

Non-technical summary
The heart is widely known to be controlled by the sympathetic nervous system, which uses the neurotransmitter noradrenaline (norepinephrine) to increase the rate and force of heart beating. We used a preparation of sympathetic nerve cells and heart cells cultured together that recapitulates this sympathetic control of the heart. Our goal was to probe the role of a particular nerve cell potassium current, called the M-current, in the control of neurotransmitter release, using the contraction rate of the co-cultured heart cells as a functional read-out of noradrenaline release. Using several drugs and receptor agonists, we manipulated the activity of M-current in the nerve cells, which were stimulated by nicotine, and monitored its effect on heart cell beating. We find that the M-type potassium current has a robust role in the control of noradrenaline release from the nerve cells, and in the response of the heart cells to increased beating frequency as a result.
Abstract
Abstract
M-type (KCNQ) K+ channels are known to regulate excitability and firing properties of sympathetic neurons (SNs), but their role in regulating neurotransmitter release is unclear, requiring further study. We sought to use a physiological preparation in which SNs innervate primary cardiomyocytes to evaluate the direct role of M-channels in the release of noradrenaline (NA) from SNs. Co-cultures of rat SNs and mouse cardiomyocytes were prepared, and the contraction rate (CR) of the cardiomyocyte syncytium monitored by video microscopy. We excited the SNs with nicotine, acting on nicotinic acetylcholine receptors, and monitored the increase in CR in the presence or absence of the specific M-channel opener retigabine, or agonists of bradykinin B2 or purinergic P2Y receptors on the SNs. The maximal adrenergic effect on the CR was determined by application of isoproterenol (isoprenaline). To isolate the actions of B2 or P2Y receptor stimulation to the neurons, we prepared cardiomyocytes from B2 receptor or P2Y2 receptor knock-out mice, respectively. We found that co-application of retigabine strongly decreased the nicotine-induced increase in CR. Conversely, co-application of bradykinin or the P2Y-receptor agonist UTP augmented the nicotine-induced increase in CR to about half of the level produced by isoproterenol. All effects on the CR were wholly blocked by propranolol. Our data support the role of M-type K+ channels in the control of NA release by SNs at functional adrenergic synapses on cardiomyocytes. We conclude that physiological receptor agonists control the heart rate via the regulation of M-current in SNs.
Introduction
Release of noradrenaline (NA) from the sympathetic nervous system exerts powerful control over cardiac beating. The effects are chronotropic, in which the heart beats faster, and inotropic, in which each contraction is stronger. Both actions are via stimulation of β-adrenergic receptors (β-ARs), which act through Gs-type G proteins and protein kinase A to phosphorylate IKs K+, If cation, and L-type Ca2+, channels (Hille, 2001). Sympathetic action on the heart arises from its innervation by cervical and upper thoracic (stellate) ganglia, as well as from circulating NA released from adrenal chromaffin cells. Thus, the mechanisms regulating NA release from sympathetic neurons are critical to cardiac function, and to cardiovascular disease. The M-type K+ current plays a dominant role in neuronal excitability and discharge properties in a variety of neurons (Robbins, 2001). It was first identified in sympathetic neurons as a non-inactivating, voltage-gated K+ current with a threshold for activation near the resting potential of neurons that is suppressed by stimulation of muscarinic acetylcholine receptors (Brown & Adams, 1980; Constanti & Brown, 1981). In the sympathetic neurons of the superior cervical ganglion (SCG) which have been most studied for investigation of M-current physiology, most neuronal M-channels are composed of KCNQ2/3 heteromers (Wang et al. 1998). M-channels also contain KCNQ5 subunits in peripheral ganglia (Schroeder et al. 2000; Passmore et al. 2003) and vascular smooth muscle (Mackie et al. 2008); KCNQ4 homomers predominate in the inner ear and auditory cortex (Kubisch et al. 1999; Kharkovets et al. 2000) and KCNQ1 is the pore-forming subunit mediating IKs currents in the heart, epithelia and inner ear (Peroz et al. 2008). Of particular note is that mutations of each KCNQ subunit are associated with specific human diseases, including epilepsy, cardiac arrhythmias and deafness (Maljevic et al. 2010).
M-current is well known for its modulation by agonists of various receptors coupled to the Gq/11 class of G proteins that activate phospholipase C, which hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol. Besides the M1 muscarinic receptors that give M-current its name, other Gq/11-coupled receptors in SNs also suppress M-channels, such as bradykinin B2, purinergic P2Y and angiotensin AT1 types (Delmas & Brown, 2005). We have characterized these receptors into two groups, based upon their intracellular mechanism of action in suppressing M-current, and their ion channel targets. Whereas stimulation of M1 and AT1 receptors inhibits both M-channels and voltage-gated Ca2+ channels (VGCCs) via depletion of PIP2 (Gamper et al. 2004; Delmas & Brown, 2005; Suh et al. 2010), bradykinin (BK) and purinergic agonists such as uridine triphosphate (UTP) do not deplete PIP2 levels, have no action on VGCCs and depress M-current via IP3-mediated signals (Zaika et al. 2007, 2011).
