

Abstract
Intestinal electroneutral Na+ absorptive processes account for most intestinal Na+ absorption in the period between meals and also for the great majority of the increase in ileal Na+ absorption that occurs post-prandially. In most diarrheal diseases, there is inhibition of neutral NaCl absorption. Elevated levels of intracellular calcium ([Ca2+]i) are known to inhibit NaCl absorption and involve multiple components of the Ca2+ signaling pathway. The BB Na+/H+ exchanger NHE3 accounts for most of the recognized digestive changes in neutral NaCl absorption, as well as most of the changes in Na+ absorption that occur in diarrheal diseases. Previous studies have examined several aspects of Ca2+ regulation of NHE3 activity. These include phosphorylation, protein trafficking and multi-protein complex formation. In addition, recent studies have demonstrated the role of the NHERF family of PDZ domain–containing proteins in Ca2+ regulation of NHE3 activity, thereby adding a new level of complexity to understanding Ca2+-dependent inhibition of Na+ absorption. In this article, we will review the current understanding of (1) Ca2+ signaling events in intestinal epithelial cells; (2) Ca2+ regulation of intestinal electroneutral sodium absorption, which includes NHE3; and (3) the role of the NHERF family of PDZ domain–containing proteins in Ca2+ regulation of NHE3 activity. We will also present new data on using advanced imaging showing rapid BB NHE3 endocytosis in response to elevated [Ca2+]i.
Keywords: NHE3, intracellular calcium, NHERF
Introduction
The sodium/hydrogen exchanger (NHE) gene family—specifically the NHE3 isoform—is a part of neutral sodium absorption in the mammalian intestine.1 Regulation of NHE3 occurs during normal digestion and is inhibited in most diarrheal diseases.2,3 In some diarrheal diseases, there is both inhibition of neutral NaCl absorption, and stimulation of electrogenic Cl− secretion. This accounts for the major GI loss of water and electrolytes in diarrhea.4,5 However, in some inflammatory diarrheal diseases, it appears that only inhibition of Na+ absorption and not stimulation of Cl− secretion occurs.
Intestinal electroneutral Na+ absorption is regulated during the postprandial state as part of the neurohumoral response in digestion.2,3 In vivo and in vitro studies have described the regulation of Na+ absorption in the GI tract using agonists/antagonists that mimic digestion as well as the second messengers through which they act.1 Cyclic AMP, cGMP and elevated intracellular calcium [Ca2+]i, as well as neurohumoral substances and bacterial toxins that cause elevation of these second messengers, inhibit neutral NaCl absorption in the ileum and colon and inhibit NHE3 activity.1 cAMP and cGMP also stimulate chloride secretion. Similarly, elevated levels of free intracellular calcium ([Ca2+]i) inhibit NHE3 activity and stimulate Cl− secretion but do not have the same prolonged time course and magnitude as cyclic nucleotide-dependent chloride secretion.6–9 Previous studies have demonstrated that elevated [Ca2+]i inhibits intestinal Na+ absorption. However, the cellular and molecular mechanisms underlying this regulation are only partially defined. This review will discuss the current understanding of the mechanisms responsible for [Ca2+]i regulation of intestinal sodium absorption.
Calcium Signaling
One major pathway of calcium signaling involves phospholipase C (PLC)–mediated release of Ca2+ from intracellular stores. Various extracellular stimuli, including hormones, growth factors, and neurotransmitters, activate receptor tyrosine kinases (RTK), which subsequently activate PLCs—both PLCβs and PLCγ.10–12 Upon activation, PLCs hydrolyze membrane-bound phosphatidylinositol 4,5-bisphosphate (PIP2), producing two second messengers, diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 induces a transient increase in [Ca2+]i by releasing Ca2+ from intracellular stores, while DAG is an activator of protein kinase C (PKC).13
In addition to the lipase-dependent functions of PLCγ, recent studies have demonstrated that PLCγ exerts lipase-independent functions during Ca2+ signaling.14–18 The PLCγ lipase-dependent increase in [Ca2+]i also activates the calmodulin/calmodulin kinase II (CaM/CaMKII) complex, which phosphorylates a wide range of downstream substrates.19–23 An example of a CaM/CaMKII downstream substrate is cytosolic phospholipase A2 (cPLA2). cPLA2 is activated by direct Ca2+ binding through its C2 domain and phosphorylation at S515 by CaM/CaMK.24 Upon activation, the C2 domain confers Ca2+-dependent translocation of cPLA2 to the plasma membrane where it facilitates arachidonic acid (AA) release.25–27
The patterns of temporal and spatial elevation of [Ca2+]i vary, especially in epithelial cells. For example, in pancreatic acinar cells, hepatocytes, lacrimal glands, and MDCK cells treated with carbachol (acting via basolateral M3 receptors), there is an initial rise in [Ca2+]i at the apical membrane that precedes gradual increases throughout the remainder of the cytosol.28–31 These results are consistent with [Ca2+]i studies in intestinal and pancreatic epithelial cells, demonstrating the presence of high affinity, type 3 IP3 receptors localized near the apical membrane.32–34 Because receptor-mediated elevation of [Ca2+]i involves multiple steps, some studies have also focused on understanding signaling events downstream of elevated [Ca2+]i by utilizing calcium ionophores. In these studies, it is presumed that elevation of [Ca2+]i occurs more evenly throughout the cell and thus may not trigger the same intracellular Ca2+ signaling events. For example, studies in the intact rabbit ileum treated with carbachol demonstrated an elevation of [Ca2+]i, which inhibited NHE3 activity by 40%.35,36 Carbachol treatment resulted in a transient increase in [Ca2+]i levels at the apical membrane which preceded a rise of [Ca2+]i throughout the cytoplasm at later time points.29 Carbachol treatment also resulted in a rapid translocation of activated PLCγ to the apical membrane, which was not tyrosine phosphorylated.37 In contrast, translocation did not occur when tissue was treated with the calcium ionophore A23187. This suggested that PLCγ translocation is linked to signaling steps initiated via activation of the M3 cholinergic receptor prior to the increase in [Ca2+]i.37 These results illustrate the necessity to define both apical and basolateral signaling events to understand Ca2+ regulation in polarized epithelial cells.
