

Non-technical summary
‘To be, or not to be’– thousands of neurons are facing this Shakespearean question in the brains of patients suffering from epilepsy or the consequences of a brain traumatism or stroke. The destiny of neurons in damaged brain depends on tiny equilibrium between pro-survival and pro-death signalling. Numerous studies have shown that the activity of the neuronal potassium chloride co-transporter KCC2 strongly decreases during a pathology. However, it remained unclear whether the change of the KCC2 function protects neurons or contributes to neuronal death. Here, using cultures of hippocampal neurons, we show that experimental silencing of endogenous KCC2 using an RNA interference approach or a dominant negative mutant reduces neuronal resistance to toxic insults. In contrast, the artificial gain of KCC2 function in the same neurons protects them from death. This finding highlights KCC2 as a molecule that plays a critical role in the destiny of neurons under toxic conditions and opens new avenues for the development of neuroprotective therapy.
Abstract
Abstract
KCC2 is a neuron-specific potassium–chloride co-transporter controlling intracellular chloride homeostasis in mature and developing neurons. It is implicated in the regulation of neuronal migration, dendrites outgrowth and formation of the excitatory and inhibitory synaptic connections. The function of KCC2 is suppressed under several pathological conditions including neuronal trauma, different types of epilepsies, axotomy of motoneurons, neuronal inflammations and ischaemic insults. However, it remains unclear how down-regulation of the KCC2 contributes to neuronal survival during and after toxic stress. Here we show that in primary hippocampal neuronal cultures the suppression of the KCC2 function using two different shRNAs, dominant-negative KCC2 mutant C568A or DIOA inhibitor, increased the intracellular chloride concentration [Cl−]i and enhanced the toxicity induced by lipofectamine-dependent oxidative stress or activation of the NMDA receptors. The rescuing of the KCC2 activity using over-expression of the active form of the KCC2, but not its non-active mutant Y1087D, effectively restored [Cl−]i and enhanced neuronal resistance to excitotoxicity. The reparative effects of KCC2 were mimicked by over-expression of the KCC3, a homologue transporter. These data suggest an important role of KCC2-dependent potassium/chloride homeostasis under neurototoxic conditions and reveal a novel role of endogenous KCC2 as a neuroprotective molecule.
Introduction
During the last decade extensive attention was given to the study of the functional role of the neuron-specific potassium-chloride co-transporter KCC2 (Blaesse et al. 2009). This transporter is expressed progressively during development in many neuronal cell types and participates in the control of neuronal chloride homeostasis both in vivo and in vitro. The suppression of the transporter functions with antisense oligonucleotides (Rivera et al. 1999) or by gene inactivation in vivo (Hubner et al. 2001) leads to an increase of the intracellular chloride concentration and consequently modifies the inhibitory action of GABA and glycine. KCC2 is also critically involved in the maintenance of the chloride homeostasis during and after different excitotoxicity-related pathologies including anoxic events, trauma, temporal lobe epilepsy, stroke and in vitro excitotoxicity (for review see Blaesse et al. 2009). Particularly, evidence show that long-lasting excitotoxic events induce suppression of the KCC2 expression both in vivo (Nabekura et al. 2002; Palma et al. 2006; Jaenisch et al. 2010) and in vitro (Rivera et al. 2004; Wake et al. 2007). Consistent with this, recent study revealed that an excitotoxic event produces KCC2 phosphorylation leading to withdrawal of the transporter from the membrane and its degradation (Lee et al. 2010). Despite important progress in the study of the expression of KCC2 under different pathological conditions related to excitotoxicity, it remains unclear how down-regulation of the KCC2 contributes to neuronal survival during and after toxic stresses.
Several molecular approaches were used to modify the expression level of endogenous KCC2 and thus to assess its functional role. At least three different lines of KCC2-deficient mice have been created including knockout of the entire KCC2 (Hubner et al. 2001), knockout of the b-isoform of KCC2 (Woo et al. 2002, see also Uvarov et al. 2007 for detailed descriptions of KCC2 isoforms) and partial knockout of both KCC2 isoforms (Tornberg et al. 2005). All these mice models strongly contributed to our understanding of KCC2 function. However, even a 20–50% change of the KCC2 expression in neurons alters neuronal network activity (Zhu et al. 2008) that, in addition to genetic compensation, might activate diverse compensatory pathways and introduce additional difficulties in the study of the functional role of KCC2. As an alternative to KCC2-deficient mice models, Rivera et al. (1999) successfully employed specific KCC2 antisense oligonucleotides that decreased the expression of KCC2 in organotypic hippocampal slices. This approach, however, did not obtain further insight due to its low efficacy. Therefore, there is a strong need for the creation of a molecular tool allowing KCC2 silencing in individual neurons without affecting the activity of the entire neuronal network.
Discovery of the evolutionary conserved RNA interference (RNAi) pathway (Fire et al. 1998) offered an opportunity of protein silencing in individual cells, including neurons, using small interfering RNA (siRNA) or short hairpin RNA (shRNA) (Brummelkamp et al. 2002; Paddison et al. 2002). Here we describe two different shRNAs designed against rat KCC2 that effectively knockdown the expression of the endogenous KCC2 protein in individual neurons in primary hippocampal cultures and organotypic hippocampal slices. Since some shRNAs show non-specific effects on neuronal functioning (Alvarez et al. 2006) we analysed in detail the efficacy and specificity of the actions of the shRNAs using different functional tests and performed rescue experiments with pcDNA-KCC2 encoding for the murine homologue. We show that the knocking down of KCC2 in 12–14 days in vitro (DIV) hippocampal neuronal cultures increases [Cl−]i and lengthens the neuronal chloride recovery time after an experimentally imposed [Cl−]i increase. Most interestingly we found that the silencing of KCC2 either using shRNAs or by over-expression of the dominant-negative KCC2 mutant led to a higher susceptibility of neurons to lipofectamine-dependent oxidative stress and NMDA receptor (NMDAR)-dependent excitotoxicity. The rescue of the KCC2 function using co-expression of rat shRNAsKCC2 and mouse KCC2 restored neuronal resistance to the toxicity. Thus, these data reveal a novel asset of the endogenous KCC2 action as a neuroprotective agent. Consequently, the decreased neurotoxic resistance of neurons with silenced KCC2 should be taken into consideration during the study of KCC2 function.
Methods
All manipulations with animals were performed in agreement with the guidelines of the Animal Care and Use Committee of INSERM (Institut National de la Santé et de la Recherche Médicale).
Primary cultures of rat hippocampal neurons, transfection
For immunocytochemistry, electrophysiology and non-invasive Cl− analysis, neuronal cultures were plated on coverslips placed in 35 mm culture dishes. For time-lapse monitoring of neuronal development, the cultures were plated directly on 4-well plastic (16 mm) plates. Twenty-four hours prior to plating, the coverslips in culture dishes and well plates were coated with poly-ethylenimine.
Neurons from 18-day-old rat embryos were dissociated using trypsin and plated at a density of 70,000 cells cm−2 in minimal essential medium (MEM) supplemented with 10% NU serum (BD Biosciences, Le Pont de Claix, France), 0.45% glucose, 1 mm sodium pyruvate, 2 mm glutamine and 10 IU ml−1 penicillin–streptomycin as previously described (Buerli et al. 2007). On days 7, 10 and 13 of culture incubation, half of the medium was changed to MEM with 2% B27 supplement (Invitrogen).
Transfections of 7–10 DIV neuronal cultures were performed as described previously (Buerli et al. 2007). For transfection of cultures growing in 35 mm dishes, 300 μl of Opti-MEM media was mixed with 7 μl of Lipofectamine reagent 2000 (Invitrogen), 1.0 μl of Magnetofection CombiMag (OZ Bioscience, France) and 1–1.5 μg of different pcDNAs pre-mixed in desired proportions. The mixture was incubated for 20 min at room temperature (RT) and thereafter distributed dropwise above the neuronal culture. Culture dishes were placed on a magnetic plate (OZ Bioscience) and incubated for 30–35 min at 37°C. Transfection was terminated by the substitution of 90% of the incubation solution with fresh culture media. Cells were used in the experiments 2–5 days after transfection. For transfection of cultures growing in 4 × 16 mm well plates, 60 μl per well of the above described mixture was used.
The majority of experiments were based on co-transfection into the same neurons of two or three different pcDNAs encoding a fluorescent marker of transfection (enhanced green fluorescent protein (eGFP) or cherry fluorescent protein (Cherry)), shRNAs and ion transporters. We specifically studied the efficacy of neuronal co-transfection with mixtures of pcDNAs in different proportions. Figure 1 illustrates that after transfection with a mixture of three pcDNAs, including eGFP (0.3 μg), Cherry (0.5 μg) and KCC2 (0.7 μg) 100% of eGFP-positive neurons express cherry and 93% of these neurons over-express KCC2. We thus used this proportion (0.3 + 0.5 + 0.7) for co-transfections of three pcDNAs (marker + shRNA + transporter) into neurons. For transfection of two pcDNAs, we used routinely mixtures of 0.3 μg of eGFP or cherry + 0.8 μg of pcDNAs of interest. Such co-transfection resulted in the exogenous expression of KCC2 in 100% of eGFP-positive neurons (not shown).
Figure 1. Co-transfection of cultured neurons with multiple pcDNAs.