M-current regulates excitability in every neuron in which it is found (Hernandez et al. 2008). The accepted mechanism is by control over axon potential generation at somato-axonal sites where firing is initiated. But does it also tune exocytosis and the release of neurotransmitter (NT) at presynaptic nerve terminals, over and above its control over action potential firing? This critical issue remains murky. Using carbon-fibre amperometry, we have shown release of NA from SCG neurons to be dependent on M-current activity (Hernandez et al. 2008), although we could not differentiate that effect from control of action potential firing. Using brain synaptosomes, the Taglialatela lab showed M-channels to regulate release of tritiated NTs by high-K+ stimulation (Martire et al. 2004, 2007). However, the data concerning this role for M-channels in measurements of tritiated NA release in SCG cells are ambiguous (Lechner et al. 2003; Kubista et al. 2009). In the case of sympathetic ganglia cells, a number of investigators have assayed NT release by culturing rodent SCG neurons together with ventricular cardiomyocytes in a dish. Using such an approach, one group of investigators showed such neurons to release a surprisingly diverse collection of NTs that include NA, acetylcholine (ACh), adenosine, or some combination of all three (Furshpan et al. 1976, 1986a,b; Potter et al. 1986; Matsumoto et al. 1987). Other labs have used this co-culture system to similarly model adrenergic control of the heart, and found the SCG neurons to nearly exclusively release NA under their experimental conditions (Conforti et al. 1991; Lockhart et al. 1997).
Recently, this approach was refined, in a way that recapitulates, in vitro, the sympathetic innervation of the heart (Shcherbakova et al. 2007). The cardiomyocytes form a syncytium that spontaneously contracts, and the neurons form functional adrenergic synapses on the heart cells that include all known components of such synaptic complexes. Thus, released NA can be observed to regulate the rate and force of contraction, providing a measure of functional NT release. We exploited this preparation to investigate the role of M-channels in control over NA release in sympathetic neurons. Importantly, the synapses are functional, providing us with a read-out that predicts the effect of M-current modulation on target organs. We find that M-current activity in the sympathetic neurons directly affects the release of NA by cholinergic stimulation of the neurons, manifested by marked effects on the chronotropic response of the co-cultured cardiomyocytes. Furthermore, stimulation of receptors that selectively depress M-current significantly increased those responses. This study directly links receptor-mediated M-channel regulation to adrenergic control of heart cell function.
Methods
Animals
Animal use and welfare adhered to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals, following a protocol reviewed and approved by the Institutional Laboratory Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio.
All mice were of the C57BL/6 strain. Wild-type and homozygous B2 knockout (k/o) mice were purchased from The Jackson Laboratory (B6; 129S7-Bdkrb2tm1Jfh/J, stock no. 2641) and bred in-house. Homozygous P2Y2 k/o mice, bred as previously described (Homolya et al. 1999), were generously given to us by Volker Vallon (UC San Diego, USA) and bred in-house.
SCG neuron/cardiomyocyte co-culture
We used previously published procedures for this preparation (Shcherbakova et al. 2007). Rats and mice were first killed by an overdose of halothane anaesthetic. Hearts were then isolated from 1-day-old neonatal mice, placed in a dish containing 137 mm NaCl, 5.4 mm KCl, 0.8 mm MgSO4, 5.6 mm glucose, 0.44 mm KH2PO4, 0.34 mm NaH2HPO4, 20 mm Hepes (CBFHH solution), and the atria removed. The hearts were quartered, and incubated in a solution containing CBFHH + collagenase (1.42 mg ml−1) for 10 min at 37°C, followed by incubation with CBFHH + papain (20.3 U ml−1) for 5 min at 37°C. The tissue was triturated 10–15 times, horse serum added to stop digestion, and cells pelleted by spinning at 160 g at room temperature (RT) for 3 min. The cells were re-suspended in medium, incubated in a culture dish at 37°C for 1 h, re-pelleted at 1000 r.p.m. at RT for 3 min, re-suspended in medium, plated onto 4 mm × 4 mm glass coverslips coated with poly-l-lysine and laminin, and incubated in a humidified incubator at 37°C (5% CO2). SCG neurons were prepared from 7- to 14-day-old Sprague–Dawley rats as previously described (Bernheim et al. 1991; Shapiro & Hille, 1993), placed on top of the cardiomyocyte cultures, and cultured for 2–4 days.
Immunostaining
Cardiomyocytes grown on poly-l-lysine-coated coverslips were fixed in 4% paraformaldehyde, washed twice with 100 mm sodium phosphate (PB, pH 7.4), three times with PB + 150 mm NaCl (PBS), and blocked with 5% goat serum and 0.1% saponin in PBS (PBS–GS). The cells were incubated for 3 h at room temperature with primary affinity-purified mouse anti-B2R (BD Biosciences) and rabbit anti-P2Y2 (Neuromics) antibodies, diluted 1:500 in PBS–GS. For the peptide controls, the primary antibodies were pre-adsorbed with a tenfold excess of the immunizing peptides used to raise the antibodies. Cells were washed six times with PBS and then incubated with goat fluorescein isothiocyanate (FITC)-conjugated anti-mouse or anti-rabbit secondary antibodies (1:500, Jackson Immunoresearch) in PBS–GS for 1 h. Cells were then washed three times with PBS, twice with PB, and three times with water. Air-dried slides were mounted on a drop of Vectashield (Vector Laboratory) and sealed with nail polish. Stained cells were viewed with an Olympus FV1000 confocal microscope, using the 488 nm line of an argon laser, and excitation/emission filters appropriate for FITC.