Intracellular Ca2+ Regulation of Intestinal Sodium Absorption
Elevated levels of [Ca2+]i have been shown to inhibit electroneutral sodium absorption in the intact intestine during normal digestion as well as in pathophysiological conditions.35,36,38,39 In animal models of diarrhea, some bacterial pathogens exert their effects through Ca2+ signaling events originating from intracellular stores. Salmonella typhimurium infection in rats resulted in increased [Ca2+]i and inhibited Na+ and Cl− absorption in a PKC-dependent manner.40 Additionally, in the rat ileum infected with Campylobacter jejuni, [Ca2+]i levels were significantly elevated and associated with reduced Na+ absorption, which was also PKC- but not calmodulin-dependent.41 Infection of the intact rabbit ileum with Shigella dysenteriae type I toxin induced a five-fold increase in [Ca2+]i, which inhibited Na+ and Cl− absorption.42 While previous studies demonstrated that bacterial pathogens induce Ca2+-mediated inhibition of electroneutral sodium absorption, the precise mechanisms involved in this regulation were not determined. In human inflammatory bowel diseases, there is inhibition of NHE3 protein amount.43 In colonic biopsies obtained from patients with ulcerative colitis, there was also a significant decrease in NHE1 and ENaC mRNA and protein expression.44 Whether elevated levels of [Ca2+]i contribute to decreased sodium absorption in patients with inflammatory bowel diseases is not known.
Mechanisms of NHE3 Regulation by Elevated [Ca2+]i
Intestinal electroneutral NaCl absorption and BB Na+/H+ exchange (e.g., NHE3) are inhibited by elevation of [Ca2+]i as induced by various physiological and pathological agonists. In addition to the above examples, these include carbachol (CCH), serotonin (5-HT), heat-stable E. coli toxin B, and the rotavirus enterotoxin, NSP5.35,39,45 In addition, elevating [Ca2+]i directly with ionophore (e.g., A23187; ionomycin) also inhibits NHE3 activity.46,47 Acute regulation of NHE3 activity is mediated predominantly by changes in maximal velocity (V max) of the exchanger.4 Such V max effects can be achieved by rapid changes either in the number of NHE3 molecules at the cell surface or in the number of exchange cycles per molecule per second (turnover number), or both. Recent studies have shown that NHE3 is often regulated by changes in its plasma membrane versus intracellular location as a result of changes in the rates of endocytosis and/or exocytosis.48–52 For example, epidermal growth factor (EGF) or clonidine stimulate NHE3 activity by increasing the rate of NHE3 exocytosis,53,54 whereas activation of PKC by phorbol myristyl acetate (PMA) inhibits NHE3 activity partly by stimulating NHE3 endocytosis.50,55 NHE3 traffics between the plasma membrane and recycling endosomes under basal and regulated conditions in all cells in which this has been studied.49,50,56 Figure 1 demonstrates NHE3 endocytosis in the BB of Caco-2BBe cells treated with 10 μM carbachol as determined by Total Internal Reflection Fluorescence (TIRF) microscopy. Delivery and removal of BB NHE3 was observed under basal conditions. However, after addition of carbachol, BB NHE3 expression was rapidly decreased (<1 min) as BB NHE3–containing vesicles were endocytosed beyond the depth of the TIRF field (<200 nm) into the cell.
Figure 1.