A, representative images of 14 DIV primary hippocampal culture transfected at DIV 11 with a mixture of pcDNAs containing 0.3 μg of eGFP, 0.5 μg of Cherry and 0.7 μg of mouse KCC2 and used for transfection of neurons in a 35 mm dish. a1, a2 and a3 illustrate the same two neurons expressing eGFP, Cherry and KCC2, respectively. KCC2 was visualized using polyclonal anti-KCC2 and Alexa CY5 antibody. a4 shows merged a1, a2 and a3 images. a5 illustrates merged low magnification image of transfected neurons. B, quantification of the co-transfection efficacy into the same neuron of three pcDNAs mixed in proportions mentioned in the key. Values are normalized to the number of eGFP-positive neurons.
Electrophysiological recording
The gramicidin-perforated whole cell patch-clamp recordings from neurons were performed according to protocols described by Rhee et al. (1994); Kyrozis & Reichling (1995); Tyzio et al. (2003). Coverslips with transfected neuronal cells were placed onto an inverted microscope and perfused with the external solution containing (in mm): 140 NaCl, 2.5 KCl, 20 Hepes, 20 d-glucose, 2.0 CaCl2, 2.0 MgCl2, 0.001 tetrodotoxin and 0.0003 strychnine, pH 7.4. Micropipettes (5 MΩ) were filled with a solution containing (in mm): KCl 150, Hepes 10, Tris 10; 20 mg ml−1 gramicidin A (dissolved in DMSO), pH 7.2. Isoguvacine (30 μm) was focally applied to soma and proximal dendrites through a micropipette connected to a picospritzer (General Valve Corporation, pulse duration 150 ms, pressure 30000 Pa). EGABA was determined as the point of crossing of the voltage–current relationship (VCR) of isoguvacine responses as illustrated in Fig. 3A. The resting membrane potential (Em) was determined by switch to the ‘I = 0’ mode.
Figure 3. Knocking down of KCC2 inhibits intracellular chloride extrusion.

Results of gramicidin-perforated patch-clamp recordings. The recordings were performed in a voltage-clamp mode with holding potential –65 mV. GABAAR-mediated currents were induced by a focal application of isoguvacine, a selective agonist of GABAAR, to neuronal soma. A, left traces show examples of recordings of reversal potentials of GABAAR-mediated currents (EGABA) from neurons transfected with scrambled shRNA and shRNAKCC2_k10. Voltage ramps (see scheme in insert) were imposed before, during and after isoguvacine application. The right plot shows net voltage–current relationships of isoguvacine-induced currents illustrated in the left part of the figure. B, example of recording of the kinetic of EGABA recovery after a [Cl−]i increase induced by a combination of neuron depolarization to 0 mV for 10 s and bath application of 30 μm of isoguvacine for 5 s. In the selected example at the beginning of the recording, brief (150 ms) focal applications of isoguvacine to soma-induced outward 20–30 pA currents. Increase of [Cl−]i provoked change of the polarity and amplitude of isoguvacine-induced test pulses that recovered to their control values 200 s after the [Cl−]i increase. The plot on the right shows the kinetic of the recovery of isoguvacine-induced responses in neurons transfected with scrambled shRNA and shRNAKCC2_k10. C, reversal potentials of GABAAR-mediated currents measured as illustrated in A from neurons co-transfected with different mixtures of pcDNAs. All pcDNA mixtures contained eGFP, one of the shRNAs (as indicated above the plot) and a pcDNA3.1 empty vector or the same vector including mouse KCC2. Non-transfected neurons were non-fluorescent neurons from cultures transfected with scrambled shRNAs. Dots show values of individual recordings. Columns show mean ± SEM values. **P < 0.01. 4 experiments, 1 to 4 neurons per experiment. D, times of the 50% recovery of isoguvacine responses after imposed [Cl−]i rise. Mean ± SEM values. *P < 0.05; **P < 0.01. 4 experiments, 1 to 4 neurons per experiment.
All experiments were performed at 22–24°C. Recordings were made using an Axopatch-200A amplifier and pCLAMP acquisition software (Axon Instruments). Data were low-pass filtered at 2 kHz and acquired at 10 kHz.
Non-invasive monitoring of intracellular chloride
Fluorescence images were acquired at RT (22–24°C) using a customized digital imaging set-up. Excitation of cells at various wavelengths was achieved using a 1 nm bandwidth polychromatic light selector equipped with a 150 W xenon lamp (Polychrome V; Till Photonics, Germany). Light intensity was attenuated using neutral density filters. A dichroic mirror (495 nm; Omega Optics, USA) was used to deflect light on to the samples. Fluorescence was visualized using an upright microscope (Axioskop) equipped with a 60× water-immersion objective (0.9 NA; LumPlanFL, Olympus, USA). Fluorescent emitted light passed to a 16-bit (Andor iXon EM+; Andor Technology PLC, Northern Ireland) electron multiplying charge-coupled device digital camera system equipped with an image intensifier. Images were acquired on a computer via a DMA serial transfer. All peripheral hardware control, image acquisition and image processing were achieved using customized software Andor iQ (Andor Technology PLC, Northern Ireland). The average fluorescence intensity of each region of interest was measured. Mean background fluorescence (measured from a non- fluorescent area) was subtracted and the ratio intensities F440/F480 were determined. The frequency of acquisition was usually 0.1 Hz and duration of excitation was 5–20 ms. The real values of [Cl−]i were obtained from the previously described calibration curves (Markova et al. 2008; Waseem et al. 2010). The increase of [Cl−]i during the experiment was achieved using focal application of NMDA to neuronal soma through a patch pipette pre-filled with 1 mm of NMDA and 10 μm glycine and connected to the picospritzer.
Monitoring of neuronal survival
The survival of transfected neurons using live imaging was studied as follows: cultures plated in 4-well plates were placed into the set-up equipped with a motorized Nikon TE300 inverted microscope and temperature and CO2 atmosphere controlling units (Princeton Instruments), and images, containing information on the fluorescence of neurons and coordinates, were taken using a Micro-MAX CCD camera and MetaMorph software (Roper Scientific). Based on their coordinates, the same neurons were localized and examined on days 2, 3, 4 and 5 after transfection. For each experimental condition 30 to 50 neurons were scanned.
Transfected neurons were assigned as ‘alive’ or ‘dead’ on the basis of their morphology (see Supplementary data for details). After live imaging for several days, the cultures were fixed and immunostained as described in the next section. Following immunostaining, culture plates were returned to the same set-up for analysis of MAP2, NeuN, cleaved caspase-3 and chromatin distribution in the same transfected neurons, whose survival was previously followed using live imaging.
The analysis of neuronal survival in non-transfected neurons was done blind in a semi-quantitative mode using Metamorph software. First, we generated a binary mask on images of NeuN-positive cells. Then, a second binary mask was applied on Hoechst-stained nuclei in order to select healthy nuclei of live neurons and exclude nuclei of dead or dying neurons containing strongly condensed chromatin or irregular chromatin clumps (see Supplementary Fig. S6 for images and Soriano et al. (2009) for detailed description of the protocol of semi-automatical nuclei selection). By combination of the two previous images (NeuN and healthy nuclei), a third binary picture was obtained and used to quantify the density of alive neurons. This semi-automatical approach was validated for every new set of experiments by comparison of manual and automatical counting of at least 100 alive neurons in order to obtain difference less than 1%.
Immunocytochemistry and quantitative immunofluorescence analysis of primary neuronal cultures
For immunocytochemical analysis cultures were fixed in Antigenfix (Diapath, Martingo, Italy) for 20 min (at RT), permeabilized with 0.3% Triton X-100, blocked by 5% goat serum, labelled overnight (4°C) with a primary antibody and for 1 h (RT) with a secondary antibody. Primary antibodies were rabbit anti-KCC2 antibody (dilution 1:1000; US Biological, Euromedex, France), rabbit cleaved caspase-3 (Asp175) antibody (dilution 1:300; Cell Signalling), mouse anti-MAP2 antibody (dilution 1:2000; Sigma-Aldrich), chicken anti-MAP2 (ab5392) antibody (dilution 1:2000; Abcam) and mouse anti-neuronal nuclei (NeuN) antibody (dilution 1:1000; Chemicon). Secondary antibodies were Cy3-conjugated goat anti-rabbit IgG, Cy3-conjugated goat anti-mouse IgG, CY5-conjugated goat anti-mouse IgG and CY5-conjugated goat anti-chicken IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Cell nuclei were revealed using 5 min staining with Hoeschst 33258 (Sigma-Aldrich, 1 mg ml−1 stock solution used at 1:1000).
For quantitative analysis images were acquired with an Olympus Fluorview-500 confocal microscope (40×; 1.0 NA). We randomly chose fields with a transfected neuron, focused on that neuron by only visualizing eGFP fluorescence and then acquired consecutively images of eGFP, KCC2 and MAP2. The intensity of KCC2 fluorescence was analysed with MetaMorph software. First, we created a binary mask of soma of eGFP-fluorescent cells and then analysed KCC2 intensity in regions overlapping with the binary mask. For analysis of the KCC2 intensity in non-transfected cells the binary masks were created around soma visualized using the MAP2 antibody. Acquisition parameters were the same for all experimental conditions. All acquisitions and analysis were done blind. After analysis, data were normalized to the mean value of KCC2 in neurons transfected with scrambled shRNA and eGFP. Five randomly chosen transfected neurons were analysed from each experiment.