Contraction rate imaging
The co-culture experiments were performed on an inverted Nikon Diaphot microscope equipped with a temperature control system (Warner Instruments) to maintain the preparation at 37°C. Cell beating was observed via a Nikon PowerHAD video camera fed to a computer via a Pinnacle video transfer adapter, and the video captured with Windows Movie Maker. The contraction rate was quantified manually in 15 s intervals. Drugs were applied using a gravity-fed chamber perfusion, controlled by solenoid valves.
Results
Controls and parameters of the approach
To examine the functional role of M-current in the release of NA from SCG neurons, we used as a model the adrenergic response of cardiomyocytes co-cultured with SCG cells. Dissociated SCG neurons plated onto coverslips pre-plated with dissociated ventricular cardiomyocytes have been shown to regulate the contraction rate (CR) of the cardiomyocytes due to release of NA and stimulation of cardiac β-ARs (Shcherbakova et al. 2007). Hence, in our assay, we stimulated the neurons and monitored the cardiomyocyte CR in response. The excitatory receptors in sympathetic ganglia are nicotinic ACh receptors (nAChRs). Thus, their stimulation should trigger NT release from the nerve terminals of the SCG cells. We first performed several control experiments to test our overall approach. Since we intended to stimulate the nAChRs on the SCG cells using nicotine, we first confirmed that nicotine does cause a large inward current through stimulation of nAChRs. Figure 1A shows an example of a perforated-patch voltage-clamp experiment on a rat SCG cell. Since we also wanted to rule out any effect of nicotine on M-current, we used the indicated ‘classic’ M-current voltage-clamp protocol. Bath-application of nicotine (1 μm) induced a large inward current that desensitized within 30 s, consistent with the properties of nAChRs. However, the large amplitude of the elicited nAChR current precluded any meaningful quantification of M-current during nicotine application. Finally, in several experiments in this paper, we compare the effects of application of nicotine alone to those from co-application of nicotine together with the Gq/11-coupled receptor agonists BK or UTP. Thus, in this experiment, we tested whether co-application of BK would alter the nAChR current elicited by nicotine. However, the current response to nicotine + BK was similar to that to nicotine alone, only somewhat smaller in amplitude, consistent with residual desensitization of the nAChRs. We then tested whether nicotine stimulation of the SCG neurons would increase the contraction rate (CR) of the co-cultured cardiomyocytes. Figure 1B shows that bath application of nicotine (1 μm) induced a robust increase in the CR of the cardiomyocytes that was fully repeatable. However, when nicotine was co-applied with the β-AR blocker propranolol (10 μm), there was no significant effect of nicotine on the CR (Fig. 1C). These data are summarized in Fig. 1D. Normalized to controls (1.00 ± 0.06, n = 4), the fold-increase in the CR by the initial nicotine application, the second nicotine application and co-application of nicotine + propranolol was 1.43 ± 0.05 (P < 0.001, n = 9), 1.40 ± 0.05 (P < 0.001, n = 4), and 1.12 ± 0.06 (not significant at P = 0.05, n = 5), respectively. Thus, we conclude that nicotine increases the cardiomyocyte CR in these co-cultures by stimulation of the nAChRs, and excitation of the SCG neurons, with consequential release of NA and stimulation of the β-ARs of the cardiomyocytes.
Figure 1. Nicotine stimulates nAChRs in SCG neurons, and increases the CR of co-cultured cardiomyocytes in a β-AR-dependent manner.
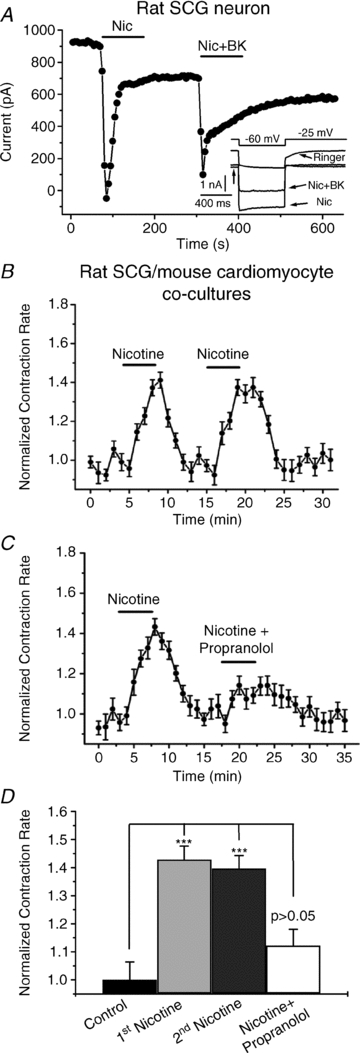
A, current amplitudes at –25 mV (arrow) are plotted from an SCG neuron studied under perforated-patch voltage-clamp, using a ‘classic’ M-current voltage protocol. Nicotine (Nic, 1 μm) or Nic + BK(250 nm) were bath-applied during the periods shown by the bars. B and C, the CR during an experiment is plotted as nicotine (1 μm), or nicotine + propranolol (10 μm) were bath-applied during the periods shown by the bars. Each point summarizes 4–5 measurements, with the SEM for each point shown. D, bars show summarized data from (B and C) showing the repeatability of the nicotine action, and its dependence upon β-AR stimulation. ***P < 0.001. The increase in CR by nicotine + propranolol is not significant at the P = 0.05 level.