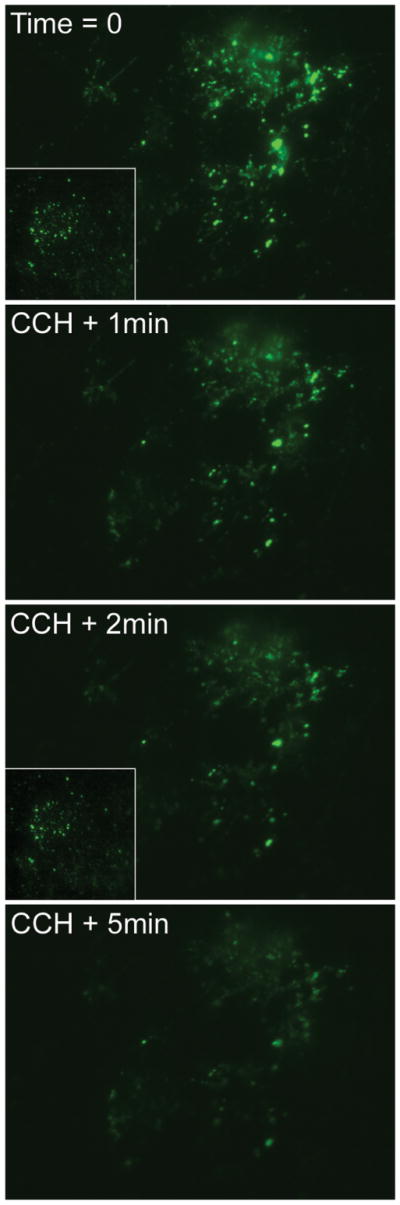
Total Internal Reflection Fluorescence (TIRF) of 3HA-NHE3 in Caco-2BBe cells treated with 10 μM carbachol (CCH). Caco2-BBe cells were grown on Anapore filters until 8 days post-confluency and infected with an adenovirus 3HA-NHE3 construct. After two days, cells were cooled at 4°C for 30 minutes and incubated with anti-HA AlexaFluor 488 primary antibody (1:100 dilution) in serum-free media for 3 hours. Cells were washed with ice-cold PBS before filter transfer to a 3 cm glass-bottom (0.17 mm) Petri dish containing pre-warmed (37°C) sodium-free buffer. NHE3 trafficking was observed on an Olympus microscope using a 100× oil (1.45 NA) TIRF objective. Caco2-BBe cells were excited with a 488 nm argon laser in 400 ms pulses and images were captured every second using a high-resolution CCD camera (Hammamatsu). Time = 0 indicates image captured 1 minute prior to addition of CCH. Remaining images demonstrate NHE3 trafficking at 1, 2 and 5 minutes after CCH treatment. Inserts illustrate BB NHE3 from untreated cells at the same time points, demonstrating lack of photobleaching.
The role of Ca2+ in signal transduction and protein trafficking remains incompletely understood. Ca2+ regulates various steps in the endocytic removal of integral membrane proteins as well as specific fusion events that take place during exocytic insertion of intracellular cargo to the plasma membrane. For example, activation of the insulin receptor results in increased surface expression of the GLUT4 transporter, which involves elevated [Ca2+]i.57,58 A role for Ca2+ has been conclusively demonstrated in the late stages of exocytosis, for instance involving the synaptotagmin family.59,60
Role of PLCγ in Ca2+ Regulation of NHE3 Activity
Decreased NHE3 activity in the intact rabbit ileum treated with carbachol was associated with decreased surface expression of NHE3 and increased size of multi-protein NHE3-containing complexes.61 Carbachol treatment in the rabbit ileum resulted in increased brush border levels of DAG and activation of brush border PLD, but not basolateral PLD, increased BB PLCγ amount and activity, as well as PKCα translocation to the BB.35 Since carbachol both elevates [Ca2+]i levels (apical initially) and translocates activated PKCα to the apical membrane, these findings suggested the involvement of PLCγ at the apical membrane. This was especially true because A23187 did not elevate apical PLCγ amount/activity, indicating a link between carbachol activation of basolateral M3 receptors and increased BB PLCγ amount/activity. Until now, it has been presumed that the sole role of PLCγ in NHE3 inhibition was to generate IP3 and DAG through its lipase activity to release calcium from intracellular stores and activate PKCα, respectively. However, PLCγ has also been shown to exert non-lipase–dependent functions in regulating agonist-induced calcium entry.14,16–18 PLCγ regulates trafficking and membrane retention of the Transient Receptor Potential Channel (TRPC3) by directly binding TRPC3 through its PH-c domain.18 Caraveo et al. demonstrated that the transcription factor TFII-I also binds to the SH2 and PH-c domains of PLCγ to prevent PLCγ binding to TRPC3 and thus prevent extracellular calcium entry.14 In addition to other studies, these results suggest that PLCγ mediates the formation of specific protein–protein interactions through its multiple protein binding domains, which contribute to the regulation of processes in addition to the lipase activity of PLCγ. In terms of NHE3 regulation, we have recently reported that PLCγ directly binds the NHE3 C-terminus through its C-terminal split PH (PH-c) domain.62
Role of NHERF Family in Ca2+ Regulation of NHE3 Activity
Acute regulation by ligands and second messengers of intestinal neutral NaCl absorption, including NHE3, is now known to depend on the NHERF family of multi-PDZ domain–containing proteins.1,63 A summary of recent studies involving the NHERF family in acute regulation of NHE3 activity is detailed in Table 1. The involvement of the NHERF family has added another level of complexity to the understanding of the post-prandial changes in Na+ absorption and the abnormalities of this process that occur in diarrheal diseases.
