2-deoxy-β-D-gluc opyranosyl-(1–3)-β-D-galactopyranosyl-(1–4)-β-D-glucopy ranoside (Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3, 6)
To a solution of compound 23 [38] (1.00 g, 2.09 mmol) in MeOH (20 mL) was added NaOMe (100 mg). The reaction mixture was stirred for overnight, neutralized by Dowex-50 (H+) resin, and evaporated to dryness. This dried product was dissolved in 10 mL of anhydrous DMF followed by the addition of PhCH(OMe)2 (1 mL, 6.57 mmol). The pH of the solution was adjusted to 2–4 using D(+)-10-camphorsulfonic acid, and was then heated at 60°C under reducing pressure at 150 mbar. After 1 hour, the reaction mixture was condensed and purified by flash column chromatography (Hexane:EtOAc = 3:2, by volume) to afford a white foam 24 (0.76 g, 83%). 1H NMR (600 MHz, CDCl3) δ 7.87–7.85 (m, 2 H), 7.72–7.71 (m, 2 H), 7.50–7.48 (m, 2 H), 7.38–7.36 (m, 3 H), 5.57 (s, 1 H), 5.40 (d, 1 H, J = 10.8 Hz), 4.65 (t, 1 H, J = 9.6 Hz), 4.39 (dd, 1 H, J = 10.2 Hz and 4.8 Hz), 4.31 (t, 1 H, J = 10.5 Hz), 3.80 (t, 1 H, J = 10.2 Hz), 3.71–3.67 (m, 1 H), 3.60 (t, 1 H, J = 9.3 Hz), 2.72–2.63 (m, 2 H), 1.19 (t, 3 H, J = 7.5 Hz). 13C NMR (150 MHz, CDCl3) δ 168.00, 167.38, 137.17, 134.46, 130.00, 129.61, 129.23, 128.62, 126.55, 102.17, 82.33, 82.10, 70.60, 69.76, 68.86, 55.73, 24.43, 15.11.
Compound 24 (0.76 g, 1.73 mmol) was coupled to trichloroacetimidate 9 (1.02 g, 2.08 mmol) using the same approach as described for compound 10. Purification by flash column chromatography (Hexane:EtOAc = 1:1, by volume) gave product 25 (1.03 g, 77%) as a white foam. 1H NMR (600 MHz, CDCl3) δ 7.90–7.77 (m, 4 H), 7.49–7.47 (m, 2 H), 7.40–7.35 (m, 3 H), 5.57 (s, 1 H), 5.28 (d, 1 H, J = 10.8 Hz), 5.19 (d, 1 H, J = 3.6 Hz), 4.99 (dd, 1 H, J = 10.2 Hz and 7.8 Hz), 4.77 (t, 1 H, J = 10.2 Hz), 4.74 (dd, 1 H, J = 10.2 Hz and 3.6 Hz), 4.55 (d, 1 H, J = 7.8 Hz), 4.42–4.37 (m, 2 H), 4.03 (dd, 1 H, J = 10.8 Hz and 7.8 Hz), 3.85–3.80 (m, 3 H), 3.72–3.69 (m, 1 H), 3.48 (dd, 1 H, J = 7.8 Hz and 6.6 Hz), 2.71–2.62 (m, 2 H), 2.07 (s, 3 H), 1.91 (s, 3 H), 1.84 (s, 3 H), 1.55 (s, 3 H), 1.17 (t, 3 H, J = 7.2 Hz). 13C NMR (150 MHz, CDCl3) δ 170.49, 170.29, 170.27, 169.11, 137.22, 129.53, 128.60, 126.31, 126.25, 101.67, 100.64, 81.93, 91.15, 76.64, 71.20, 70.87, 70.52, 69.40, 68.90, 66.85, 61.02, 54.40, 24.09, 20.85, 20.79, 20.67, 20.33, 15.01.
A solution of glycosyl donor 25 (350 mg, 0.46 mmol) and glycosyl acceptor 26 [39] (400 mg, 0.38 mmol) in anhydrous CH2Cl2 (20 mL) was stirred with activated 4 A molecular sieves (0.80 g) under argon for 30 min. The reaction mixture was cooled to −20 °C and NIS (368 mg, 1.64 mmol) was added. After 10 min, TfOH (10 μL) was added. The reaction mixture was quenched with Et3N (0.5 mL) after 1 hour and filtered through celite. The filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (Hexanes:EtOAc = 2:1, by volume) to afford tetrasaccharide 27 as an amorphous solid (428 mg, 64%). 1H NMR (600 MHz, CDCl3) δ 8.10 (dd, 1 H, J = 8.4 Hz and 1.2 Hz), 7.99 (dd, 1 H, J = 8.4 Hz and 1.2 Hz), 7.90–7.86 (m, 4 H), 7.74 (dd, 1 H, J = 8.4 Hz and 1.2 Hz), 7.65–7.60 (m, 3 H), 7.55–7.31 (m, 20 H), 7.25–7.17 (m, 4 H), 6.87 (d, 2 H, J = 7.8 Hz), 5.61 (t, 1 H, J = 9.6 Hz), 5.51 (d, 1 H, J = 3.0 Hz), 5.45 (s, 1 H), 5.35–5.31 (m, 2 H), 5.20 (d, 1 H, J = 8.4 Hz), 5.10 (dd, 1 H, J = 3.6 Hz and 1.2 Hz), 4.80 (dd, 1 H, J = 10.2 Hz and 1.8 Hz), 4.61 (dd, 1 H, J = 10.2 Hz and 3.6 Hz), 4.57 (d, 1 H, J = 7.8 Hz), 4.55 (d, 1 H, J = 7.8 Hz), 4.51 (dd, 1 H, J = 10.2 Hz and 9.0 Hz), 4.35–4.33 (m, 2 H), 4.28 (dd, 1 H, J = 10.8 Hz and 4.8 Hz), 4.23 (dd, 1 H, J = 12.6 Hz and 4.5 Hz), 4.07 (dd, 1 H, J = 10.2 Hz and 8.4 Hz), 4.04 (t, 1 H, J = 9.6 Hz), 3.97–3.93 (m, 3 H), 3.81–3.78 (m, 1 H), 3.74–3.58 (m, 5 H), 3.51–3.46 (m, 2 H), 3.37 (t, 1 H, J = 6.6 Hz), 3.25 (dd, 1 H, J = 11.4 Hz and 7.8 Hz), 3.17–3.10 (m, 2 H), 1.99 (s, 3 H), 1.83 (s, 3 H), 1.73 (s, 3 H), 1.721.60 (m, 2 H), 1.37 (s, 3 H). 13C NMR (150 MHz, CDCl3) δ 177.19, 170.29, 170.03, 168.73, 166.02, 165.99, 165.61, 165.57, 165.34, 164.21, 137.33, 133.54–132.88, 130.10–128.45, 128.05, 126.24, 101.55, 101.34, 100.85, 100.27, 99.15, 80.63, 77.41, 75.60, 75.11, 73.25, 72.75, 72.14, 71.88, 71.74, 71.19, 70.46, 69.84, 69.32, 68.64, 66.90, 66.72, 66.52, 62.54, 62.14, 61.00, 55.29, 48.10, 29.81, 20.61, 20.59, 20.46, 20.01.