We wished to assay the effects of both up- and down-regulation of M-current activity on the CR in the co-cultures. For the former, we used the anti-convulsant retigabine (RTG), which strongly increases the activity of M-channels (Wickenden et al. 2000), but is inactive against the KCNQ1-containing channels that underlie cardiac IKs (Tatulian et al. 2001; Schenzer et al. 2005). In a variety of neurons, including in SCG, RTG decreases excitability, action-potential firing and release of NT (Hernandez et al. 2008; Maljevic et al. 2008). Both of the M-channel blockers, linopirdine and XE991, also block KCNQ1-containing channels (Zaczek et al. 1998), making them unsuitable for our studies here. Thus, we tested the effect on cardiomyocyte beating of M-current depression by stimulation of the B2 and P2Y receptors of the SCG neurons, using simultaneous application of nicotine and BK, or nicotine and UTP, as B2 and P2Y-receptor agonists that we know inhibit M-channels, but not VGCCs. However, it is important to tease apart effects of agonists on the Gq/11-coupled receptors of the neurons vs. those of the cardiomyocytes. Since mouse cardiomyocytes express the same B2 receptors as do SCG cells, and the cardiac P2Y2 receptors of heart cells will also respond to UTP as do the P2Y6 receptors of SCG cells (Emanueli et al. 1999; Burnstock & Knight, 2004), we must use some strategy to isolate the effects of receptor stimulation to the receptors of the neurons, not of the cardiomyocytes. Our approach was to exploit B2 and P2Y2 receptor k/o mice. To verify that the heart cells from these mice do indeed lack B2 or P2Y2 receptors, we performed immunostaining of cardiomyocytes isolated from B2 and P2Y2 receptor k/o, or wild-type, mice, using specific anti-B2 receptor or anti-P2Y2-receptor primary antibodies. Slide-mounted fixed cells were imaged under confocal microscopy. Figure 2A shows fluorescence images of cardiomyocytes from wild-type, B2 receptor k/o or P2Y2 receptor k/o mice, demonstrating robust labelling of B2 and P2Y2 receptors in cells from wild-type mice, but no expression in cells from the corresponding k/o mice. As controls, when the primary antibodies were omitted, or pre-adsorbed with the immunizing peptide used to raise the antibodies, there was no labelling. Thus, the B2 and P2Y2 k/o mice should be suitable for our studies.
Figure 2. Knock-out mouse control experiments.
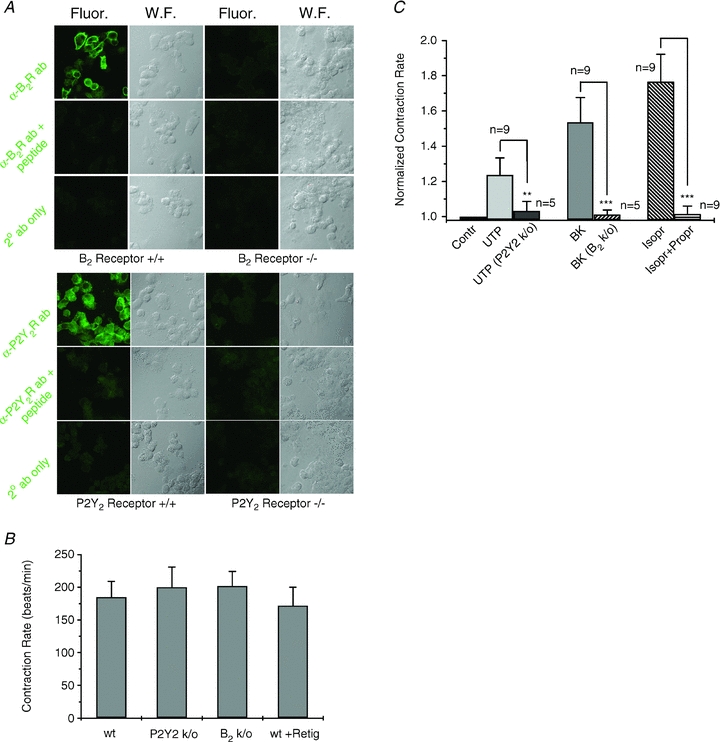
A, confocal images of cardiomyocytes from wild-type (+/+) or B2R or P2Y2R knock-out (−/−) animals. Fluorescence (Fluor) or transmitted light wide-field (W.F.) micrographs are shown from cells treated with the indicated antibody (ab), the ab pre-adsorbed with the immunizing peptide, or secondary (2°) ab only. B, bars are the tonic contraction rate of cardiomyocytes-only cultured from wild-type (wt), P2Y2R k/o, or B2R k/o mice, or wt cells in the presence of retigabine (10 μm). C, bars show the normalized effect on the contraction rate of a cardiomyocyte-only preparation, in response to application of UTP (10 μm) to wt or P2Y2R k/o cells, BK (250 nm) on wt or B2R k/o cells, and isoproterenol (Isopr, 10 μm) or Isopr + propranalol (Propr, both 10 μm) on wt cells. **P < 0.01, ***P < 0.001.