TABLE 1.
Role of the NHERF Proteins in Acute Regulation of NHE3 Activity by Second Messengers
| 2nd Messenger | NHERF1 | NHERF2 | NHERF3 | NHERF4 |
|---|---|---|---|---|
| cAMP | Yes | Yes | No | No |
| cGMP | No | Yes | No | No |
| ↑[Ca2+]i | No | Yes (inhibit) | Yes (inhibit) | Yes (stimulate) |
Studies in cell culture models and mouse KO models have demonstrated differing roles for each NHERF protein in second-messenger regulation of NHE3 activity. Although NHERF2–4 reconstitute elevated [Ca2+]i regulation of NHE3, NHERF4 is associated with stimulated NHE3 activity, while NHERF2 and NHERF3 are associated with NHE3 inhibition. However, NHERF2 and NHERF3 inhibit NHE3 activity by different mechanisms. NHERF2 is involved in NHE3 complex formation and endocytosis while NHERF3 appears to anchor NHE3 in the BB under basal conditions and releases NHE3 after elevated [Ca2+]i.
Several approaches have been taken to understand the contribution of individual NHERF proteins to NHE3 regulation. Cell lines lacking endogenous NHERF family members have been transfected with individual NHERF proteins, and knockout mouse models of individual NHERFs have been created. Studies performed in the proximal tubule of NHERF1 KO mice confirmed previous cell line studies suggesting a role for NHERF1 in cAMP-mediated inhibition of NHE3 activity but indicating, however, no role for NHERF2 that could compensate. In addition, these studies demonstrated that cAMP regulation via NHERF1 was tissue-dependent (i.e., NHERF1 was necessary for cAMP inhibition of NHE3 activity in the renal proximal tubule but not in the distal ileum).64,65 Ileal studies with the NHERF2 KO mouse gave a different picture, with reduced basal NHE3 activity and failure of cAMP, cGMP and [Ca2+]i to inhibit NHE3 activity, while colonic studies demonstrated dependence on NHERF2 for [Ca2+]i inhibition of NHE3.66 Moreover, studies of the NHERF3/PDZK1 KO mouse demonstrated that while these mice appeared healthy, basal colonic NHE3 activity was significantly reduced and these mice failed to respond to both cAMP and elevated [Ca2+]i with NHE3 inhibition.67,68 While these studies strongly suggest a role for NHERF proteins in NHE3 regulation, the cellular and molecular mechanisms responsible for NHERF-dependent regulation remain unclear. However, some insights into the role of NHERF3 and NHERF4 in NHE3 regulation have been recently reported. In Caco-2BBe cells, shRNA knockdown (KD) of endogenous NHERF3 (by ~60%) significantly reduced basal NHE3 activity by ~40% and abolished carbachol-mediated NHE3 inhibition (Zachos et al., unpublished results). Additionally, confocal microscopy illustrated that BB NHE3 expression was significantly reduced in Caco-2BBe cells with NHERF3 KD, suggesting that NHERF3 anchors NHE3 to the BB of polarized intestinal epithelial cells (Zachos et al., unpublished results). In PS120 cells stably expressing NHE3 and NHERF4, elevated [Ca2+]i increased NHE3 activity through stimulated exocytosis via NHERF4 release of NHE3 from the recycling endosome.69 In addition, studies of NHERF170,71 and NHERF2 KO72 mice have shown changes in expression of other NHERF protein family members, making it important to identify which NHERF protein accounts for changes in function. Intestinal studies of NHERF3/PDZK1 KO mice have not yet addressed whether changes in NHE3 regulation could also be due to compensatory changes of the other NHERF proteins.67,68
Previous studies examining acute NHE3 regulation by cAMP, cGMP and elevated [Ca2+]i identified a role for members of the NHERF family in forming NHE3-containing, multi-protein complexes.63,73 Of these identified regulatory processes, the most complicated appears to be Ca2+ inhibition. In addition to studies described above, cell model studies have indicated a role for NHERF proteins in NHE3 regulation, including Ca2+ inhibition. NHERF2 and NHERF3 can reconstitute Ca2+ inhibition in fibroblasts that lack all endogenous NHERFs other than a small amount of NHERF1 expression. Mechanistic studies have suggested that the role of NHERF2 in Ca2+ regulation of NHE3 involves the formation of NHE3-containing complexes that include α-actinin-4 and PKCα46 and dynamic fixation of NHE3 to the cytoskeleton.74 Recently, we have demonstrated that NHERF3 also reconstitutes elevated [Ca2+]i inhibition in PS120 cells but in a manner different from NHERF2, involving dynamic changes in plasma membrane fixation.47 That both NHERF2 and NHERF3 are involved in NHE3 inhibition with elevated [Ca2+]i indicates the complexity of this regulatory process.