Compound 27 (428 mg, 0.24 mmol) was dissolved in 80% acetic acid (10 mL) and heated to 60 °C. After 3 hours, the solvent was removed and the residue was dissolved in ethanol (5 mL). Hydrazine hydrate (10 mL, 66.7% in water) was added. The reaction mixture was refluxed at 100 °C for 3 hours, cooled to room temperature and evaporated to dryness. To the dry product in pyridine (10 mL) was added acetic andydride (1 mL). After overnight stirring, the mixture was condensed and applied to flash column chromatography (Hexanes:Acetone = 1:1, by volume) to give the O-acetyl/benzoyl protected tetrasaccharide. The O-acetyl and O-benzoyl were then removed by treating with NaOMe (50 mg) in methanol (10 mL) to afford Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (6) as a white foam (102 mg, 54%). 1H NMR (600 MHz, D2O) δ 4.74 (d, 1 H, J = 8.4 Hz), 4.50 (d, 1 H, J = 7.8 Hz), 4.45 (d, 2 H, J = 7.8 Hz), 4.16 (d, 1 H, J = 3.6 Hz), 4.03–3.98 (m, 2 H), 3.93–3.89 (m, 3 H), 3.84–3.71 (m, 10 H), 3.67–3.46 (m, 11 H), 3.34–3.30 (m, 1 H), 2.04 (s, 3 H), 1.92 (pentalet, 2 H, J = 6.6 Hz). 13C NMR (75 MHz, D2O) δ 175.19, 103.72, 103.18, 102.75, 102.36, 82.37, 82.22, 78.69, 75.52, 75.44, 75.13, 75.00, 74.62, 73.03, 72.73, 70.93, 70.24, 68.78, 68.72, 68.55, 67.61, 62.81, 61.26, 61.18, 60.78, 60.36, 54.94, 48.14, 28.48, 22.49. HRMS (ESI) m/z calcd for C29H50N4O21 (M-H) 789.2884, found 789.2875.
2.3 One-pot three-enzyme synthesis of α2–3-linked sialosides
General Procedure
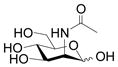
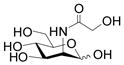
A prospective acceptor for a multi-functional Pasteurella multocida α2–3-sialyltransferase PmST1 (1–6, 0.1 mmol), a sialic acid precursor (ManNAc or ManNGc, 1.2 eq), sodium pyruvate (1.8 equiv.), and CTP (1.2 equiv.) were dissolved in water. Stock solutions of Tris-HCl buffer (1 M, pH 8.8, 1 mL) and MgCl2·6H2O (0.2 M, 1 mL) were added. After the addition of a recombinant E. coli sialic acid aldolase [40] (1.96 mg), an N. meningitidis CMP-sialic acid synthetase [40] (1.08 mg), and PmST1 [31] (0.62 mg), water was added until the total volume of the reaction mixture reached 10 mL. The reaction was carried out by incubating the solution in an isotherm incubator for 2 h at 37 °C (or for 15 h at room temperature) with agitation at 140 rpm. The product formation was monitored by TLC developed with EtOAc:MeOH:H2O:HOAc = 5:2:1:0.2 (by volume, for substrates 1, 2, 4, 5) or 4:2:1:0.2 (by volume, for substrates 3 and 6). TLC plates were stained with p-anisaldehyde sugar stain. The reaction was quenched by adding the same volume (10 mL) of ice-cold EtOH and incubation at 4 °C for 30 min. The mixture was then centrifuged to remove precipitates. The supernatant was concentrated and passed through a BioGel P-2 gel filtration column with water as the eluant to obtain the crude product. Further purification was performed using flash column chromatography to afford the desired α2–3-linked sialosides.
Neu5Acα2–3Galβ1–3GlcNAcβProN3 (30)
From disaccharide 1 (46.6 mg, 0.1 mmol), compound 30 was obtained as a white foam (66 mg, 85%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 8.4 Hz), 4.47 (d, 1 H, J = 7.8 Hz), 4.06 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 3.97–3.45 (m, 20 H), 3.37–3.34 (m, 2 H), 2.73 (dd, 1 H, J = 12.3 Hz and 4.5 Hz), 2.01 (s, 3 H), 2.00 (s, 3 H), 1.82 (pentalet, 2 H, J = 6.3 Hz), 1.76 (t, 1 H, J = 12.3 Hz). 13C NMR (75 MHz, D2O)δ 175.08, 174.70, 174.04, 103.57, 101.02, 99.75, 82.63, 75.72, 75.49, 75.21, 72.92, 71.96, 69.20, 68.84, 68.51, 68.14, 67.36, 67.27, 62.56, 61.14, 60.83, 54.56, 51.77, 47.90, 39.87, 28.22, 22.43, 22.18. HRMS (ESI) m/z calcd for C28H46N5O19Na (M-Na) 756.2782, found 756.2783.