Organotypic hippocampal slices: electroporation, immunocytochemistry and image analysis
Organotypic slices were prepared from hippocampi of postnatal day 7 (P7) Wistar rats according to the procedure described by Stoppini et al. (1991) with minor changes. Briefly, rats were decapitated after chilling on ice. Following the removal of the brain, hippocampal sections, 400 μm thick, were prepared using a tissue chopper (McIlwain, USA) and collected in PBS solution supplemented with 0.5% glucose at 4°C.
The expression of exogenous pcDNAs into organotypic slices was achieved using an electroporation technique developed according to the procedure described by Kawabata et al. (2004) with several modifications. A plasmid solution (1.5 μg μl−1) was injected onto the pyramidal cells layer in CA1 using a glass micropipette and a picospritzer. An electroporator (CUY21EDIT, Nepa gene, Japan) was used to apply five square pulses (25 V, 5 ms duration with an interval of 995 ms).
Electroporated slices were placed onto Millicell-CM culture inserts (0.4 μm; Millipore) and incubated in 35 mm culture dishes with 1 ml of culture medium: MEM (Sigma-Aldrich) supplemented with 150 mm Hepes, insulin 0.1 mg l−1 (solution 5 mg ml−1) and horse serum (20%, Invitrogen) at pH 7.25–7.29.
Slices were maintained for 2–7 days in a cell culture incubator at 37°C, in an atmosphere containing 5% CO2. The medium was changed 3 times per week.
For immunocytochemistry analysis, organotypic slices were fixed in Antigenfix (Diapath, Martingo, Italy) overnight at 4°C. The slices were then washed in PBS and dehydrated through a graded series of methanol in PBS. Thereafter, the slices were incubated in Dent's fixative (80% methanol–20% DMSO; Sigma-Aldrich), washed in TBSTD (Tris-buffered saline (TBS), 0.1% Tween, 5% DMSO), and incubated in 5% BSA, 0.4% sheep serum in TBSTD overnight at 4°C. Afterward, slices were incubated with a primary antibody at 4°C for 48 h. Rabbit anti-KCC2 antibody (Ludwig et al. 2003) was diluted 1:500. After washing in TBSTD, slices were incubated overnight with a secondary antibody (donkey anti-rabbit CY5, Jackson ImmunoResearch Laboratories, dilution 1:400), then washed in TBSTD and mounted on Superfrost Plus slides in Vectashield mounting medium (Vector Laboratories).
Fluorescence images were acquired with a Leica TCS SP5 confocal microscope using an HCX APO 63× glycerol objective. Images were analysed using Image Pro Plus image analysis software (Media Cybernetics). Due to the substantial thickness of organotypic slices, the transfected neurons were located in different optical sections across the stack. For each transfected neuron an image with maximal GFP cell body fluorescent intensity was selected from the entire stack and the cell body was manually outlined. Cell bodies of the neighbouring non-transfected neurons were also manually outlined. Mean intensity of KCC2 immunoreactivity in the outlined area was measured. Mean intensity of KCC2 immunoreactivity in transfected neurons was normalized to the one in the non-transfected neurons to compensate for possible intensity variations between different image stacks. For background correction, the mean fluorescent intensity of three cell-free areas in each stack was measured, averaged and subtracted from KCC2 intensity values.
Constructs and materials
shRNA constructs for KCC2 knockdown were created with the mU6pro vector system (Yu et al. 2002) to target the following sequences in rat KCC2 mRNA (accession no. U55816):
shRNAKCC2_k10; GCTGTGAATGGCAATCTGG;
shRNAKCC2_k11; GACATTGGTAATGGAACAACG;
shRNAKCC2_12; GTCCTTAGTTACTGTCTCC;
shRNAKCC2_13; GGAGAGGATCAGTTCTTTCC.
A control construct with a scrambled sequence (GATGAACCTGATGACGTTC) lacked homology to any known mammalian mRNAs. Vectors encoding eGFP and cherry were from Clontech. The CMV promoter in original vectors was replaced with an ubiquitin promoter. A Cl− Sensor was inserted into the gw1 vector as described previously (Waseem et al. 2010). Rat KCC2 in the Clontech pIRES2 vector and rat KCC3 in the pcDNA1 vector were a gift from Dr C. Hubner. Mouse HA-KCC2 in the pcDNA3.1 vector was a gift from Dr C. Rivera. To create the eGFP–KCC2 fusion constructs, we first removed the EcoR1 restriction site by making a silent mutation (A3267G in rat coding sequence), thereafter introduced BglII and EcoR1 restriction sites using PCR at the beginning and at the end of coding rat KCC2 sequence and subcloned the obtained cDNA into eGFP-C1 vector (Clontech).
All mutations were generated using the QuikChange (Stratagene) site-directed method and verified by DNA sequencing.
The potassium–chloride transporter antagonist DIOA (R-(+)-[(dihydroindenyl)oxy] alkanoic acid) (20 μm) was purchased from Sigma and pre-dissolved in 0.1 n NaOH. Trolox ((±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid), a hydrosoluble vitamin-E analogue, was used at 100 μm and purchased from Sigma-Aldrich.
Statistical analysis
All population data were expressed as mean ± SEM, unless otherwise specified. The one-way ANOVA or non-parametric Mann–Whitney tests were employed to examine the statistical significance of the differences between groups of data.
Results
shRNAKCC2 constructs
In order to knockdown the expression of KCC2 in rat neurons we designed and created several short hairpin RNAs (shRNAs) targeting different regions of rat KCC2 mRNA (see Methods for details). The efficacy of the created shRNAs was verified using immunocytochemistry analysis of the KCC2 protein expression in cultured hippocampal neurons transfected at 10 DIV with a mixture of vectors encoding shRNA and enhanced green fluorescent protein (eGFP) as the marker of transfection. A special methodological study was performed to ensure the co-expression of different vectors in the same neurons (Fig. 1, see Methods for details). We found that two constructs (called shRNAKCC2_k10 and shRNAKCC2_k11, Fig. 2A) effectively decreased the expression of the KCC2 protein already 3 days after culture transfection (Fig. 2B and C). Two other constructs, shRNAKCC2_k12 (Fig. 2C) and shRNAKCC2_k13, did not show significant decrease of KCC2 protein expression 3 or 4 days after transfection (not shown). The efficacy of shRNAKCC2_k11 was also verified in organotypic hippocampal slices to confirm that the shRNA-induced decrease of KCC2 immunofluorescence is not limited to dissociated neuronal cultures (Supplementary Fig. S1).
Figure 2. shRNAs silencing rat KCC2.

A, schematic representation of the location of rat mRNA regions targeted with shRNAs. Aligned sequences show two nucleotide mismatches of both mRNA regions with mouse KCC2. B, images of the cultured hippocampal neurons co-transfected with eGFP plus scrambled shRNA (upper row) or eGFP plus shRNAKCC2_K10 and immunostained with anti-MAP2 monoclonal antibody and anti-KCC2 polyclonal antibody. To better illustrate the change of the intensity of the KCC2 staining the images were coloured using a three-colour gradual palette shown below the images. Arrowheads indicate the location of soma of transfected neurons. Dotted squares in low magnification images are dendrite regions shown in high magnification images. Dotted lines in high magnification images (KCC2 staining) outline dendrites of transfected neurons. Note the low level of KCC2 staining in soma and dendrites of neurons transfected with shRNAKCC2_k10. C, quantification of the average intensity of KCC2 immunofluorescence in soma of neurons transfected with different shRNAs + eGFP. Non-transfected neurons are neurons that did not show eGFP fluorescence in cultures transfected with scrambled shRNA. Asterisks indicate significantly different values, **P < 0.01. n = 4 experiments, 5 randomly selected transfected pyramidal neurons per each condition and experiment.
shRNA-induced silencing of KCC2 modifies neuronal Cl− homeostasis
One of the major functions of KCC2 in mature neurons is its ability to control the [Cl−]i and produce a shift in EGABA (Rivera et al. 1999; Zhu et al. 2005). To study if a transfection with the shRNAs modifies [Cl−]i extrusion in neurons we employed a gramicidin-perforated patch-clamp recording and estimated EGABA by measuring the voltage dependence of neuronal responses to isoguvacine, a selective GABAA receptor (GABAAR) agonist (Fig. 3A). We found that transfection with shRNAKCC2_k10 produced a significant shift of EGABA as compared to neurons transfected with scrambled shRNA and non-transfected neurons (Fig. 3A and C). Similar EGABA values were obtained after the expression of shRNAKCC2_k11 (Fig. 3C). In the same neurons transfected with shRNAKCC2_k10 or shRNAKCC2_k11, the level of the resting membrane potential (Em) was similar and did not differ from the Em measured in neurons transfected with scrambled shRNA (−61.0 ± 4.2 mV, −58.8 ± 4.6 mV and −57.8 ± 5.2 mV, respectively (n = 8)).
The co-expression together with shRNAs of the mouse KCC2 that possess two nucleotide mismatches with both shRNAs created against rat KCC2 mRNA (Fig. 2A) restored the level of EGABA (Fig. 3C). Additional information on the ability of KCC2 shRNAs to modify the neuronal chloride extrusion system was obtained using analysis of the time of the recovery of isoguvacine responses to their basal level after an event producing a strong [Cl−]i rise (Fig. 3B). The increase of [Cl−]i in recorded neurons was achieved by the combination of neuron depolarization to 0 mV and prolonged 10 s bath application of 30 μm isoguvacine. Analysis showed that the time of the recovery of isoguvacine responses to basal level was systematically longer in neurons transfected with KCC2 shRNAs (Fig. 3D). As in the case of resting [Cl−]i, the co-transfection of mouse KCC2 with rat shRNAs effectively restored the neuronal [Cl−]i extrusion system. Taken together the findings, which show that two different shRNAs produce similar modifications of the neuronal [Cl−]i extrusion system and that these modifications are abolished by over-expression of KCC2 from other species, strongly indicate that the shRNA-dependent increase of [Cl−]i is due to the knockdown of endogenous KCC2.