Furthermore, other studies in the rabbit ileum have demonstrated that elevation of [Ca2+]i through activation of the muscarinic receptor (M3R) inhibits electroneutral NaCl absorption by a PKC-dependent mechanism, with NHE3 being the major transporter involved.35 Although the role of elevated [Ca2+]i is to inhibit sodium absorption, studies of OK cells (polarized renal epithelial cell model) treated with lysophosphatidic acid (LPA) demonstrated increased NHE3 activity and increased plasma membrane NHE3 by a NHERF2-dependent mechanism that was associated with elevated [Ca2+]i and was calcium-dependent (prevented by BAP-TAM).75 The NHERF2-requiring step identified was activation of PLCβ3 (bound to NHERF2) by LPA with a higher and more prolonged elevated Ca2+ response occurring in cells expressing NHERF2. Thus, the results of these studies provide a precedent for involvement of an NHERF protein in calcium-mediated stimulation of NHE3 activity under conditions in which proteins required for Ca2+ inhibition of NHE3 activity were absent.
Summary
In summary, previous studies have demonstrated that elevated levels of [Ca2+]i inhibit electroneutral sodium absorption through inhibition of the BB NHE3. Ca2+ regulation of NHE3 activity occurs through a variety of mechanisms, including protein trafficking, mobility in the plasma membrane, and multi-protein complex formation as well as phosphorylation (not of NHE3 but rather of the endocytic machinery). While eukaryotic cells possess multiple pathways that carry out specific biological events in response to elevated [Ca2+]i, recent studies of NHE3 regulation by the NHERF family of PDZ domain–containing proteins have added an additional level of complexity to understanding Ca2+ of NHE3 activity. Moreover, emerging evidence of previously undiscovered functions of Ca2+ signaling proteins (e.g., PLCγ) may provide new insights into the mechanisms responsible for inhibition of sodium absorption by elevated [Ca2+]i in diarrheal diseases as well as offering new strategies by which to develop potential therapeutic agents.
Acknowledgments
Supported in part by NIH NIDDK Grants, R01-DK26523, R01-DK61765, PO1-DK72084, R24-DK64388 (The Hopkins Basic Research Digestive Diseases Development Core Center), The Hopkins Center for Epithelial Disorders, and T32-DK07632.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 2.Maher MM, et al. The Na+/H+ exchange isoform NHE3 regulates basal canine ileal Na+ absorption in vivo. Gastroenterology. 1997;112:174–183. doi: 10.1016/s0016-5085(97)70232-4. [DOI] [PubMed] [Google Scholar]
- 3.Maher MM, et al. Role of brush border Na+/H+ exchange in canine ileal absorption. Dig Dis Sci. 1996;41:651–659. doi: 10.1007/BF02213119. [DOI] [PubMed] [Google Scholar]
- 4.Donowitz M, Tse CM. Molecular physiology of mammalian epithelial Na/H exchangers NHE2 and NHE3. Curr Topics Membr. 2001;50:437–498. [Google Scholar]
- 5.Donowitz M, Welsh MJ. Ca2+ and cyclic AMP in regulation of intestinal Na, K, and Cl transport. Annu Rev Physiol. 1986;48:135–150. doi: 10.1146/annurev.ph.48.030186.001031. [DOI] [PubMed] [Google Scholar]
- 6.Kachintorn U, et al. Activation by calcium alone of chloride secretion in T84 epithelial cells. Br J Pharmacol. 1993;109:510–517. doi: 10.1111/j.1476-5381.1993.tb13599.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells. Implications for carbachol-stimulated chloride secretion. J Biol Chem. 1998;273:27111–27117. doi: 10.1074/jbc.273.42.27111. [DOI] [PubMed] [Google Scholar]
- 8.Uribe JM, et al. Epidermal growth factor inhibits Ca(2+)-dependent Cl− transport in T84 human colonic epithelial cells. Am J Physiol. 1996;271:C914–C922. doi: 10.1152/ajpcell.1996.271.3.C914. [DOI] [PubMed] [Google Scholar]
- 9.Uribe JM, et al. Phosphatidylinositol 3-kinase mediates the inhibitory effect of epidermal growth factor on calcium-dependent chloride secretion. J Biol Chem. 1996;271:26588–26595. doi: 10.1074/jbc.271.43.26588. [DOI] [PubMed] [Google Scholar]
- 10.Kim HK, et al. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell. 1991;65:435–441. doi: 10.1016/0092-8674(91)90461-7. [DOI] [PubMed] [Google Scholar]