Neu5Gcα2–3Galβ1–3GlcNAcβProN3 (31)
From disaccharide 1 (46.6 mg, 0.1 mmol), compound 31 was obtained as a white foam (65 mg, 83%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 8.4 Hz), 4.48 (d, 1 H, J = 7.8 Hz), 4.10 (s, 2 H), 4.07 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 3.98–3.44 (m, 20 H), 3.37–3.34 (m, 2 H), 2.75 (dd, 1 H, J = 12.3 Hz and 4.5 Hz), 2.02 (s, 3 H), 1.82 (pentalet, 2 H, J = 6.3 Hz), 1.78 (t, 1 H, J = 12.3 Hz). 13C NMR (75 MHz, D2O)δ 175.87, 174.71, 174.08, 103.59, 101.04, 99.77, 82.63, 75.73, 75.51, 75.22, 72.64, 72.04, 69.21, 68.84, 68.25, 68.07, 67.36, 67.28, 62.53, 61.15, 60.85, 54.58, 51.48, 47.92, 39.97, 28.24, 22.44. HRMS (ESI) m/z calcd for C28H46N5O20Na (M-Na) 772.2731, found 772.2728.
Neu5Acα2–3Galβ1–4GlcNAcβProN3 (32)
From disaccharide 2 (46.6 mg, 0.1 mmol), compound 32 was obtained as a white foam (65 mg, 84%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 7.8 Hz), 4.50 (d, 1 H, J = 7.8 Hz), 4.09 (dd, 1 H, J = 9.9 Hz and 3.3 Hz), 3.99–3.93 (m, 3 H), 3.88–3.81 (m, 4 H), 3.73–3.53 (m, 13 H), 3.37–3.33 (m, 2 H), 2.73 (dd, 1 H, J = 12.6 Hz and 4.2 Hz), 2.02 (s, 3 H), 2.01 (s, 3 H), 1.82 (pentalet, 2 H, J = 6.3 Hz), 1.78 (t, 1 H, J = 12.6 Hz). 13C NMR (75 MHz, D2O) δ 175.12, 174.61, 174.03, 102.69, 101.28, 99.93, 78.37, 75.58, 75.28, 74.86, 73.00, 72.46, 71.89, 69.50, 68.48, 68.20, 67.59, 67.25, 62.68, 61.15, 60.13, 55.20, 51.80, 47.89, 39.74, 28.23, 22.29, 22.17. HRMS (ESI) m/z calcd for C28H46N5O19Na (M-Na) 756.2782, found 756.2789.
Neu5Gcα2–3Galβ1–4GlcNAcβProN3 (33)
From disaccharide 2 (46.6 mg, 0.1 mmol), compound 33 was obtained as a white foam (64 mg, 80%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 7.8 Hz), 4.50 (d, 1 H, J = 8.4 Hz), 4.11 (dd, 1 H, J = 9.3 Hz and 3.3 Hz), 4.10 (s, 2 H), 3.99–3.54 (m, 20 H), 3.38–3.34 (m, 2 H), 2.75 (dd, 1 H, J = 12.6 Hz and 4.8 Hz), 2.02 (s, 3 H), 1.82 (pentalet, 2 H, J = 6.0 Hz), 1.80 (t, 1 H, J = 12.6 Hz). 13C NMR (75 MHz, D2O)δ 175.90, 174.19, 174.06, 102.68, 101.27, 99.92, 78.36, 75.57, 75.28, 74.86, 72.71, 72.45, 71.94, 69.51, 68.21, 68.12, 67.57, 67.24, 62.64, 61.08, 60.12, 55.20, 51.49, 47.88, 39.79, 28.22, 22.29. HRMS (ESI) m/z calcd for C28H46N5O20Na (M-Na) 772.2731, found 772.2730.
Neu5Acα2–3Galβ1–4GlcNAc6SβProN3 (34)
From disaccharide 3 (56.8 mg, 0.1 mmol), compound 34 was obtained as a white foam (77 mg, 88%). 1H NMR (400 MHz, D2O) δ 4.61 (d, 1 H, J = 7.6 Hz), 4.54 (d, 1 H, J = 8.0 Hz), 4.42–4.32 (m, 2 H), 4.12 (dd, 1 H, J = 9.8 Hz and 3.0 Hz), 3.99–3.53 (m, 18 H), 3.39–3.54 (m, 2 H), 2.74 (dd, 1 H, J = 12.4 Hz and 4.4 Hz), 2.04 (s, 3 H), 2.03 (s, 3 H), 1.84 (pentalet, 2 H, J = 6.2 Hz), 1.80 (t, 1 H, J = 12.0 Hz). 13C NMR (75 MHz, D2O) δ 175.09, 174.67, 174.21, 102.27, 101.36, 99.89, 77.29, 75.47, 75.24, 72.99, 72.66, 72.37, 71.64, 69.60, 68.59, 68.21, 67.62, 67.44, 66.40, 62.65, 61.21, 55.21, 51.86, 47.94, 39.74, 28.26, 22.34, 22.24. HRMS (ESI) m/z calcd for C28H45N5O22SNa2 (M-Na) 858.2174, found 858.2151.