This conclusion was further confirmed using non-invasive fluorescence monitoring of [Cl−]i with a genetically encoded probe, Cl−Sensor, and ratiometric analysis (Markova et al. 2008; Waseem et al. 2010). Analysis revealed that [Cl−]i was systematically higher in soma than in dendrites (Fig. 4A and B). Neurons transfected with shRNAKCC2_k10 exhibited significant increase in the basal level of [Cl−]i (Fig. 4B and C) and a decreased capability to restore the [Cl−]i level after its strong rise caused by a 10 s application of NMDA + glycine (Fig. 4C and D). Contrary to shRNA, the over-expression of KCC2 significantly decreased [Cl−]i and shortened the time of [Cl−]i recovery after NMDA application (Fig. 4C and D).
Figure 4. Non-invasive monitoring of [Cl−]i in cultured hippocampal neurons.

A, fluorescence micrograph of hippocampal neuron (14 DIV) expressing Cl-Sensor. Numbers indicate [Cl−]i estimated in soma and dendritic branch. Note higher [Cl−]i value in somatic region. B, mean ± SEM values of the basal level of [Cl−]i in soma and dendrites (measured after the first bifurcation at the distance of >50 μm from the soma) in neurons transfected with different constructs. Note that in neurons transfected with KCC2 [Cl−]i was significantly lower than in control ones (transfected with scrambled shRNA), while cells expressing shRNAKCC2_k10 possess much higher [Cl−]i. Data from 6 to 7 neurons per condition (3 transfections). *P < 0.05; **P < 0.01; ANOVA test. C, examples of [Cl−]i transients induced by 10 s focal application of NMDA. Records from three different neurons (indicated above traces). Note different basal levels of [Cl−]i and different kinetics of [Cl−]i recovery in neurons that express or do not express KCC2. D, mean ± SEM values of time required for 50% recovery of [Cl−]i after change induced by 10 s application of NMDA + glycine (half-decay time). Data were from 7 controls, 7 KCC2- and 6 shRNAKCC2_k10-transfected neurons. Note that KCC2 over-expression accelerated the [Cl−]i extrusion from the cell, while KCC2 silencing with shRNAKCC2_k10 strongly delayed it. **P < 0.01; Mann–Whitney non-parametric test.
Endogenous KCC2 protects neurons from transfection-induced toxicity
During the study of the effectiveness of the KCC2 shRNAs we noticed that in cultures transfected with either shRNAKCC2_k10 or shRNAKCC2_11 there were more dying transfected neurons than in control cultures transfected with scrambled shRNA. However, a quantification of the percentage of dying transfected neurons with condensed chromatin nuclei (Soriano et al. 2006), propidium iodide incorporation (Medina et al. 1999) or cleaved caspase-3-positive immunoreactivity (Adamec et al. 2001) did not reveal a significant difference between cultures transfected with scrambled shRNA and shRNAKCC2_k10. Therefore, to monitor more precisely the neuronal loss after transfection with different constructs we employed the previously described time-lapse approach to image the same transfected neurons during several consecutive days (Buerli et al. 2007). The advantage of this approach is that it allows us to take into account dead neurons that cannot be detected in fixed samples (see Supplementary material for details). Figure 5A and B illustrate the progressive loss of transfected neurons after transfection (number of alive transfected neurons at day 2 after transfection is taken as 100%). Remarkably, neurons transfected with shRNAKCC2_k10 died more rapidly in comparison to neurons transfected with scrambled shRNA. Overall, 4 days after transfection the difference between the percentages of surviving neurons in the control condition (scrambled shRNA, 79.3 ± 4.8%) and neurons expressing shRNAKCC2_k10 (59.4 ± 3.2%) or shRNAKCC2_11 (58.3 ± 2.4%) was statistically significant (P < 0.01, n = 6) (Fig. 5C). The survival of neurons transfected with non-silencing shRNAKCC2_k12 or shRNAKCC2_k13 was not different from that of control neurons (not shown). Thus, neuronal transfection with shRNAs silencing KCC2 is toxic.
Figure 5. Knocking down of the KCC2 expression reduces neuronal survival.

A, images of the same fluorescent neurons transfected with scrambled shRNA + eGFP (upper row) or with shRNAKCC2_k11 and eGFP (bottom row) taken at different days after transfection (ptd, post-transfection days). Arrows indicate neurons that died during the period of monitoring (1 out of 5 neurons transfected with scrambled shRNA and 2 out of 3 neurons transfected with shRNAKCC2_k11). B, percentage of surviving neurons as function of time after transfection. Example of an individual experiment (same experiment as in A). 35–40 neurons per experimental condition were analysed. C, mean ± SEM of the percentage of surviving neurons 4 days after transfection with indicated shRNA constructs. n = 6, 30–50 transfected neurons per condition and experiment were analysed. Similar experiments to those depicted in A and B. All data are normalized to the number of living neurons documented 48 h after transfection. Asterisks indicate significantly different values, **P < 0.01. D, application of Trolox (100 μm) to the culture media reduces death of neurons with knocked-down KCC2. Neuronal death was monitored 4 days after transfection. Data are normalized to the values obtained with transfection of scrambled shRNA under control condition. n = 5.
To explore whether shRNA-induced toxicity indeed relies on KCC2 knockdown or represents a side-effect of shRNAs action, we performed rescue experiments using the simultaneous expression of rat shRNAsKCC2 and mouse KCC2. Such a co-expression fully abolished the toxic effects of shRNAs (Fig. 5B and C) suggesting that shRNAKCC2-induced toxicity is due to decreased KCC2 expression.
One of the factors that might contribute to neuronal loss during the transfection procedure using lipofectamine reagents is oxidative stress (Dokka et al. 2000; Kongkaneramit et al. 2008). To verify whether reactive oxygen species are, at least in part, responsible for neuronal death in our experimental conditions, we incubated neurons during and after transfection with 100 μm Trolox, an analogue of vitamin E, frequently used to assess the role of oxidative injury in neuronal cell death (Vergun et al. 2001; Boland et al. 2002; Quintanilla et al. 2005). The application of Trolox significantly reduced loss of neuronal cells expressing either scrambled or silencing shRNAs (Fig. 5D), confirming that the transfection procedure itself is an important factor affecting neuronal survival.
C568A mutant of KCC2 increases Cl− and reduces neuronal viability
To further confirm the importance of endogenous KCC2 in neuronal resistance to the transfection-induced toxicity (i.e. oxidative stress) we suppressed the KCC2 function using an alternative approach to shRNA. Studying the properties of non-active KCC2 mutants C568A (Reynolds et al. 2008) and Y1087D (Akerman & Cline, 2006) we noticed that over-expression in 14 DIV cultures of construct encoding eGFP-KCC2C568A, but not eGFP-KCC2Y1087D, produced a strong depolarizing shift of EGABA and elongated EGABA recovery after an event inducing [Cl−]i rise as compared to control neurons transfected with eGFP or neurons transfected with eGFP-KCC2 (Fig. 6A) (see Methods and Supplementary Fig. S5 for details on creation and study of the effectiveness of mutant constructs). The changes in EGABA, produced by over-expression of eGFP-KCC2C568A were similar to those produced by expression of shRNAKCC2 and suggested that eGFP-KCC2C568A might act as a dominant-negative molecule. Although the exact mechanisms of eGFP-KCC2C568A action should be the subject of a separate study, we used an advantage of this construct to verify if an increase of [Cl−]i obtained using an alternative to the shRNA approach will modify neuronal survival. Analysis of neuronal survival 2–5 days after transfection showed that over-expression of the eGFP-KCC2C568A mutant progressively decreased the percentage of surviving neurons, whereas over-expression of another non-active mutant, eGFP-KCC2Y1087D, and wild-type eGFP-KCC2 construct did not affect neuronal viability (Fig. 6B). These data obtained using the shRNA-unrelated approach further support the hypothesis that endogenous KCC2 contributes to neuronal survival under oxidative stress conditions.
Figure 6. Chloride homeostasis and survival of neurons expressing different KCC2 mutants.

A, the basal level of EGABA (left panel) and times of 50% recovery of isoguvacine responses after imposed [Cl−]i rise (right panel) in neurons expressing different KCC2 mutants. Transfection at DIV 11, recording at DIV 14. Mean ± SEM values. *P < 0.05; **P < 0.01; 5 experiments, 1 to 3 neurons per experiment. Gramicidin-perforated patch. Vh=−65 mV. Similar protocol of experiment as in Fig. 3. For the sake of comparison, the column shRNAKCC2_K10 shows values from experiments described in Fig. 3C and D. Notice that expression of KCC2C568A induces a 20 mV positive shift of EGABA and elongates twofold the time of the recovery of [Cl−]i. These effects are of the same magnitude as those induced by expression of shRNAKCC2_K10. B, left panel shows an example for quantification of neuronal survival from an individual experiment (same protocol as illustrated in Fig. 5A). 35 to 40 neurons per experimental condition were analysed. Right panel shows mean ± SEM of the percentage of surviving neurons 4 days after transfection with indicated KCC2 mutants. For the sake of comparison, the column shRNAKCC2_K10 shows values from experiments described in Fig. 5C. 5 to 8 experiments for different mutants, 30–50 transfected neurons per condition and experiment were analysed. **P < 0.01. Notice that KCC2C568A induces a 1.8-fold decrease of the number of neurons surviving during 4 days after transfection.