- 11.Ronnstrand L, et al. Identification of two C-terminal autophosphorylation sites in the PDGF beta-receptor: involvement in the interaction with phospholipase C-gamma. Embo J. 1992;11:3911–3919. doi: 10.1002/j.1460-2075.1992.tb05484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rotin D, et al. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. Embo J. 1992;11:559–567. doi: 10.1002/j.1460-2075.1992.tb05087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wahl M, Carpenter G. Selective phospholipase C activation. Bioessays. 1991;13:107–113. doi: 10.1002/bies.950130303. [DOI] [PubMed] [Google Scholar]
- 14.Caraveo G, et al. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science. 2006;314:122–125. doi: 10.1126/science.1127815. [DOI] [PubMed] [Google Scholar]
- 15.Choi JH, et al. Phospholipase C-gamma1 is a guanine nucleotide exchange factor for dynamin-1 and enhances dynamin-1-dependent epidermal growth factor receptor endocytosis. J Cell Sci. 2004;117:3785–3795. doi: 10.1242/jcs.01220. [DOI] [PubMed] [Google Scholar]
- 16.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 17.Patterson RL, et al. Phospholipase C-gamma: diverse roles in receptor-mediated calcium signaling. Trends Biochem Sci. 2005;30:688–697. doi: 10.1016/j.tibs.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.van Rossum DB, et al. Phospholipase Cgamma1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
- 19.Dolmetsch RE, et al. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 20.Hook SS, Means AR. Ca(2+)/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 21.Meyer T, Stryer L. Calcium spiking. Annu Rev Biophys Biophys Chem. 1991;20:153–174. doi: 10.1146/annurev.bb.20.060191.001101. [DOI] [PubMed] [Google Scholar]
- 22.Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- 23.Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–2352. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- 24.Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131:285–292. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]
- 25.Balboa MA, et al. Expression and function of phospholipase A(2) in brain. FEBS Lett. 2002;531:12–17. doi: 10.1016/s0014-5793(02)03481-6. [DOI] [PubMed] [Google Scholar]
- 26.Evans JH, et al. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J Biol Chem. 2001;276:30150–30160. doi: 10.1074/jbc.M100943200. [DOI] [PubMed] [Google Scholar]
- 27.Gijon MA, et al. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol. 1999;145:1219–1232. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickinson KE, Frizzell RA, Sekar MC. Activation of T84 cell chloride channels by carbachol involves a phosphoinositide-coupled muscarinic M3 receptor. Eur J Pharmacol. 1992;225:291–298. doi: 10.1016/0922-4106(92)90102-2. [DOI] [PubMed] [Google Scholar]
- 29.Kasai H, Augustine GJ. Cytosolic Ca2+ gradients triggering unidirectional fluid secretion from exocrine pancreas. Nature. 1990;348:735–738. doi: 10.1038/348735a0. [DOI] [PubMed] [Google Scholar]
- 30.Nathanson MH, et al. Mechanism of Ca2+ wave propagation in pancreatic acinar cells. J Biol Chem. 1992;267:18118–18121. [PubMed] [Google Scholar]
- 31.Thorn P, et al. Ca2+ oscillations in pancreatic acinar cells: spatiotemporal relationships and functional implications. Cell Calcium. 1993;14:746–757. doi: 10.1016/0143-4160(93)90100-k. [DOI] [PubMed] [Google Scholar]
- 32.Kasai H, Li YX, Miyashita Y. Subcellular distribution of Ca2 +release channels underlying Ca2+ waves and oscillations in exocrine pancreas. Cell. 1993;74:669–677. doi: 10.1016/0092-8674(93)90514-q. [DOI] [PubMed] [Google Scholar]
- 33.Nathanson MH, et al. Localization of the type 3 inositol 1,4,5-trisphosphate receptor in the Ca2+ wave trigger zone of pancreatic acinar cells. J Biol Chem. 1994;269:4693–4696. [PubMed] [Google Scholar]
- 34.Siefjediers A, et al. Characterization of inositol 1,4,5-trisphosphate (IP3) receptor subtypes at rat colonic epithelium. Cell Calcium. 2007;41:303–315. doi: 10.1016/j.ceca.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Cohen ME, et al. Carbachol- and elevated Ca(2+)-induced translocation of functionally active protein kinase C to the brush border of rabbit ileal Na+ absorbing cells. J Clin Invest. 1991;88:855–863. doi: 10.1172/JCI115387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emmer E, et al. Role of calcium and calmodulin in the regulation of the rabbit ileal brush-border membrane Na+/H+ antiporter. J Membr Biol. 1989;108:207–215. doi: 10.1007/BF01871735. [DOI] [PubMed] [Google Scholar]