As a verification of this approach, we tested the effect of BK and UTP on the CR of wild-type, B2 or P2Y2 receptor k/o mouse cardiomyocytes only (without SCG neurons) and the effect of RTG on the cardiomyocytes. There were no differences between the tonic CR of cardiomyocytes from wild-type and either type of k/o mouse and RTG had no effect on the CR of cardiomyocytes from wild-type mice (Fig. 2B). Application of UTP or BK significantly increased the CR of cardiomyocytes from control mice, but those responses were completely absent in cardiomyocytes from B2 or P2Y2 receptor k/o mice, respectively (Fig. 2C). As a test of the maximal effect of adrenergic stimulation, we applied the β-AR receptor agonist isoproterenol (Isopr, 10 μm), which causes the maximal increase in CR that can be achieved by β-AR stimulation. This increase was fully reversed by application of the β-AR receptor antagonist, propranolol (Propr, 10 μm). Normalized to control, the fold-increase by UTP on the CR of cells from wild-type or P2Y2 k/o mice was 1.24 ± 0.10 (n = 9) and 1.03 ± 0.05 (n = 5, P < 0.01), respectively; that by BK on the CR of cells from wild-type or B2 k/o mice was 1.54 ± 0.14 (n = 9) and 1.01 ± 0.02 (n = 5, P < 0.001), respectively, and that by Isopr or Isopr + Propr on the CR of cells from wild-type mice was 1.76 ± 0.15 (n = 9) and 1.02 ± 0.04 (n = 9, P < 0.001), respectively. Thus, we have a system for manipulating M-current in the SCG neurons, in which applied drugs or agonists do not directly affect the CR of the heart cells taken from the appropriate mouse line.
Agonists of B2 or P2Y receptors, and RTG, have opposing effects on nicotine-induced stimulation of CR
We then began to examine the role of M-channels in controlling the response to nicotine stimulation of the SCG neurons. Figure 3A shows an example of an experiment in which rat SCG neurons were co-cultured with cardiomyocytes isolated from B2-receptor k/o mice. The tonic CR was ∼180 beats min−1, which was reversibly increased to almost 230 beats min−1 by application of nicotine (1 μm), presumably due to stimulation of the SCG neurons, and release of NA. However, when nicotine was co-applied with retigabine (10 μm), the increase in CR induced by nicotine was markedly reduced, suggesting that RTG-induced up-regulation of M-current prevented most NA release from the SCG cells. Conversely, when BK (250 nm) was co-applied with nicotine, the increase in CR was much greater than by nicotine alone, to ∼250 beats min−1. Finally, we calibrated the system by application of Isopr (10 μm), resulting in an increase in the CR to ∼275 beats min−1. That this effect is mediated by β-ARs is demonstrated by its full blockade when Isopr was co-applied with propranolol (10 μm). Figure 3B summarizes all such experiments in which RTG action was tested, and Fig. 3C summarizes all such experiments in which BK action was tested, with the mean ± SEM given for each 15 s time point during the experiments. We then performed similar experiments on co-cultures of rat SCG neurons and cardiomyocytes isolated from P2Y2-receptor k/o mice. A representative experiment is shown in Fig. 4A. Again, nicotine application significantly increased the cardiomyocyte CR, and the response was augmented when nicotine was co-applied with UTP (10 μm). The response was again calibrated by Isopr application, and also was fully blocked by co-application of Propr. Such data are summarized in Fig. 4B, with the mean ± SEM given for each 15 s time point during the experiments.
Figure 3. Retigabine and bradykinin affect the chronotropic response of the cardiomyocytes to nicotine stimulation of the co-cultured SCG neurons.
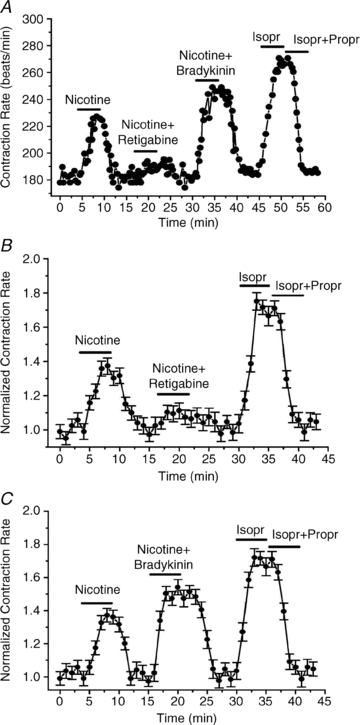
A, CR is plotted during an experiment as nicotine (1 μm), nicotine (1 μm) + retigabine (10 μm), bradykinin (250 nm), Isopr (10 μm), or Isopr + Propr (both 10 μm) were bath-applied to co-cultured rat SCG and B2 k/o mouse cardiomyocytes. B, summarized experiments as in A, in co-cultured rat SCG and wt mouse cardiomyocytes, showing the inhibitory effect of retigabine. C, summarized experiments as in A, in co-cultured rat SCG and B2 k/o mouse cardiomyocytes, showing the potentiating effect of bradykinin on the response to nicotine. In B and C, each point summarizes 4–5 measurements, with the SEM for each point shown.
Figure 4. UTP affects the chronotropic response of the cardiomyocytes to nicotine stimulation of co-cultured SCG neurons.
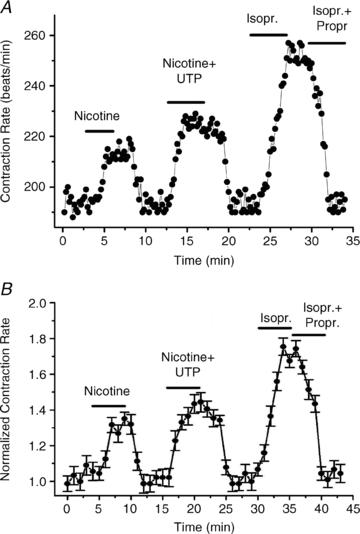
A, the contraction rate is plotted during an experiment as nicotine (1 μm), nicotine (1 μm) + UTP (10 μm), Isopr (10 μm), or Isopr + Propr (both 10 μm) were bath-applied to co-cultured rat SCG and P2Y2 k/o mouse cardiomyocytes. B, summarized experiments as in A, showing the potentiating effect of UTP on the response to nicotine. Each point summarizes 4–5 measurements, with the SEM for each point shown.