Pharmacological blockage of KCC2 reduces viability of transfected neuronal cultures
When eGFP-KCC2C568A mutant and shRNAKCC2 modify neuronal survival by decreasing the activity of KCC2, then a pharmacological blockade of KCC2 should have a similar effect. To test this hypothesis, we examined the effects of an inhibitor of the KCC co-transporter [(dihydroindenyl)oxy]alkanoic acid (DIOA) (Taouil & Hannaert, 1999; Shen et al. 2000) on properties of cultured hippocampal neuronal cells. We found that incubation of eGFP-transfected neurons with DIOA (20 μm) for 1 h significantly depolarized EGABA (Fig. 7A) without any detectable change of Em (Fig. 7B). Application of DIOA also increased the frequency of spontaneous neuronal firing reflecting DIOA-dependent change in the neuronal network activity and/or neuronal excitability (Fig. 7C). In addition, exposure of EGFP-transfected cultures to DIOA for 48 h reduced the density of alive neuronal cells by 26% (Fig. 7D). In control cultures, that were not transfected, DIOA had no significant effect on neuronal survival (Fig. 7E). Thus, the incubation of the transfected cultures with DIOA produces a change in [Cl−]i and an increase of the neuronal loss similar to those observed after transfection of neurons with shRNA-silencing KCC2. These data are consistent with the hypothesis that both genetically and pharmacologically suppressed activity of KCC2 reduces neuronal resistance to toxicity induced by oxidative stress during a transfection procedure with lipofectamine.
Figure 7. Effects of DIOA (20 μm) on electrophysiological properties and survival of eGFP-positive neurons in cultures transfected using lipofectamine reagent.

A and B, effect of DIOA on EGABA (A) and resting membrane potential (Em, B) measured using gramicidin-perforated patch-clamp recording. EGABA and Em were determined as described in Methods. 3 experiments, 2–3 neurons per experiment and condition. C, traces illustrate example of the spontaneous neuronal firing recorded using conventional cell-attached mode of patch-clamp recording (no gramicidin included) at pipette potential 60 mV applied in order to prevent local membrane depolarization. Plot shows mean ± SEM of spike frequency recorded from neurons growing in control cultures and cultures incubated with DIOA during 1 h. 4 experiments, 2–3 neurons per experiment and condition. D, normalized number of eGFP-positive alive neurons determined using time-lapse recording 48 h after transfection. DIOA or H2O were applied 30 min before transfection and kept in media during experiment (48 h). EGFP-positive cells were monitored 24 and 48 h after transfection. n = 3, 30–50 neurons per experiment. E, density of alive neurons (NeuN-positive cells with healthy nuclei) in control (non-transfected) and transfected cultures. n = 4, 20 optical fields per experiment. All data are shown as mean ± SEM. *P < 0.05; **P < 0.01; Mann–Whitney non-parametric test.
NMDA-induced excitotoxicity and expression of the endogenous KCC2
Next we studied whether KCC2 contributes to neuronal survival in the well-characterized and widely used model of the excitotoxicity induced in primary neuronal cultures following treatment with NMDA, an agonist of NMDA subtype of glutamate receptors (Hardingham & Bading, 2010) (Supplementary Figs S6 and S7). First, we determined the dose-dependency of NMDA-induced toxicity in our culture model. Notably, we found that culture treatment with 20 or 40 μm NMDA for 30 min induced neuronal loss of 48.5 ± 5.3% and 63.3 ± 3.5%, respectively, 24 h later (Supplementary Fig. S7B). These doses were used in further experiments to study whether excitotoxicity modifies KCC2 expression in neuronal cultures and whether modulation of the KCC2 expression affects the self-protective neuronal resistance. Note, in all excitotoxicity experiments NMDA was applied together with 10 μm of glycine, a co-agonist of the NMDARs.
Previous studies showed the rapid decrease of the KCC2 expression under excitotoxic conditions in hippocampal slices (Rivera et al. 2004) and primary cultures of hippocampal neurons (Wake et al. 2007). We therefore next studied how excitotoxic treatment modifies expression of the endogenous KCC2 in our experiments. We found that 40 μm NMDA induced strong re-organization of the KCC2 expression pattern analysed 30 min after beginning of the treatment with NMDA (Fig. 8A and B). In the majority of neurons that were considered alive, based on their unaltered chromatin distribution, KCC2 immunofluorescence disappeared from tiny dendritic structures and remained concentrated in soma and varicosities formed on principal dendrites (76.8 ± 4.7%, n = 4, 100 neurons per experiment, see Fig. 8B panels b2 and b3, for illustration). In 18.5 ± 3.8% of neurons KCC2 fluorescence in soma decreased below mean 2SD value determined in control (0 μm NMDA) (Fig. 8B, panels b4 and b5) and in only 4.8 ± 1.4% of alive neurons KCC2 staining remained similar to control conditions (not shown). Five hours after NMDA withdrawal KCC2 fluorescence returned to normal cluster-like distribution in 53.8 ± 7.2% of neurons (Fig. 8C, panels c4 and c5), in 34.8 ± 8.3% it remained in varicosities (Fig. 8C, panels c2 and c3), and in 11.5 ± 1.7% of alive neurons it was below the level of the mean 2SD value. Twenty-four hours after excitotoxic insult, the cluster-like organization of KCC2 immunofluorescence in dendrites was observed in all alive neurons (>1000 neurons analysed after exposure to NMDA) (Fig. 8D). Although the obtained results do not allow any conclusion to be made about change in the transport capacity of KCC2 in neurons exposed to NMDA, our observation of the NMDA-dependent re-distribution of the transporter together with recent findings that excitotoxic activity can regulate the phosphorylation of the KCC2 and its membrane insertion (Rinehart et al. 2009; Lee et al. 2010) suggest that in cultured hippocampal neurons excitotoxicity might compromise the KCC2 functioning. We therefore next studied whether excitotoxic events affect neuronal ability to extrude chloride.
Figure 8. The distribution of KCC2 staining in cultures fixed at different times after 30 min treatment with 40 μm of NMDA and indicated above each set of images.

In control experiments (A) equivalent volume of H2O was applied instead of NMDA. Left column shows low magnification images of KCC2-positive neurons. Central column shows high magnification images of KCC2 immunofluorescence in alive neurons. Alive neurons were identified as MAP2-positive neurons (right column) with homogeneous distribution of chromatin (not shown, see also Supplementary Figs S6 and S7 describing neuronal survival after NMDA treatment). High magnification images in B illustrate two major types of neurons detected immediately after incubation with NMDA: neurons with KCC2 translocated to varicosities (b2 and b3) and neuron with low level of KCC2 expression (b4 and b5). White dotted line indicates boundaries of neuron for better illustration. High magnification images in C illustrate two major types of neurons observed 5 h after NMDA withdrawal: neurons with remaining varicosities (c2 and c3) and neuron recovered to control-like distribution of KCC2 and MAP2 (c5 and c6). Notice that all neurons surviving excitotoxic event for more than 24 h had high level of KCC2 protein expression (d1 and d2). We do not provide, however, the quantification of the intensity of the immunofluorescence since protein expression does not guarantee the functional activity of the KCC2 and constitutes a subject for special study.
[Cl−]i in NMDA-treated neurons
To determine [Cl−]i non-invasively we used Cl-Sensor. Analysis showed that in neurons expressing Cl-Sensor and empty pcDNA 3.1 vector (mock transfected) the long 10 min application of NMDA induced a sustained rise of [Cl−]i to 60–80 mm. After washout of the NMDA, [Cl−]i started to decline in 4 out of 5 neurons (Fig. 9A and B), but the kinetic of the recovery was twofold slower as compared to the recovery after brief NMDA application described in Fig. 4D. The [Cl−]i remained at the high saturated level for at least 5–10 min of the recording. In one neuron showing distinct varicosities after NMDA application, the Cl− recovery was not obtained and neuron disintegrated during recording (Fig. 9A). To verify if KCC2 might contribute to the Cl− extrusion under excitotoxic conditions, we measured [Cl−]i in neurons over-expressing KCC2 and found that exogenous KCC2 starts extruding intracellular Cl− already during NMDA application (Fig. 9A). Overall, the kinetics of [Cl−]i recovery was twofold faster than in mock-transfected neurons (Fig. 9B). Thus, the excitotoxic insult strongly reduces neuronal Cl− extrusion ability and expression of the exogenous KCC2 improves recovery to normal Cl− homeostasis.
Figure 9. Non-invasive monitoring of [Cl−]i in NMDA-treated neurons.

A, examples of [Cl−]i transients induced by 10 min focal application of NMDA. Recordings from three different neurons: two controls (top and middle traces) and one KCC2-expressing (bottom trace). Note different basal levels of [Cl−]i and different kinetics of [Cl−]i recovery in neurons expressing or not expressing KCC2. B, mean ± SEM values of time required for 50% recovery of [Cl−]i after change induced by 10 min application of NMDA + glycine (half-decay time). Data from 4 control and 4 KCC2-transfected neurons. Note that KCC2 over-expression strongly accelerates the [Cl−]i extrusion from the cell. *P < 0.05; Mann–Whitney non-parametric test. C, mean ± SEM values of basal level of [Cl−]i in soma of intact control neurons and NMDA exposed (30 min, 40 μm) mock and KCC2-transfected neurons. Recordings were started 4–6 h after NMDA withdrawal. Data from 11 neurons per condition. Note the identical [Cl−]i values for all conditions.