- 37.Khurana S, et al. Asymmetric signal transduction in polarized ileal Na(+)-absorbing cells: carbachol activates brush-border but not basolateral-membrane PIP2-PLC and translocates PLC-gamma 1 only to the brush border. Biochem J. 1996;313(Pt 2):509–518. doi: 10.1042/bj3130509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Donowitz M, et al. Elevated intracellular Ca2+ acts through protein kinase C to regulate rabbit ileal NaCl absorption. Evidence for sequential control by Ca2+/calmodulin and protein kinase C. J Clin Invest. 1989;83:1953–1962. doi: 10.1172/JCI114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris AP, Estes MK. Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VIII. Pathological consequences of rotavirus infection and its enterotoxin. Am J Physiol Gastrointest Liver Physiol. 2001;281:G303–G310. doi: 10.1152/ajpgi.2001.281.2.G303. [DOI] [PubMed] [Google Scholar]
- 40.Khurana S, et al. Studies on the mechanism of Salmonella typhimurium enterotoxin-induced diarrhoea. Biochim Biophys Acta. 1991;1097:171–176. doi: 10.1016/0925-4439(91)90031-4. [DOI] [PubMed] [Google Scholar]
- 41.Kanwar RK, et al. Calcium and protein kinase C play an important role in Campylobacter jejuni-induced changes in Na+ and Cl− transport in rat ileum in vitro. Biochim Biophys Acta. 1995;1270:179–192. doi: 10.1016/0925-4439(95)00045-6. [DOI] [PubMed] [Google Scholar]
- 42.Kaur T, et al. Role of enteric nervous system in Shigella dysenteriae type 1 toxin-induced fluid secretion in rabbit ileum. J Diarrhoeal Dis Res. 1995;13:159–165. [PubMed] [Google Scholar]
- 43.Li X, et al. Na transporters NHE3 and Na/K ATPase, NHE3-regulatory factor 1 (NHERF1) and the intracellular chloride channel Clc5 are down regulated in IBD: Probable contribution to IBD diarrhea. DDW. 2006;130:T1999. [Google Scholar]
- 44.Khan I, et al. Role of Na+/H +exchanger isoform-1 in human inflammatory bowel disease. Can J Gastroenterol. 2003;17:31–36. doi: 10.1155/2003/673819. [DOI] [PubMed] [Google Scholar]
- 45.Goyal J, et al. Role of Ca2(+)-calmodulin and protein kinase C in the secretory action of heat-labile enterotoxin of Escherichia coli in mice. Biochem Int. 1989;19:1007–1017. [PubMed] [Google Scholar]
- 46.Kim JH, et al. Ca(2+)-dependent inhibition of Na+/H+ exchanger 3 (NHE3) requires an NHE3-E3KARP-alpha-actinin-4 complex for oligomerization and endocytosis. J Biol Chem. 2002;277:23714–23724. doi: 10.1074/jbc.M200835200. [DOI] [PubMed] [Google Scholar]
- 47.Zachos NC, et al. Elevated intracellular calcium ([Ca2+]i) inhibits NHE3 activity by a PDZK1 (NHERF3) dependent process in which NHE3 and PDZK1 bind basally and dissociate with elevated [Ca2+]i. DDW. 2006;130:A332. [Google Scholar]
- 48.Akhter S, et al. C-terminal domains of Na(+)/H(+) exchanger isoform 3 are involved in the basal and serum-stimulated membrane trafficking of the exchanger. Biochemistry. 2000;39:1990–2000. doi: 10.1021/bi991739s. [DOI] [PubMed] [Google Scholar]
- 49.D’Souza S, et al. The epithelial sodium-hydrogen antiporter Na+/H+ exchanger 3 accumulates and is functional in recycling endosomes. J Biol Chem. 1998;273:2035–2043. doi: 10.1074/jbc.273.4.2035. [DOI] [PubMed] [Google Scholar]
- 50.Janecki AJ, et al. Subcellular redistribution is involved in acute regulation of the brush border Na+/H+ exchanger isoform 3 in human colon adenocarcinoma cell line Caco-2. Protein kinase C-mediated inhibition of the exchanger. J Biol Chem. 1998;273:8790–8798. doi: 10.1074/jbc.273.15.8790. [DOI] [PubMed] [Google Scholar]
- 51.Kurashima K, et al. Identification of sites required for down-regulation of Na+/H+ exchanger NHE3 activity by cAMP-dependent protein kinase. Phosphorylation-dependent and -independent mechanisms. J Biol Chem. 1997;272:28672–28679. doi: 10.1074/jbc.272.45.28672. [DOI] [PubMed] [Google Scholar]
- 52.Yip KP, et al. Redistribution of Na+/H+ exchanger isoform NHE3 in proximal tubules induced by acute and chronic hypertension. Am J Physiol. 1998;275:F565–F575. doi: 10.1152/ajprenal.1998.275.4.F565. [DOI] [PubMed] [Google Scholar]
- 53.Janecki AJ, et al. Quantitation of plasma membrane expression of a fusion protein of Na/H exchanger NHE3 and green fluorescence protein (GFP) in living PS120 fibroblasts. J Histochem Cytochem. 2000;48:1479–1492. doi: 10.1177/002215540004801105. [DOI] [PubMed] [Google Scholar]
- 54.Li X, et al. Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J Physiol. 2001;537:537–552. doi: 10.1111/j.1469-7793.2001.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu MC, et al. Dopamine acutely stimulates Na+/H+ exchanger (NHE3) endocytosis via clathrin-coated vesicles: dependence on protein kinase A-mediated NHE3 phosphorylation. J Biol Chem. 2001;276:26906–26915. doi: 10.1074/jbc.M011338200. [DOI] [PubMed] [Google Scholar]