Figure 5 summarizes the normalized action of each treatment on the cardiomyocyte CR in the above co-culture experiments. The fold increases in CR by application of nicotine, nicotine + BK, nicotine + UTP, nicotine + RTG, Isopr or Isopr + Propr were 1.33 ± 0.01 (n = 18), 1.55 ± 0.05 (n = 6, P < 0.001), 1.44 ± 0.05 (n = 5, P < 0.05), 1.09 ± 0.05 (n = 7, P < 0.001), 1.75 ± 0.05 (n = 18) and 1.03 ± 0.04 (n = 18), respectively. It is important to note that the effects of RTG and BK must be due to actions on the neurons, and not the cardiomyocytes, since the latter do not contain B2 receptors, and their IKs channels are not sensitive to RTG. Thus, release of NA from the SCG cells induced by nicotine is enough to stimulate the heart cells about half of that maximally possible, as defined by Isopr application. The nicotine-induced NA release is highly dependent on M-current amplitude, since the CR response was mostly blunted when M-channels were maximally opened by co-application of RTG, but was augmented when many M-channels were closed by inclusion of BK or UTP. We conclude that M-channels control the release of NT at functional adrenergic synapses, and that M-current in sympathetic ganglia is very likely to affect cardiac function through innvervation of the heart by sympathetic nerves.
Figure 5. Summary of the normalized action of each treatment on the cardiomyocyte CR in the co-culture experiments.
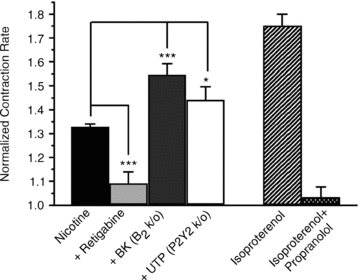
Bars show effect of the indicated treatment on the cardiomyocyte contraction rate, normalized to its value in control. All cells for the experiments labelled Nicotine, Retigabine, Isoproterenol and Isoproterenol + Propranolol were cultured from wild-type rodents. *P < 0.05, ***P < 0.001.
Discussion
Most studies on control of NT release by M-current have used more reduced preparations such as tritiated NT released from synaptosomes (Martire et al. 2004, 2007; Luisi et al. 2009), sympathetic neurons in a culture dish (Lechner et al. 2003, 2004; Edelbauer et al. 2005; Kubista & Boehm, 2006; Kubista et al. 2009), or carbon-fibre amperometry (Koh & Hille, 1997; Hernandez et al. 2008). Although these studies have yielded important and persuasive information, they might not represent what transpires at functional synapses or from the results of more physiological stimulation. Since our work has focused on signalling pathways in sympathetic neurons, we desired to investigate this issue in the milieu of a prototypic tissue that is regulated by sympathetic innervation. Previous work indicates that the adrenergic synapses formed in the co-culture contain much of the same organization of such synapses in vivo, including multiple exocytotic machinery proteins in the presynaptic terminals, and accumulations of β-ARs and the scaffolding proteins SAP97 and AKAP79/150 in the postsynaptic cardiomyocyte membranes (Shcherbakova et al. 2007). We realize that the heart is not innervated by neurons of the SCG in vivo (other sympathetic ganglia do so), and that the region of the heart primarily responsible for pacemaking is the sino-atrial node, not the ventricles as used here; however, this co-culture preparation is challenging to perfect, and we felt it necessary to exploit this carefully worked-out preparation that presents an ideal model preparation to answer the questions addressed in this paper.
For the purposes of this study, it is fortuitous that the B2 and P2Y types of Gq/11-coupled receptors in the SCG neurons selectively inhibit M-channels, without any action on VGCCs, since Ca2+ influx through VGCCs is the main driver for exocytosis and NT release in neurons. Although it would have been very desirable to depress M-current via stimulation of the muscarinic receptors that give M-current its name, there seemed no straightforward way to do this. Certainly we could treat the cultures with pertussis toxin, which would selectively block the ‘fast,’ voltage-dependent actions of Go/i-coupled M2 and M4 receptors (Hille, 1994), or use mice with the M2 receptors that dominate in rodent heart knocked-out, but still the same Gq/11-coupled M1 receptors responsible for M-current depression are also responsible for the slower, PIP2-mediated, depression of VGCCs that would obfuscate the results (Shapiro et al. 1999; Gamper et al. 2004; Suh et al. 2010). Indeed, Koh & Hille (1997), using carbon-fibre amperometry on cultured SCG cells, found stimulated NA release to be unaltered by muscarinic agonist applied to cells pre-treated with pertussis toxin, probably because the increased excitability caused by M-current depression was counterbalanced by the ‘slow-pathway’ mechanism of VGCC inhibition. However, since RTG does not affect KCNQ1-containing channels, we could selectively augment M current in the neurons, without affecting cardiac IKs, and we found RTG to have no effect by itself on the CR of cardiomyocyte-only cultures. We also note that the observed CR was similar in cardiomyocyte-only cultures and in the co-cultures, suggesting that sympathetic release from resting neurons in this system does not seem to participate in the tonic regulation of CR.