Next we analysed the level of [Cl−]i in neurons that restored normal dendritic morphology 4–6 h after 30 min exposure to 40 μm NMDA. Surprisingly, we found that in all NMDA-exposed mock-transfected neurons, as well as in neurons over-expressing KCC2, the level of [Cl−]i was identical to those in neurons not exposed to NMDA (Fig. 9C). The analysis of Cl− in neurons with distinct varicosities, similar to those shown in Supplementary Fig. S2C (i.e. neurons that are presumably damaged and will die) revealed a high level of [Cl−]i ranging from 50 to 70 mm (n > 20, not shown). Taken together, these data suggest that maintenance of the normal intracellular chloride concentration is essential for neuronal survival after excitotoxic insult and suggest that KCC2 can contribute to this process.
KCC2 and neuronal survival after excitotoxic insult
The importance of KCC2 expression for neuronal survival after excitotoxic insult was further verified in experiments involving transfection of different constructs modifying expression of chloride–potassium co-transporters as well as using a pharmacological approach. In experiments involving genetic modification we over-expressed desired constructs under conditions minimizing the transfection-induced toxicity (adding of Trolox to the transfection mixture, 100% media change after transfection, 50% media change 2 days after transfection). On day 3 after transfection (a steady-state level of the survival of transfected neurons) we exposed cultures for 30 min to different concentrations of NMDA (Fig. 10A).
Figure 10. Effects of the modulation of KCC2 expression on neuronal survival after excitotoxic insult.
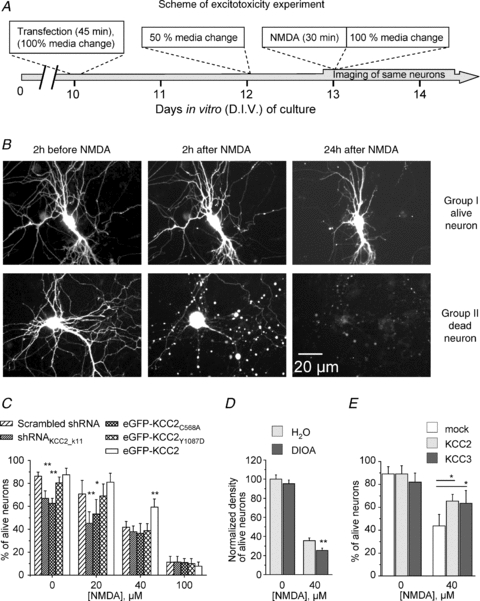
A, scheme of experiment. Transfections were performed at DIV 10 under conditions minimizing oxidative stress (see Methods). Three days after transfection cultures were incubated with NMDA for 30 min. Images of same neurons were taken 2 h before application of NMDA and different times after NMDA wash-out. B, images illustrate kinetic of changes of neuronal morphology after exposure to NMDA (40 μm, 30 min). Group I corresponds to alive neurons expressing eGFP protein; 24 h after treatment the fluorescence is still evenly distributed. Dendrites are well preserved. Group II represents neurons that disappeared completely from the optical field 24 h after exposure to NMDA. Usually these cells showed large varicosities in dendrites already 2 h after NMDA treatment. C, survival rate of neurons transfected with different constructs and exposed to indicated concentrations of NMDA for 30 min. The number of surviving neurons was determined 24 h after NMDA application. Pooled data from 4–6 experiments per condition. *P < 0.05; **P < 0.01; one-way ANOVA test. D, effect of DIOA on survival of neurons exposed to 0 or 40 μm NMDA for 30 min. DIOA (20 μm) or H2O were applied 30 min before application of NMDA. Cultures were fixed 24 h after NMDA wash-out. Density of neurons was determined as detailed in Methods. 3 experiments, 20 optical fields per experiment. Non-parametric Mann–Whitney test. E, effect of the over-expression of KCC2 and KCC3 on neuronal survival 24 h after exposure to NMDA. Mock-transfected neurons were neurons transfected with eGFP and pcDNA3.1. 4 experiments. Non-parametric Mann–Whitney test.
Using time-lapse monitoring of the same neurons (Fig. 10B and C) we found that neuronal transfection with either shRNAKCC2_k11 or mutant eGFP-KCC2C568A, but not eGFP-KCC2Y1087D, led to a significant decrease of neuronal survival after 30 min exposure to 20 μm NMDA, but not to 40 or 100 μm of NMDA. In contrast, the over-expression of the wild-type EGFP-KCC2 increased the number of surviving neurons after 30 min culture treatment with 20 and 40 μm, but not 100 μm of NMDA.
Consistent with the above observations, application of DIOA (20 μm) significantly decreased the number of neurons surviving after excitotoxic insult induced by 30 min treatment with 40 μm NMDA (Fig. 10D). Thus, taken together, the above results strongly support the hypothesis of the neuroprotective action of KCC2 during excitotoxicity.
Neuroprotective action of the KCC2 requires active Cl−–K+ co-transport
Previous studies showed that some regulatory actions of KCC2 do not require active transport of the KCC2 and rely on interaction of its C-terminus with the cytoskeleton (Li et al. 2007; Horn et al. 2010). The experiments involving eGFP-KCC2C568A and eGFP-KCC2Y1087D mutants (Fig. 10B) suggest that the neuroprotective effects of KCC2 require the active form of KCC2. However, it remains unclear if it involves also the KCC2-specific interactions with the cytoskeleton. To answer this question, we over-expressed KCC3 (an analogue of KCC2 that has a different structure of C-terminus end, does not interact with neuronal cytoskeleton (Li et al. 2007), but effectively decreases neuronal [Cl−]i to a similar extent as does exogenous KCC2 (not shown)) and found that over-expression of this transporter significantly increased the neuronal survival after 30 min treatment with 40 μm NMDA (Fig. 10E). Thus, the maintenance of neuronal potassium–chloride co-transport contributes to resistance of neuronal cells to excitotoxic insults.
Discussion
The main finding of this study is that silencing of endogenous KCC2 reduces the neuronal resistance for toxicity. Previous studies reported an important role of KCC2 in neuronal migration (Bortone & Polleux, 2009), development (Cancedda et al. 2007), formation of the inhibitory and excitatory synapses (Chudotvorova et al. 2005; Akerman & Cline, 2006; Li et al. 2007) and prevention of the spasticity after spinal cord injury (Boulenguez et al. 2010). The present work extends this list by attributing a neuroprotective role for endogenous KCC2 during injury of hippocampal neurons induced by transfection-dependent oxidative stress or NMDA-dependent excitotoxicity.
In our study the functional suppression of endogenous KCC2 was achieved using three different approaches – shRNAs silencing rat KCC2, over-expression of the mutant KCC2C568A that, as we suggest, acts as a dominant-negative molecule, and using DIOA, an inhibitor of the KCC2. Although each of the molecular and pharmacological tools employed in the present study might induce neuronal death or exert a neuroprotective action by a variety of mechanisms independent on KCC2, it is unlikely that two shRNAs targeting different regions of KCC2, the KCC2C568A mutant and an inhibitor of KCC2, would co-incidentally produce similar changes in chloride homeostasis and neuronal survival.
The siRNA approach is one of the most effective ways of protein knocking down that was used to study the functional role of thousands of molecules. However, considering the complex mechanism of siRNA action, some siRNAs produce non-specific effects including cellular toxicity (Bauer et al. 2009) and modulation of neuronal growth and synapse formation (Alvarez et al. 2006). Therefore, when we found that shRNAs directed against the rat KCC2 transporter induce neuronal death, an action reminiscent of non-specific siRNAs effects, we studied in detail whether shRNA-induced toxicity was a result of non-specific shRNA action or a specific consequence of KCC2 silencing.
Obtained evidence favoured the hypothesis that shRNA-induced toxicity was a result of KCC2 silencing. Indeed, we found that out of five different shRNAs used in the present work (one scrambled shRNA and four shRNAs complementary to different regions of rat KCC2 mRNA) neuronal death was only induced by shRNAs effectively silencing KCC2. Other shRNAs not modifying the KCC2 expression did not affect neuronal survival. In addition to this we found that the toxic action of shRNAKCC2_K10 and shRNAKCC2_K11 was rescued after over-expression of mouse (Fig. 5), but not rat, KCC2 (not shown).