- 56.Donowitz M, et al. Short-term regulation of NHE3 by EGF and protein kinase C but not protein kinase A involves vesicle trafficking in epithelial cells and fibroblasts. Epithelial Transport and Barrier Function. 2000;915:30–42. doi: 10.1111/j.1749-6632.2000.tb05221.x. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, et al. Regulation of insulin secretion and GLUT4 trafficking by the calcium sensor synaptotagmin VII. Biochem Biophys Res Commun. 2007;362:658–664. doi: 10.1016/j.bbrc.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whitehead JP, et al. The role of Ca2+ in insulin-stimulated glucose transport in 3T3-L1 cells. J Biol Chem. 2001;276:27816–27824. doi: 10.1074/jbc.M011590200. [DOI] [PubMed] [Google Scholar]
- 59.Bommert K, et al. Inhibition of neurotransmitter release by C2-domain peptides implicates synaptotagmin in exocytosis. Nature. 1993;363:163–165. doi: 10.1038/363163a0. [DOI] [PubMed] [Google Scholar]
- 60.DeBello WM, Betz H, Augustine GJ. Synaptotagmin and neurotransmitter release. Cell. 1993;74:947–950. doi: 10.1016/0092-8674(93)90716-4. [DOI] [PubMed] [Google Scholar]
- 61.Li X, et al. Carbachol regulation of rabbit ileal brush border Na+-H+ exchanger 3 (NHE3) occurs through changes in NHE3 trafficking and complex formation and is Src dependent. J Physiol. 2004;556:791–804. doi: 10.1113/jphysiol.2004.060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zachos NC, et al. Phospholipase Cgamma (PLCg) Directly Binds to the Na+/H+ Exchanger 3 (NHE3) C-terminus Which Is Necessary for Basal and Calcium-Mediated Inhibition of NHE3 Activity. DDW. 2008;134:A744. [Google Scholar]
- 63.Donowitz M, et al. NHERF family and NHE3 regulation. J Physiol. 2005;567:3–11. doi: 10.1113/jphysiol.2005.090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murtazina R, et al. Tissue-specific regulation of sodium/proton exchanger isoform 3 activity in Na(+)/H(+) exchanger regulatory factor 1 (NHERF1) null mice. cAMP inhibition is differentially dependent on NHERF1 and exchange protein directly activated by cAMP in ileum versus proximal tubule. J Biol Chem. 2007;282:25141–25151. doi: 10.1074/jbc.M701910200. [DOI] [PubMed] [Google Scholar]
- 65.Yun CH, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murtazina R, et al. NHERF2 is Necessary for Basal and All Second Messenger Regulation of NHE3 Activity in Intact Mouse Ileal Na Absorptive Cells. DDW. 2008;134:A749. [Google Scholar]
- 67.Cinar A, et al. NHE3 inhibition by cAMP and Ca2+ is abolished in PDZ-domain protein PDZK1-deficient murine enterocytes. J Physiol. 2007;581:1235–1246. doi: 10.1113/jphysiol.2007.131722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hillesheim J, et al. Down regulation of small intestinal ion transport in PDZK1- (CAP70/NHERF3) deficient mice. Pflugers Arch. 2007;454:575–586. doi: 10.1007/s00424-007-0239-x. [DOI] [PubMed] [Google Scholar]
- 69.Zachos NC, et al. Brush Border (BB) PDZ Proteins PDZK1 and IKEPP (Intestinal and Kidney Epithelial PDZ Domain Protein) Differentially Regulate NHE3 in Response to Elevated Ca2+: Inhibition with PDZK1 and Stimulation with IKEPP. DDW. 2005;128:A177. [Google Scholar]
- 70.Morales FC, et al. Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc Natl Acad Sci USA. 2004;101:17705–17710. doi: 10.1073/pnas.0407974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Donowitz M, et al. Proteome of the Small Intestinal Brush Border of the NHERF1 Knock Out Mouse: Regulation of Transporters and Pro-Proliferation Molecules. Gastroenterology. 2008;134:S1770. [Google Scholar]
- 72.Murtazina R, et al. NHERF2 is Necessary for Basal and All Second Messenger Regulation of NHE3 Activity in Intact Mouse Ileal Na Absorptive Cells. Gastroenterology. 2008;134:A749. [Google Scholar]
- 73.Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825–872. doi: 10.1152/physrev.00030.2006. [DOI] [PubMed] [Google Scholar]
- 74.Cha B, et al. The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J Cell Sci. 2004;117:3353–3365. doi: 10.1242/jcs.01180. [DOI] [PubMed] [Google Scholar]
- 75.Lee-Kwon W, et al. Lysophosphatidic acid stimulates brush border Na+/H+ exchanger 3 (NHE3) activity by increasing its exocytosis by an NHE3 kinase A regulatory protein-dependent mechanism. J Biol Chem. 2003;278:16494–16501. doi: 10.1074/jbc.M300580200. [DOI] [PubMed] [Google Scholar]