M-channels have gained much notoriety for their critical role in the nervous system. However, the function of M-type channels in cardiac function demonstrated here adds to the recent literature documenting the powerful role of KCNQ channels in control over the cardiovascular system. M-channels are well expressed in nodose ganglia sensory neurons, whose input reflects the status of visceral organs in the thorax, and baroreceptor function, and whose output is necessary for proper homeostasis (Wladyka & Kunze, 2006; Wladyka et al. 2008). In nodose ganglia neurons, the same collection of Gq/11-coupled M1, B2, P2Y and AT1 receptors are expressed as in SCG neurons, and agonists of these four receptors likewise depress M-current in those cells (O. Zaika, G. Tolstykh, J. Zhang & M. S. Shapiro, unpublished observations), making it likely that receptor-mediated regulation of M-channels play a part in central cardiovascular regulation. We also find M-currents in the nucleus of the tractus solitarius, which integrates vital cardiac, respiratory and gastrointestinal inputs and is a main projection site of nodose ganglia afferents in the brain stem (G. Tolstykh & M. S. Shapiro, unpublished observations). Recently, a powerful role of KCNQ4 and KCNQ5 channels has been documented in vascular smooth muscle cells (VSMCs), where they help set the resting potential, and control the extent of depolarization, much like their role in stabilization of the negative resting potential in neurons (Mackie & Byron, 2008). In addition, the mechanism of vasopressin in constriction of blood vessels has been revealed to be heavily dependent on phosphorylation of KCNQ4 and KCNQ5 in VSMCs. Taken together, M-channels in the sympathetic system, in visceral sensory neurons, and in the vasculature appear to represent powerful and as yet untapped novel targets for cardiovascular therapeutics (Mackie et al. 2008), and M-currents may emerge as equally important players in cardiovascular disease, as they have been in syndromes of nervous dysfunction.
Acknowledgments
We thank Pamela Reed for expert technical assistance. This work was supported by NIH grants R01 NS43394 and ARRA R01 NS065138 to M.S.S.
Glossary
Abbreviations
- β-AR
β-adrenergic receptor
- BK
bradykinin
- CR
contraction rate
- Isopr
isoproterenol
- k/o
knockout
- NA
noradrenaline
- nAChR
nicotinic ACh receptor
- NT
neurotransmitter
- Propr
propranolol
- RTG
retigabine
- SCG
superior cervical ganglion
- SN
sympathetic neuron
- VGCC
voltage-gated Ca2+ channel
- wt
wild-type
Author contributions
O.Z. and J.Z. conceived, designed and performed the experiments, and analysed the data, and M.S.S. wrote the manuscript. All authors approved the final version of the manuscript.
Author's present address
O. Zaika: Univ. Texas Health Science Center-Houston, Dept of Integrative Biology and Pharmacology, 6431 Fannin St, MSB 4.220, Houston, TX 77030, USA. Email: oleg.l.zaika@uth.tmc.edu
References
- Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- Conforti L, Tohse N, Sperelakis N. Influence of sympathetic innervation on the membrane electrical properties of neonatal rat cardiomyocytes in culture. J Dev Physiol. 1991;15:237–246. [PubMed] [Google Scholar]
- Constanti A, Brown DA. M-currents in voltage-clamped mammalian sympathetic neurones. Neurosci Lett. 1981;24:289–294. doi: 10.1016/0304-3940(81)90173-7. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Edelbauer H, Lechner SG, Mayer M, Scholze T, Boehm S. Presynaptic inhibition of transmitter release from rat sympathetic neurons by bradykinin. J Neurochem. 2005;93:1110–1121. doi: 10.1111/j.1471-4159.2005.03084.x. [DOI] [PubMed] [Google Scholar]
- Emanueli C, Maestri R, Corradi D, Marchione R, Minasi A, Tozzi MG, Salis MB, Straino S, Capogrossi MC, Olivetti G, Madeddu P. Dilated and failing cardiomyopathy in bradykinin B2 receptor knockout mice. Circulation. 1999;100:2359–2365. doi: 10.1161/01.cir.100.23.2359. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, Landis SC, Matsumoto SG, Potter DD. Synaptic functions in rat sympathetic neurons in microcultures. I. Secretion of norepinephrine and acetylcholine. J Neurosci. 1986a;6:1061–1079. doi: 10.1523/JNEUROSCI.06-04-01061.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci U S A. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, Potter DD, Matsumoto SG. Synaptic functions in rat sympathetic neurons in microcultures. III. A purinergic effect on cardiac myocytes. J Neurosci. 1986b;6:1099–1107. doi: 10.1523/JNEUROSCI.06-04-01099.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer and Associates; 2001. [Google Scholar]