Neu5Gcα2–3Galβ1–4GlcNAc6SβProN3 (35)
From disaccharide 3 (56.8 mg, 0.1 mmol), compound 35 was obtained as a white foam (74 mg, 82%). 1H NMR (400 MHz, D2O) δ 4.60 (d, 1 H, J = 7.6 Hz), 4.53 (d, 1 H, J = 7.6 Hz), 4.40–4.31 (m, 2 H), 4.12 (dd, 1 H, J = 9.6 Hz and 2.4 Hz), 4.10 (s, 2 H), 3.98–3.52 (m, 18 H), 3.38–3.32 (m, 2 H), 2.75 (dd, 1 H, J = 12.6 Hz and 4.6 Hz), 2.03 (s, 3 H), 1.83 (pentalet, 2 H, J = 6.0 Hz), 1.80 (t, 1 H, J = 12.6 Hz). 13C NMR (100 MHz, D2O) δ 175.90, 174.69, 174.23, 102.36, 101.38, 99.95, 77.46, 75.53, 75.26, 72.73, 72.42, 71.70, 69.63, 68.34, 68.21, 67.66, 67.45, 66.44, 62.64, 61.22, 55.24, 51.63, 47.96, 39.85, 27.27, 22.38. HRMS (ESI) m/z calcd for C28H45N5O23SNa2 (M-Na) 874.2124, found 874.2107.
Neu5Acα2–3Galβ1–3GalNAcαProN3 (36)
From disaccharide 4 (46.6 mg, 0.1 mmol), compound 36 was obtained as a white foam (68 mg, 87%). 1H NMR (600 MHz, D2O) δ 4.88 (d, 1 H, J = 3.6 Hz), 4.52 (d, 1 H, J = 7.8 Hz), 4.29 (dd, 1 H, J = 10.8 Hz and 3.6 Hz), 4.22 (d, 1 H, J = 3.0 Hz), 4.05 (dd, 1 H, J = 10.8 Hz and 3.6 Hz), 4.02 (dd, 1 H, J = 10.8 Hz and 3.0 Hz), 3.97 (t, 1 H, J = 6.0 Hz), 3.91 (d, 1 H, J = 3.6 Hz), 3.87–3.41 (m, 17 H), 2.73 (dd, 1 H, J = 12.3 Hz and 4.5 Hz), 2.01 (s, 3 H), 1.88 (pentalet, 2 H, J = 6.3 Hz), 1.76 (t, 1 H, J = 12.3 Hz). 13C NMR (150 MHz, D2O) δ 175.12, 174.71, 174.05, 104.60, 99.85, 97.31, 77.52, 75.78, 74.90, 72.93, 71.96, 70.76, 69.22, 68.70, 68.51, 68.17, 67.52, 65.04, 62.63, 61.35, 61.11, 51.78, 48.81, 48.30, 39.84, 28.10, 22.18. HRMS (ESI) m/z calcd for C28H46N5O19Na (M-Na) 756.2782, found 756.2801.
Neu5Gcα2–3Galβ1–3GalNAcαProN3 (37)
From disaccharide 4 (46.6 mg, 0.1 mmol), compound 37 was obtained as a white foam (60 mg, 76%). 1H NMR (600 MHz, D2O) δ 4.88 (d, 1 H, J = 3.6 Hz), 4.52 (d, 1 H, J = 8.4 Hz), 4.30 (dd, 1 H, J = 11.1 Hz and 3.9 Hz), 4.22 (d, 1 H, J = 3.0 Hz), 4.10 (s, 2 H), 4.06 (dd, 1 H, J = 9.6 Hz and 3.6 Hz), 3.97 (t, 1 H, J = 6.0 Hz), 3.93–3.41 (m, 18 H), 2.75 (dd, 1 H, J = 12.6 Hz and 4.8 Hz), 2.01 (s, 3 H), 1.88 (pentalet, 2 H, J = 6.3 Hz), 1.78 (t, 1 H, J = 12.3 Hz). 13C NMR (75 MHz, D2O) δ 175.98, 174.79, 174.13, 104.67, 99.98, 97.42, 77.60, 75.91, 75.03, 72.78, 72.12, 70.87, 69.34, 68.80, 68.34, 68.24, 67.64, 65.21, 62.74, 61.45, 61.23, 51.62, 48.93, 48.44, 40.05, 28.19, 22.30. HRMS (ESI) m/z calcd for C28H46N5O20Na (M-Na) 772.2731, found 772.2758.
Neu5Acα2–3Galβ1–3GalNAcβProN3 (38)
From disaccharide 5 (46.6 mg, 0.1 mmol), compound 38 was obtained as a white foam (63 mg, 81%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 7.8 Hz), 4.52 (d, 1 H, J = 8.4 Hz), 4.19 (d, 1 H, J = 3.0 Hz), 4.08 (dd, 1 H, J = 10.2 Hz and 3.0 Hz), 4.04–3.98 (m, 2 H), 3.90 (d, 1 H, J = 2.4 Hz), 3.87–3.54 (m, 16 H), 3.40 (t, 2 H, J = 6.6 Hz), 2.77 (dd, 1 H, J = 12.3 Hz and 4.5 Hz), 2.05 (s, 3 H), 2.04 (s, 3 H), 1.86 (pentalet, 2 H, J = 6.0 Hz), 1.80 (t, 1 H, J = 12.3 Hz). 13C NMR (75 MHz, D2O) δ 175.24, 174.95, 174.14, 104.80, 101.64, 99.95, 80.24, 75.82, 74.98, 73.04, 72.80, 72.04, 69.26, 68.59, 68.32, 68.12, 67.64, 67.27, 62.89, 62.77, 61.19, 51.93, 51.43, 48.07, 39.99, 28.36, 22.56, 22.31. HRMS (ESI) m/z calcd for C28H46N5O19Na (M-Na) 756.2782, found 756.2783.