Complementary data confirming the importance of endogenous KCC2 expression for neuronal resistance to neurotoxicity was obtained using expression of the KCC2C568A mutant. We found that over-expression of this mutant (but not the other non-active mutant KCC2Y1087D) modifies a neuronal chloride homeostasis and decreases neuronal survival in a manner similar to those induced by shRNA. The C568A mutant of KCC2 was employed for the first time in the study of Reynolds et al. (2008), where it was used as a negative control for the effects observed after expression of KCC2 in zebrafish embryos. The same mutant was successfully used as a negative control in the in vivo study of KCC2 effects on the development of immature rat cortical neurons (Cancedda et al. 2007). Consistent with these works, we found that in developing (9 DIV) hippocampal cultures both the C568A and the Y1087D point mutations effectively prevented the KCC2-dependent hyperpolarizing shift of EGABA (Supplementary Fig. S5). An unexpected finding was that in more mature hippocampal neurons at 14 DIV (a stage characterized by extensive expression of endogenous KCC2; Khirug et al. 2005), the over-expression of eGFP-KCC2C568A (but not eGFP-KCC2Y1087D) inhibited the neuronal chloride extrusion capacity (Fig. 6A) to a similar extent as observed after expression of KCC2-selective shRNAs (Fig. 3C). This observation is compatible with the suggestion that eGFP-KCC2C568A acts as a dominant-negative construct. Previous findings showed that, in a neuronal membrane, the KCC2 transporter forms heteromers (Blaesse et al. 2006; Uvarov et al. 2006). Thus, one of the mechanisms of eGFP-KCC2C568A-dependent inhibition of chloride extrusion could be an interaction with endogenous KCC2, rendering the newly formed complex inactive. The unusual properties of the KCC2C568A mutant were recently mentioned in a study by Horn et al. (2010) who found that, unlike wild-type KCC2 and the transport inactive N-terminal-deleted KCC2, KCC2C568A does not interact with the cytoskeleton-associated protein 4.1N. The authors suggest that the KCC2C568A mutation might cause changes in the tertiary structure of KCC2, yielding a protein inactive both as an ion transporter and as an interacting partner of 4.1N. Additional investigations are needed to characterize changes in the KCC2 protein structure and function caused by the C568A mutation, and clarify the mechanisms leading to KCC2C568A-dependent lowering of the chloride extrusion capacity.
Finally, we found that culture treatment with DIOA, a molecule effectively blocking potassium–chloride co-transporters (Taouil & Hannaert, 1999; Shen et al. 2000), produces a depolarizing shift of EGABA and reduction of the neuronal survival, similar to shRNAKCC2 and KCC2C568A mutants. Previous work has shown that DIOA in micromolar concentrations might induce a wide spectrum of side-effects including depletion of the ATP from acute hippocampal slices and cytochrome c release from isolated mitochondria (Pond et al. 2004), inhibition of P-type ATPases (Fujii et al. 2007) and swelling-activated chloride channels (Zierler et al. 2008). Our data suggest that in primary hippocampal cultures within the studied time scale, DIOA acts mainly as an inhibitor of potassium–chloride co-transporters. This observation is consistent with the report by Boulenguez et al. (2010) showing that in the in vitro spinal cord isolated from neonatal rat, DIOA (30 μm) produced an increase of [Cl−]i of similar extent to that observed in preparations from KCC2-deficient animals. An alternative explanation could be that in our model the effects of DIOA on other than KCC2 targets may induce an increase of [Cl−]i and reduce neuronal survival in a manner similar to that suggested for the inhibitor of KCC2.
Our work also provides important methodological suggestions to study properties of small populations of genetically modified neurons. We illustrate a striking difference in results of analysis of neuronal death using traditional quantification of the number of alive/dead neurons at a given fixed point of culture development and also using a time-lapse approach. The analysis of the fixed cultures underestimated sixfold the percentage of dying neurons and most importantly, led to wrong conclusions of the absence of neurotoxic action of constructs silencing KCC2. A time-lapse approach revealed a strong and significant decrease in survival of neurons with suppressed function of KCC2. Our results illustrate the importance of choosing an appropriate analytical method when studying small populations of transfected neurons. The time-lapse approach is one way of characterizing the fate of every genetically modified neuron, easy determining the time point of highest effectiveness of the molecule of interest and avoiding possible artefacts related to the variability in rates of cultures transfection with different constructs. In the present study the time-lapse approach allowed us to determine the contribution of endogenous KCC2 to maintenance of the neuronal pro-survival ability.
Numerous previous studies have shown that KCC2 is one of the neuronal molecules whose activity and expression change during different physiological and pathological processes. The physiological pattern of the activity (Ganguly et al. 2001; Ludwig et al. 2003) and single epileptiform discharge (Khirug et al. 2010) potentiate KCC2 expression, whereas long-lasting excitotoxic events (Rivera et al. 2004; Jin et al. 2005; Wake et al. 2007; Nardou et al. 2009) strongly suppress KCC2 expression on functional, transcriptional and translational levels. Numerous data showed also that down-regulation of the KCC2 contributes to the formation of epileptiform discharges (Ben-Ari et al. 2007). However, it remained unclear if decreased KCC2 expression affects neuronal survival in damaged tissue. Some results suggested that KCC2 might exert a neuroprotective action in different neuronal injury models. Thus, Papp et al. (2008) and Jaenisch et al. (2010) reported strong membrane expression of KCC2 in neurons surviving transient brain ischaemia. Wake et al. (2007) found that KCC2 over-expression protected cultured hippocampal neurons against peroxide-induced toxicity. Our study provides the first direct evidence on the importance of the endogenous KCC2 for survival of the hippocampal neuronal after oxidative stress and excitotoxic insults.
The resistance to various neurotoxic signals is one of the most important basic properties of neuronal cells. During the last decade numerous physiological processes and signalling cascades have been described to contribute to pro-survival neuronal signalling (for recent reviews see Medina, 2007; Hardingham & Bading, 2010). A particular importance has been attributed to the ongoing neuronal activity and local calcium influxes through synaptic NMDARs (Hardingham et al. 2002; Ivanov et al. 2006; Soriano et al. 2006; Tauskela et al. 2008). According to the theory of neuronal health (Isacson, 1993), neurons can exist in a spectrum of states ranging from fully functioning and resilient at one extreme to dysfunctional and vulnerable to insults at the other extreme. The decrease of neuronal self-protective strength caused by the suppression of the KCC2 function might lead to a shift in the equilibrium between pro-survival and pro-death signalling and change neuronal sensitivity to different external stimuli. Indeed, we found that silencing of the KCC2 enhances neurotoxic action of NMDA and vice versa, the over-expression of the KCC2 exerts a neuroprotective effect only under experimental conditions leading to loss of less than 60% of neuronal cells (Fig. 10E). Under conditions of strong excitotoxic events producing death of more than 80% of neurons, we could not detect any regulatory action of the KCC2 (Fig. 10E). One explanation of this could be that a strong excitotoxicity event rapidly inactivates both endogenous or over-expressed KCC2 making negligible difference in potassium–chloride homeostasis between neurons with knocked-down or over-expressed KCC2. An alternative possibility could be that the rise of the intracellular calcium during strong excitotoxic events largely overpasses the threshold required for activation of the pro-death signalling and, respectively, the KCC2-dependent modulation of ion homeostasis is ineffective under such conditions.
What are the mechanisms mediating neuroprotective action of KCC2 during toxicity insults? Our data on decrease of the viability of neurons over-expressing KCC2 mutant and successful rescue of shRNA transfected neurons with over-expression of the KCC3, strongly suggest that it is KCC2-mediated ion transport which is crucial for neuronal survival. Multiple studies have shown that one of the main functions of KCC2 in mature neurons is maintenance of the low neuronal [Cl−]i and inhibitory strength of GABA/glycine (for reviews see Ben-Ari et al. 2007; Blaesse et al. 2009). The reduction of the inhibitory strength of GABA leads to activation of the voltage-gated calcium channels and facilitation of the calcium influxes through NMDARs (Leinekugel et al. 1997; Akerman & Cline, 2006). Under toxic conditions the endogenous KCC2 might protect neurons from excessive influx of Ca2+, whereas knocking down of the KCC2 would facilitate activation of the NMDA-dependent pro-death signalling cascades. One additional mechanism of the neuroprotective action of KCC2 might rely on reduction of the Cl−-dependent swelling occurring under toxic conditions, which is considered as an important event contributing to cell death (Rothman, 1985; Pond et al. 2006). However, the contribution of the KCC2 to cell swelling during toxic insult has not yet been elucidated and constitutes an important direction for future studies.
Acknowledgments
We are grateful to Drs Genevieve Chazal and Claudio Rivera for critical reading of the manuscript, Dr Natalia Kashchak for helpful discussions, Galina Medyna for help in manuscript editing, Thomas Marissal for assistance in the creation of some pcDNA constructs used in this study and Miika Palviainen for technical assistance in some sets of experiments. This work was supported by an ANR grant (French National Research Council) attributed to I.M., an EMBO fellowship for S.S. and A.L., an IBRO grant for Y.S., the French Foundation for Medical Research grant for I.C. and European FP-7 grant no. HEALTH-F2-2008-202088 ‘NeuroCypres’ for M.M. and P.B.
Glossary
Abbreviations
- DIV
days in vitro
- shRNA
short hairpin RNA
- RT
room temperature
- TBSTD
tris-buffered saline, 0.1% Tween, 5% DMSO
Author contributions
C.P. created and validated shRNA constructs, prepared all primary cultures, performed characteristic of the excitotoxicity models and contributed to the study of organotypic cultures. M.S. performed patch-clamp analysis of the Cl− extrusion ability of KCC3 and contributed to the analysis of the neuroprotective action of KCC3. H.B. and A.L. performed analysis of shRNA effectiveness in hippocampal organotypic slices. O.G., I.C. and S.S. performed time-lapse imaging of transfected hippocampal neurons in culture and analysis of neuronal survival. M.M. and P.B. completed the measurement of [Cl−]i using Cl− Sensor in cultured hippocampal neurons. S.C. created mutants of KCC2. Y.S. performed quantitative immunocytochemistry analysis of KCC2 in culture. I.M. was in charge of electrophysiology, management of the project and writing the manuscript. All authors approved the final version of the manuscript.