- Homolya L, Watt WC, Lazarowski ER, Koller BH, Boucher RC. Nucleotide-regulated calcium signaling in lung fibroblasts and epithelial cells from normal and P2Y2 receptor (–/–) mice. J Biol Chem. 1999;274:26454–26460. doi: 10.1074/jbc.274.37.26454. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ. KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci U S A. 2000;97:4333–4338. doi: 10.1073/pnas.97.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Hille B. Modulation by neurotransmitters of catecholamine secretion from sympathetic ganglion neurons detected by amperometry. Proc Natl Acad Sci U S A. 1997;94:1506–1511. doi: 10.1073/pnas.94.4.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- Kubista H, Boehm S. Molecular mechanisms underlying the modulation of exocytotic noradrenaline release via presynaptic receptors. Pharmacol Ther. 2006;112:213–242. doi: 10.1016/j.pharmthera.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Kubista H, Kosenburger K, Mahlknecht P, Drobny H, Boehm S. Inhibition of transmitter release from rat sympathetic neurons via presynaptic M1 muscarinic acetylcholine receptors. Br J Pharmacol. 2009;156:1342–1352. doi: 10.1111/j.1476-5381.2009.00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Dorostkar MM, Mayer M, Edelbauer H, Pankevych H, Boehm S. Autoinhibition of transmitter release from PC12 cells and sympathetic neurons through a P2Y receptor-mediated inhibition of voltage-gated Ca2+ channels. Eur J Neurosci. 2004;20:2917–2928. doi: 10.1111/j.1460-9568.2004.03760.x. [DOI] [PubMed] [Google Scholar]
- Lechner SG, Mayer M, Boehm S. Activation of M1 muscarinic receptors triggers transmitter release from rat sympathetic neurons through an inhibition of M-type K+ channels. J Physiol. 2003;553:789–802. doi: 10.1113/jphysiol.2003.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart ST, Turrigiano GG, Birren SJ. Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci. 1997;17:9573–9582. doi: 10.1523/JNEUROSCI.17-24-09573.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisi R, Panza E, Barrese V, Iannotti FA, Viggiano D, Secondo A, Canzoniero LM, Martire M, Annunziato L, Taglialatela M. Activation of pre-synaptic M-type K+ channels inhibits [3H]d-aspartate release by reducing Ca2+ entry through P/Q-type voltage-gated Ca2+ channels. J Neurochem. 2009;109:168–181. doi: 10.1111/j.1471-4159.2009.05945.x. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, Byron KL. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie AR, Byron KL. Cardiovascular KCNQ (Kv7) potassium channels: physiological regulators and new targets for therapeutic intervention. Mol Pharmacol. 2008;74:1171–1179. doi: 10.1124/mol.108.049825. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Wuttke TV, Lerche H. Nervous system Kv7 disorders: breakdown of a subthreshold brake. J Physiol. 2008;586:1791–1801. doi: 10.1113/jphysiol.2008.150656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch. 2010;460:277–288. doi: 10.1007/s00424-010-0831-3. [DOI] [PubMed] [Google Scholar]
- Martire M, Castaldo P, D'Amico M, Preziosi P, Annunziato L, Taglialatela M. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire M, D'Amico M, Panza E, Miceli F, Viggiano D, Lavergata F, Iannotti FA, Barrese V, Preziosi P, Annunziato L, Taglialatela M. Involvement of KCNQ2 subunits in [3H]dopamine release triggered by depolarization and pre-synaptic muscarinic receptor activation from rat striatal synaptosomes. J Neurochem. 2007;102:179–193. doi: 10.1111/j.1471-4159.2007.04562.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto SG, Sah D, Potter DD, Furshpan EJ. Synaptic functions in rat sympathetic neurons in microcultures. IV. Nonadrenergic excitation of cardiac myocytes and the variety of multiple-transmitter states. J Neurosci. 1987;7:380–390. doi: 10.1523/JNEUROSCI.07-02-00380.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroz D, Rodriguez N, Choveau F, Baro I, Merot J, Loussouarn G. Kv7.1 (KCNQ1) properties and channelopathies. J Physiol. 2008;586:1785–1789. doi: 10.1113/jphysiol.2007.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter DD, Landis SC, Matsumoto SG, Furshpan EJ. Synaptic functions in rat sympathetic neurons in microcultures. II. Adrenergic/cholinergic dual status and plasticity. J Neurosci. 1986;6:1080–1098. doi: 10.1523/JNEUROSCI.06-04-01080.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, Schwake M. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Hille B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron. 1993;10:11–20. doi: 10.1016/0896-6273(93)90237-l. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc Natl Acad Sci U S A. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova OG, Hurt CM, Xiang Y, Dell'Acqua ML, Zhang Q, Tsien RW, Kobilka BK. Organization of β-adrenoceptor signaling compartments by sympathetic innervation of cardiac myocytes. J Cell Biol. 2007;176:521–533. doi: 10.1083/jcb.200604167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Leal K, Hille B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M- type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- Wladyka CL, Feng B, Glazebrook PA, Schild JH, Kunze DL. The KCNQ/M-current modulates arterial baroreceptor function at the sensory terminal in rats. J Physiol. 2008;586:795–802. doi: 10.1113/jphysiol.2007.145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Fisher BN, Rominger DH, Earl RA. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther. 1998;285:724–730. [PubMed] [Google Scholar]
- Zaika O, Tolstykh GP, Jaffe DB, Shapiro MS. Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y-receptor regulation of neuronal ion channels. J Neurosci. 2007;27:8914–8926. doi: 10.1523/JNEUROSCI.1739-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Zhang J, Shapiro MS. Combined phosphoinositide and Ca2+ signals mediating receptor specificity toward neuronal Ca2+ channels. J Biol Chem. 2011;286:830–841. doi: 10.1074/jbc.M110.166033. [DOI] [PMC free article] [PubMed] [Google Scholar]