Neu5Gcα2–3Galβ1–3GalNAcβProN3 (39)
From disaccharide 5 (46.6 mg, 0.1 mmol), compound 39 was obtained as a white foam (63 mg, 79%). 1H NMR (600 MHz, D2O) δ 4.53 (d, 1 H, J = 7.8 Hz), 4.52 (d, 1 H, J = 8.4 Hz), 4.19 (d, 1 H, J = 3.6 Hz), 4.14 (s, 2 H), 4.10 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 4.04–3.55 (m, 19 H), 3.41 (t, 2 H, J = 6.3 Hz), 2.79 (dd, 1 H, J = 12.3 Hz and 4.5 Hz), 2.05 (s, 3 H), 1.87 (pentalet, 2 H, J = 6.3 Hz), 1.82 (t, 1 H, J = 12.3 Hz). 13C NMR (150 MHz, D2O) δ 175.92, 174.92, 174.16, 104.79, 101.60, 99.88, 80.21, 75.76, 74.97, 74.93, 72.70, 72.07, 69.19, 68.31, 68.17, 68.06, 67.54, 67.20, 62.64, 61.17, 61.14, 51.56, 51.35, 47.99, 39.99, 28.30, 22.50. HRMS (ESI) m/z calcd for C28H46N5O20Na (M-Na) 772.2731, found 772.2729.
Neu5Acα2–3Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (40)
From tetrasaccharide 6 (30 mg, 0.038 mmol), compound 40 was obtained as a white foam (37 mg, 88%). 1H NMR (600 MHz, D2O) δ 4.71 (d, 1 H, J = 8.4 Hz), 4.49 (d, 1 H, J = 7.8 Hz), 4.46 (d, 1 H, J = 7.8 Hz), 4.42 (d, 1 H, J = 8.4 Hz), 4.13 (d, 1 H, J = 3.6 Hz), 4.06 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 4.00–3.43 (m, 32 H), 3.30–3.27 (m, 1 H), 2.73 (dd, 1 H, J = 12.6 Hz and 4.8 Hz), 2.01 (s, 6 H), 1.89 (pentalet, 2 H, J = 6.3 Hz), 1.76 (t, 1 H, J = 12.6 Hz). 13C NMR (75 MHz, D2O) δ 175.08, 174.05, 103.51, 103.06, 102.65, 102.23, 99.75, 82.24, 82.05, 78.46, 75.71, 75.31, 75.21, 75.02, 74.89, 74.48, 72.90, 71.97, 70.12, 69.20, 68.51, 68.41, 68.14, 67.48, 67.36, 62.71, 62.56, 61.08, 60.61, 60.14, 54.70, 51.77, 47.97, 39.88, 28.35, 22.42, 22.16. HRMS (ESI) m/z calcd for C40H66N5O29Na (M-Na) 1080.3849, found 1080.3827.
Neu5Gcα2–3Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (41)
From tetrasaccharide 6 (30 mg, 0.038 mmol), compound 41 was obtained as a white foam (35 mg, 88%). 1H NMR (600 MHz, D2O) δ 4.71 (d, 1 H, J = 8.4 Hz), 4.49 (d, 1 H, J = 8.4 Hz), 4.46 (d, 1 H, J = 7.8 Hz), 4.42 (d, 1 H, J = 7.8 Hz), 4.13 (d, 1 H, J = 3.6 Hz), 4.10 (s, 2 H), 4.07 (dd, 1 H, J = 9.6 Hz and 3.0 Hz), 3.99–3.51 (m, 29 H), 3.47–3.43 (m, 3 H), 3.30–3.27 (m, 1 H), 2.75 (dd, 1 H, J = 12.6 Hz and 4.8 Hz), 2.01 (s, 3 H), 1.89 (pentalet, 2 H, J = 6.6 Hz), 1.78 (t, 1 H, J = 12.6 Hz). 13C NMR (75 MHz, D2O) δ 175.91, 175.09, 174.10, 103.55, 103.11, 102.67, 102.28, 99.83, 99.77, 82.29, 82.10, 78.56, 75.71, 75.33, 75.25, 75.03, 74.94, 74.48, 72.95, 72.69, 72.07, 70.18, 69.26, 68.57, 68.48, 68.25, 68.15, 67.49, 62.59, 61,13, 60.62, 60.18, 54.76, 51.54, 47.98, 39.97, 28.40, 22.48. HRMS (ESI) m/z calcd for C40H66N5O30Na (M-Na) 1096.3787, found 1096.3783.
3 Results and discussion
3.1 Synthesis of sialyltransferase acceptors
Six sialyltransferase acceptors (1–6) including type 1 (Galβ1–3GlcNAcβProN3, 1), type 2 (Galβ1–4GlcNAcβProN3, 2 and 6-sulfo-Galβ1–4GlcNAcβProN3, 3), type 3 or core 1 (Galβ1–3GalNAcαProN3, 4), and type 4 (Galβ1–3GalNAcβProN3, 5) disaccharides as well as type 1 tetrasaccharide (Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3, 6) were chemically synthesized.
Disaccharide Galβ1–3GalNAcβProN3 (1) was prepared as shown in Scheme 1. Deacetylation of glucosamine derivative 7 with NaOMe/MeOH [35] followed by 4,6-O-benzylidene protection with benzaldehyde dimethyl acetal produced 8 (87%), which was coupled with 2,3,4,6-O-acetyl-α-D-galactopyranosyl trichloroacetimidate donor 9 [36] in the presence of TMSOTf to afford disaccharide 10 in a moderate 53% yield. Removal of benzylidene protection group from 10 by hydrogenation followed by SN2 substitution of the chlorine by azido group gave 11 in 95% yield. Removal of the phthalimido group by treating with hydrazine, followed by acetylation and deacetylation, the disaccharide Galβ1–3GlcNAcβProN3 (1) was produced in 84% yield over three steps.
Scheme 1.

Reagents and yields: (a) (i) NaOMe, MeOH; (ii) PhCH(OMe)2, CSA, DMF, 87%; (b) TMSOTf, CH2Cl2, 53%; (c) (i) Pd/C, H2; (ii) NaN3, TBAI, DMF, 95%; (d) (i) N2H4-H2O, EtOH; (ii) Ac2O, pyridine; (iii) NaOMe, MeOH, 84%.