Authors' present addresses
I. Chudotvorova: Brandeis University, 415 South Street, Waltham, MA 02453, USA.
S. Salozhin: Molecular Neurobiology Group, Institute of Higher Nervous Activity and Neurophysiology RAS, 117485, Butlerova 5A, Moscow, Russia.
M. Schaefer: Institute of Anatomy and Cell Biology, Center for Neurosciences, University of Freiburg, Germany.
M. Mukhtarov and P. Bregestovski: INSERM U 751, Marseille, France.
Supplementary material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Adamec E, Yang F, Cole GM, Nixon RA. Multiple-label immunocytochemistry for the evaluation of nature of cell death in experimental models of neurodegeneration. Brain Res Brain Res Protoc. 2001;7:193–202. doi: 10.1016/s1385-299x(01)00072-1. [DOI] [PubMed] [Google Scholar]
- Akerman CJ, Cline HT. Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci. 2006;26:5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez VA, Ridenour DA, Sabatini BL. Retraction of synapses and dendritic spines induced by off-target effects of RNA interference. J Neurosci. 2006;26:7820–7825. doi: 10.1523/JNEUROSCI.1957-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Kinkl N, Meixner A, Kremmer E, Riemenschneider M, Forstl H, Gasser T, Ueffing M. Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther. 2009;16:142–147. doi: 10.1038/gt.2008.123. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Blaesse P, Guillemin I, Schindler J, Schweizer M, Delpire E, Khiroug L, Friauf E, Nothwang HG. Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J Neurosci. 2006;26:10407–10419. doi: 10.1523/JNEUROSCI.3257-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland A, Gerardy J, Mossay D, Delapierre D, Seutin V. Pirlindole and dehydropirlindole protect rat cultured neuronal cells against oxidative stress-induced cell death through a mechanism unrelated to MAO-A inhibition. Br J Pharmacol. 2002;135:713–720. doi: 10.1038/sj.bjp.0704519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron. 2009;62:53–71. doi: 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Buerli T, Pellegrino C, Baer K, Lardi-Studler B, Chudotvorova I, Fritschy JM, Medina I, Fuhrer C. Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat Protoc. 2007;2:3090–3101. doi: 10.1038/nprot.2007.445. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hubner CA, Pellegrino C, Ben Ari Y, Medina I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol. 2005;566:671–679. doi: 10.1113/jphysiol.2005.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000;17:521–525. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Fujii T, Ohira Y, Itomi Y, Takahashi Y, Asano S, Morii M, Takeguchi N, Sakai H. Inhibition of P-type ATPases by [(dihydroindenyl)oxy]acetic acid (DIOA), a K+-Cl− cotransporter inhibitor. Eur J Pharmacol. 2007;560:123–126. doi: 10.1016/j.ejphar.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–532. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Horn Z, Ringstedt T, Blaesse P, Kaila K, Herlenius E. Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur J Neurosci. 2010;31:2142–2155. doi: 10.1111/j.1460-9568.2010.07258.x. [DOI] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Isacson O. On neuronal health. Trends Neurosci. 1993;16:306–308. doi: 10.1016/0166-2236(93)90104-t. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the ERK activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–e159. doi: 10.1161/STROKEAHA.109.570424. [DOI] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired Cl− extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Kawabata I, Umeda T, Yamamoto K, Okabe S. Electroporation-mediated gene transfer system applied to cultured CNS neurons. Neuroreport. 2004;15:971–975. doi: 10.1097/00001756-200404290-00008. [DOI] [PubMed] [Google Scholar]
- Khirug S, Ahmad F, Puskarjov M, Afzalov R, Kaila K, Blaesse P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J Neurosci. 2010;30:12028–12035. doi: 10.1523/JNEUROSCI.3154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Kongkaneramit L, Sarisuta N, Azad N, Lu Y, Iyer AK, Wang L, Rojanasakul Y. Dependence of reactive oxygen species and FLICE inhibitory protein on lipofectamine-induced apoptosis in human lung epithelial cells. J Pharmacol Exp Ther. 2008;325:969–977. doi: 10.1124/jpet.107.136077. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, Reichling DB. Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J Neurosci Methods. 1995;57:27–35. doi: 10.1016/0165-0270(94)00116-x. [DOI] [PubMed] [Google Scholar]
- Lee HH, Jurd R, Moss SJ. Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol Cell Neurosci. 2010;45:173–179. doi: 10.1016/j.mcn.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben Ari Y, Khazipov R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABAA and NMDA receptors in the neonatal hippocampus. Neuron. 1997;18:243–255. doi: 10.1016/s0896-6273(00)80265-2. [DOI] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinanen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Markova O, Mukhtarov M, Real E, Jacob Y, Bregestovski P. Genetically encoded chloride indicator with improved sensitivity. J Neurosci Methods. 2008;170:67–76. doi: 10.1016/j.jneumeth.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Medina I. Extrasynaptic NMDA receptors reshape gene ranks. Sci STKE. 2007;2007:e23. doi: 10.1126/stke.3862007pe23. [DOI] [PubMed] [Google Scholar]
- Medina I, Ghose S, Ben Ari Y. Mobilization of intracellular calcium stores participates in the rise of [Ca2+]i and the toxic actions of the HIV coat protein GP120. Eur J Neurosci. 1999;11:1167–1178. doi: 10.1046/j.1460-9568.1999.00550.x. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci. 2002;22:4412–4417. doi: 10.1523/JNEUROSCI.22-11-04412.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Ben-Ari Y, Khalilov I. Bumetanide, an NKCC1 antagonist, does not prevent formation of epileptogenic focus but blocks epileptic focus seizures in immature rat hippocampus. J Neurophysiol. 2009;101:2878–2888. doi: 10.1152/jn.90761.2008. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Amici M, Sobrero F, Spinelli G, Di AS, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F. Anomalous levels of Cl− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD. The chloride transporter Na+-K+-Cl− cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci. 2006;26:1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond BB, Galeffi F, Ahrens R, Schwartz-Bloom RD. Chloride transport inhibitors influence recovery from oxygen-glucose deprivation-induced cellular injury in adult hippocampus. Neuropharmacology. 2004;47:253–262. doi: 10.1016/j.neuropharm.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Quintanilla RA, Munoz FJ, Metcalfe MJ, Hitschfeld M, Olivares G, Godoy JA, Inestrosa NC. Trolox and 17β-estradiol protect against amyloid β-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J Biol Chem. 2005;280:11615–11625. doi: 10.1074/jbc.M411936200. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Brustein E, Liao M, Mercado A, Babilonia E, Mount DB, Drapeau P. Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci. 2008;28:1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Ebihara S, Akaike N. Gramicidin perforated patch-clamp technique reveals glycine-gated outward chloride current in dissociated nucleus solitarii neurons of the rat. J Neurophysiol. 1994;72:1103–1108. doi: 10.1152/jn.1994.72.3.1103. [DOI] [PubMed] [Google Scholar]
- Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138:525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci. 1985;5:1483–1489. doi: 10.1523/JNEUROSCI.05-06-01483.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MR, Chou CY, Ellory JC. Volume-sensitive KCI cotransport associated with human cervical carcinogenesis. Pflugers Arch. 2000;440:751–760. doi: 10.1007/s004240000338. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Bell KF, Hardingham GE. Role of histone acetylation in the activity-dependent regulation of sulfiredoxin and sestrin 2. Epigenetics. 2009;4:152–158. doi: 10.4161/epi.4.3.8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Taouil K, Hannaert P. Evidence for the involvement of K+ channels and K+-Cl− cotransport in the regulatory volume decrease of newborn rat cardiomyocytes. Pflugers Arch. 1999;439:56–66. doi: 10.1007/s004249900117. [DOI] [PubMed] [Google Scholar]
- Tauskela JS, Fang H, Hewitt M, Brunette E, Ahuja T, Thivierge JP, Comas T, Mealing GA. Elevated synaptic activity preconditions neurons against an in vitro model of ischemia. J Biol Chem. 2008;283:34667–34676. doi: 10.1074/jbc.M805624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–1337. doi: 10.1111/j.1460-9568.2005.03959.x. [DOI] [PubMed] [Google Scholar]
- Tyzio R, Ivanov A, Bernard C, Holmes GL, Ben Ari Y, Khazipov R. Membrane potential of CA3 hippocampal pyramidal cells during postnatal development. J Neurophysiol. 2003;90:2964–2972. doi: 10.1152/jn.00172.2003. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Pruunsild P, Kaila K, Delpire E, Timmusk T, Rivera C, Airaksinen MS. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem. 2007;282:30570–30576. doi: 10.1074/jbc.M705095200. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Rivera C, Airaksinen MS. Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J Neurosci. 2006;26:13463–13473. doi: 10.1523/JNEUROSCI.4731-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR. Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol. 2001;531:147–163. doi: 10.1111/j.1469-7793.2001.0147j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem T, Mukhtarov M, Buldakova S, Medina I, Bregestovski P. Genetically encoded Cl-Sensor as a tool for monitoring of Cl-dependent processes in small neuronal compartments. J Neurosci Methods. 2010;193:14–23. doi: 10.1016/j.jneumeth.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–268. doi: 10.1002/hipo.10014. [DOI] [PubMed] [Google Scholar]
- Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
- Zhu L, Polley N, Mathews GC, Delpire E. NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res. 2008;79:201–212. doi: 10.1016/j.eplepsyres.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierler S, Frei E, Grissmer S, Kerschbaum HH. Chloride influx provokes lamellipodium formation in microglial cells. Cell Physiol Biochem. 2008;21:55–62. doi: 10.1159/000113747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.