Disaccharides 2 and 3 were synthesized as shown in Scheme 2. Monosaccharide acceptor 12 was synthesized from 7 in 84% yield by deacetylation followed by selective silylation at O-6 with tert-butyldimethylsilyl chloride. Coupling of trichloroacetimidate 9 with acceptor 12 was performed in dichloromethane using TMSOTf as a promoter to give 13 (79%). Removal of the tert-butyldimethylsilyl and transformation of chlorine to azide afforded 14 in 76% yield. Deprotection of 14 as described for 11 afforded disaccharide Galβ1–4GlcNAcβProN3 (2) in 82% yield over three steps. Acetylation of 13 with Ac2O in pyridine, followed by removal of TBS group and transformation of chlorine to azide groups produced 15 (85%). Sulfation at C-6 of 15 with SO3. Pyinpyridine, removal of the 2-N-phthalyl group, followed by acetylation and deacetylation produced disaccharide Galβ1–4GlcNAc6SβProN3 (3) in 66% overall yield.
Scheme 2.

Reagents and yields: (a) (i) NaOMe, MeOH (ii) TBSCl, imidazole, CH2Cl2, 84%; (b) 9, TMSOTf, CH2Cl2, 79%; (c) (i) TABF, THF; (ii) NaN3, TBAI, DMF, 76%; (d) (i) Ac2O, pyridine; (ii) TABF, THF; (iii) NaN3, TBAI, DMF, 85%; (e) (i)) N2H4-H2O, EtOH; (ii) Ac2O, pyridine; (iii) NaOMe/MeOH, 82%; (f) (i) Py-SO3, pyridine; (ii) N2H4-H2O, EtOH; (iii) Ac2O, pyridine; (iv) NaOMe, MeOH, 66%.
To prepare disaccharides 4 and 5, GalNAc derivative 16 [37] was treated with 3-chloro-1-propanol and acetyl chloride followed by 4,6-O-benzylidenation to produce a mixture of 17 (21%) and 18 (57%). Coupling of trichloroacetimidate 9 with 17 and 18, respectively, using TMSOTf as a promoter produced disaccharides 19 and 20 in 62% and 48% yields, respectively. Substitution of the chlorine by azide using sodium azide and subsequent removal of benzylidene using acetic acid afforded 21 (94%) and 22 (91%). Deprotection of 21 and 22 as described for 11 afforded disaccharides Galβ1–4GalNAcαProN3 (4) and Galβ1–4GalNAcβProN3 (5) in 85% and 76% yields, respectively.
For the synthesis of type 1 LNnT tetrasaccharide 6, compound 23 [38] was deacetylated and selectively protected with benzylidene to produce 24 (88%). Disaccharide donor 25 was synthesized by coupling the monosaccharide 24 with trichloroacetimidate donor 9 in the presence of TMSOTf with 77% yield. Glycosylation of 25 with a disaccharide acceptor 26 [39] using NIS TfOH as the promoters produced tetrasaccharide 27 (64%). Global deprotection leaded to the production of the desired LNT tetrasaccharide Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (6) in 54% yield.
3.2 One-pot three-enzyme approach for the synthesis of sialylated glycans
With chemically synthesized glycan acceptors 1–6 in hands, the preparation of α2–3-linked sialyl oligosaccharides was carried out using an efficient one-pot three-enzyme approach [32] (Figure 2) developed in our laboratory. In this system, N-acetylmannosamine (ManNAc) or N-glycolylmannosamine (ManNGc) was coupled with pyruvate to give sialic acids Neu5Ac or Neu5Gc by a recombinant Escherichia coli K12 sialic acid aldolase-catalyzed reaction [40]. The sialic acid formed was then activated by a recombinant Neisseria meningitidis CMP-sialic acid synthetase [40], and transferred to a suitable sialyltransferase acceptor by a Pasteurella multocida multifunctional α2–3-sialyltransferase [31] for the formation of sialosides.
Figure 2.

One-pot three-enzyme synthesis of sialosides containing different sialic acid forms and various internal glycans. Abbreviations: ManNAc, N-acetylmannosamine; ManNGc, N-glycolylmannosamine; CTP, cytidine 5′-triphosphate. Enzymes: E. coli aldolase, Escherichia coli K12 sialic acid aldolase; NmCSS, Neisseria meningitidis CMP-sialic acid synthetase; PmST1, Pasteurella multocida sialyltransferase for the formation of α2–3-linked sialosides.
As shown in Table 1, all six oligosaccharides 1–6 prepared were very good acceptor substrates for Pasteurella multocida α2–3-sialyltransferase. Sialic acids (Neu5Ac or Neu5Gc) were successfully transferred to the galactose (Gal) residue through an α2–3-sialyl linkage by the one-pot three-enzyme reaction system. Twelve sialosides representing commond naturally occurring sialic acid-containing carbohydrate epitopes including α2–3-linked sialy l type 1 Neu5Acα2–3Galβ1–3GlcNAcβProN3 (30, 85%) and Neu5Gcα2–3Galβ1–3GlcNAcβProN3 (31, 83%); type 2 Neu5Acα2–3Galβ1–4GlcNAcβProN3 (32, 8–4 % ), Neu5Gcα2–3Galβ1–4GlcNAcβProN3 (33, 8–0 % ), Neu5Acα2–3Galβ1–4GlcNAc6SβProN3 (34, 88%), and Neu5Gcα2–3Galβ1–4GlcNAc6SβProN3 (35, 82%); type 3 Neu5Acα2–3Galβ1–3GalNAcαProN3 (36, 87%) and Neu5Gcα2–3Galβ1–3GalNAcαProN3 (37, 76%); type 4 Neu5Acα2–3Galβ1–3GalNAcβProN3 (38, 81%), and Neu5Gcα2–3Galβ1–3GalNAcβProN3 (39, 79%) glycans as well as α2–3-linked sialyllacto-N-tetraose Neu5Acα2–3Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (40, 86%) and Neu5Gcα2–3Galβ1–3GlcNAcβ1–3Galβ1–4GlcβProN3 (41, 81%) were obtained in high yields.
Table 1.
α2–3-Linked sialosides synthesized using one-pot three-enzyme system shown in Figure 2
| entry | acceptor | sialic acid precurso | α2–3-linked sialosides (yield %) |
|---|---|---|---|
| a |
1 |
 ManNAc, 28 |
30 (85%) |
| b |
 ManNGc, 29 |
31 (83%) |
|
| c |
2 |
28 |
32 (84%) |
| d | 29 |
33 (80%) |
|
| e |
3 |
28 |
34 (88%) |
| f | 29 |
35 (82%) |
|
| g |
 4 |
28 |
36 (87%) |
| h | 29 |
37 (76%) |
|
| i |
5 |
28 |
38 (81%) |
| j | 29 |
39 (79%) |
|
| k |
6 |
28 |
40 (86%) |
| l | 29 |
41 (81%) |
4 Conclusions
In summary, chemoenzymatic method which combines chemical and enzymatic synthetic approaches, was demonstrated once again to be a general and efficient strategy to obtain a library of homogenous sialosides containing different sialic acid forms and various internal carbohydrate structures. The α2–3-linked sialosides obtained in this work are valuable probes to study the biological importance of naturally occurring diverse sialic acid-containing carbohydrate epitopes.
Scheme 3.

Reagents and yields: (a) (i) AcCl, 3-chloro-1-propanol; (ii) PhCH(OMe)2, CSA, DMF, 17:21%; 18: 57%; (b) 9, TMSOTf, CH2Cl2, 19:62%; 20: 48%; (c) (i) NaN3, TBAI, DMF; (ii) 80% HOAc, 21:94%; 22: 91%; (d) (i) N2H4-H2O, EtOH; (ii) Ac2O, pyridine; (iii) NaOMe, MeOH, 4:85%; 5: 76%.
Scheme 4.

Reagents and yields: (a) (i) NaOMe, MeOH; (ii) PhCH(OMe)2, CSA, 88%; (b) 9, TMSOTf, CH2Cl2, 77%; (c) NIS, TfOH, CH2Cl2, 64%; (d) (i) 80% HOAc; ii) N2H4-H2O, EtOH; (iii) Ac2O, pyridine; (iv) NaOMe, MeOH, 54%.
Acknowledgments
This work was supported by NIH grants R01GM076360 and U01CA128442. X. Chen is an Alfred P. Sloan Research Fellow, a Camille Dreyfus Teacher-Scholar, and a UC-Davis Chancellor’s Fellow.
References
- 1.Tsuji S, Datta AK, Paulson JC. Systematic nomenclature for sialyltransferases. Glycobiology. 1996;6:v–vii. doi: 10.1093/glycob/6.7.647. [DOI] [PubMed] [Google Scholar]
- 2.Fukuda MN, Dell A, Oates JE, Fukuda M. Embryonal lactosaminoglycan. The structure of branched lactosaminoglycans with novel disialosyl (sialyl alpha 2–9 sialyl) terminals isolated from PA1 human embryonal carcinoma cells. J Biol Chem. 1985;260:6623–6631. [PubMed] [Google Scholar]
- 3.Schauer R. Achievements and challenges of sialic acid research. Glycoconj J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Varki A. Advances in the biology and chemistry of sialic acids. ACS Chem Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomiya N, Narang S, Lee YC, Betenbaugh MJ. Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines. Glycoconj J. 2004;21:343–360. doi: 10.1023/B:GLYC.0000046275.28315.87. [DOI] [PubMed] [Google Scholar]
- 7.Betenbaugh MJ, Tomiya N, Narang S, Hsu JT, Lee YC. Biosynthesis of human-type N-glycans in heterologous systems. Curr Opin Struct Biol. 2004;14:601–606. doi: 10.1016/j.sbi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2008. [PubMed] [Google Scholar]
- 9.Vimr E, Lichtensteiger C, Steenbergen S. Sialic acid metabolism’s dual function in Haemophilus influenzae. Mol Microbiol. 2000;36:1113–1123. doi: 10.1046/j.1365-2958.2000.01925.x. [DOI] [PubMed] [Google Scholar]
- 10.Fuller TE, Kennedy MJ, Lowery DE. Identification of Pasteurella multocida virulence genes in a septicemic mouse model using signature-tagged mutagenesis. Microb Pathog. 2000;29:25–38. doi: 10.1006/mpat.2000.0365. [DOI] [PubMed] [Google Scholar]
- 11.Ringenberg M, Lichtensteiger C, Vimr E. Redirection of sialic acid metabolism in genetically engineered Escherichia coli. Glycobiology. 2001;11:533–539. doi: 10.1093/glycob/11.7.533. [DOI] [PubMed] [Google Scholar]
- 12.Yamaji T, Teranishi T, Alphey MS, Crocker PR, Hashimoto Y. A small region of the natural killer cell receptor, Siglec-7, is responsible for its preferred binding to alpha 2,8-disialyl and branched alpha 2,6-sialyl residues. A comparison with Siglec-9. J Biol Chem. 2002;277:6324–6332. doi: 10.1074/jbc.M110146200. [DOI] [PubMed] [Google Scholar]
- 13.Seymour JL, Costello CE, Zaia J. The influence of sialylation on glycan negative ion dissociation and energetics. J Am Soc Mass Spectrom. 2006;17:844–854. doi: 10.1016/j.jasms.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaila N, Thomas BE. Design and synthesis of sialyl Lewis(x) mimics as E- and P-selectin inhibitors. Med Res Rev. 2002;22:566–601. doi: 10.1002/med.10018. [DOI] [PubMed] [Google Scholar]
- 15.Stevens J, Blixt O, Paulson JC, Wilson IA. Glycan microarray technologies: tools to survey host specificity of influenza viruses. Nat Rev Microbiol. 2006;4:857–864. doi: 10.1038/nrmicro1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, Sasisekharan V, Sasisekharan R. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- 17.Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science. 2004;303:1838–1842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 18.Ha Y, Stevens DJ, Skehel JJ, Wiley DC. X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc Natl Acad Sci U S A. 2001;98:11181–11186. doi: 10.1073/pnas.201401198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell RJ, Stevens DJ, Haire LF, Gamblin SJ, Skehel JJ. Avian and human receptor binding by hemagglutinins of influenza A viruses. Glycoconj J. 2006;23:85–92. doi: 10.1007/s10719-006-5440-1. [DOI] [PubMed] [Google Scholar]
- 20.Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol. 2006;355:1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312:404–410. doi: 10.1126/science.1124513. [DOI] [PubMed] [Google Scholar]
- 22.Crocker PR. Siglecs in innate immunity. Curr Opin Pharmacol. 2005;5:431–437. doi: 10.1016/j.coph.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 24.Crocker PR, Varki A. Siglecs in the immune system. Immunology. 2001;103:137–145. doi: 10.1046/j.0019-2805.2001.01241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McMillan SJ, Crocker PR. CD33-related sialic-acid-binding immunoglobulin-like lectins in health and disease. Carbohydr Res. 2008;343:2050–2056. doi: 10.1016/j.carres.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Campanero-Rhodes MA, Childs RA, Kiso M, Komba S, Le Narvor C, Warren J, Otto D, Crocker PR, Feizi T. Carbohydrate microarrays reveal sulphation as a modulator of siglec binding. Biochem Biophys Res Commun. 2006;344:1141–1146. doi: 10.1016/j.bbrc.2006.03.223. [DOI] [PubMed] [Google Scholar]
- 27.Bochner BS, Alvarez RA, Mehta P, Bovin NV, Blixt O, White JR, Schnaar RL. Glycan array screening reveals a candidate ligand for Siglec-8. J Biol Chem. 2005;280:4307–4312. doi: 10.1074/jbc.M412378200. [DOI] [PubMed] [Google Scholar]
- 28.Tateno H, Crocker PR, Paulson JC. Mouse Siglec-F and human Siglec-8 are functionally convergent paralogs that are selectively expressed on eosinophils and recognize 6′-sulfo-sialyl Lewis X as a preferred glycan ligand. Glycobiology. 2005;15:1125–1135. doi: 10.1093/glycob/cwi097. [DOI] [PubMed] [Google Scholar]
- 29.Rapoport EM, Pazynina GV, Sablina MA, Crocker PR, Bovin NV. Probing sialic acid binding Ig-like lectins (siglecs) with sulfated oligosaccharides. Biochemistry (Mosc) 2006;71:496–504. doi: 10.1134/s0006297906050051. [DOI] [PubMed] [Google Scholar]
- 30.Boons GJ, Demchenko AV. Recent advances in O-sialylation. Chem Rev. 2000;100:4539–4566. doi: 10.1021/cr990313g. [DOI] [PubMed] [Google Scholar]
- 31.Yu H, Chokhawala H, Karpel R, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. A multifunctional Pasteurella multocida sialyltransferase: a powerful tool for the synthesis of sialoside libraries. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Chokhawala HA, Huang S, Chen X. One-pot three-enzyme chemoenzymatic approach to the synthesis of sialosides containing natural and non-natural functionalities. Nat Protoc. 2006;1:2485–2492. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural alpha-2,6-linked sialosides: a P. damsela alpha-2,6-sialyltransferase with extremely flexible donor-substrate specificity. Angew Chem Int Ed Engl. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H, Cheng J, Ding L, Khedri Z, Chen Y, Chin S, Lau K, Tiwari VK, Chen X. Chemoenzymatic synthesis of GD3 oligosaccharides and other disialyl glycans containing natural and non-natural sialic acids. J Am Chem Soc. 2009;131:18467–18477. doi: 10.1021/ja907750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao H, Huang S, Cheng J, Li Y, Muthana S, Son B, Chen X. Chemical preparation of sialyl Lewis x using an enzymatically synthesized sialoside building block. Carbohydr Res. 2008;343:2863–2869. doi: 10.1016/j.carres.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H, Chen X. Aldolase-catalyzed synthesis of beta-D-Galp-(1–9)-D-K D N: A novel accept or for sialyltransferases. Org Lett. 2006;8:2393–2396. doi: 10.1021/ol060736m. [DOI] [PubMed] [Google Scholar]
- 37.Karpiesiuk W, Banaszek A. Highly stereoselective synthesis of alpha,beta-linked, nonreducing disaccharides related to tunicamycin. Carbohydr Res. 1994;261:243–253. doi: 10.1016/0008-6215(94)84021-0. [DOI] [PubMed] [Google Scholar]
- 38.Agnihotri G, Tiwari P, Misra AK. One-pot synthesis of per-O-acetylated thioglycosides from unprotected reducing sugars. Carbohydr Res. 2005;340:1393–1396. doi: 10.1016/j.carres.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 39.Yudina ON, Sherman AA, Nifantiev NE. Synthesis of propyl and 2-aminoethyl glycosides of alpha-D-galactosyl-(1–3)-beta-lactoside. Carbohydr Res. 2001;332:363–371. doi: 10.1016/s0008-6215(01)00097-0. [DOI] [PubMed] [Google Scholar]
- 40.Yu H, Yu H, Karpel R, Chen X. Chemoenzymatic synthesis of CMP-sialic acid derivatives by a one-pot two-enzyme system: comparison of substrate flexibility of three microbial CMP-sialic acid synthetases. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]