

Abstract
Background
The border zone of healing myocardial infarcts is an arrhythmogenic substrate partly due to structural and functional remodeling of the ventricular gap junction protein, Connexin43 (Cx43). Cx43 in arrhythmogenic substrates is a potential target for antiarrhythmic therapy.
Methods and Results
We characterized Cx43 remodeling in the epicardial border zone (EBZ) of healing canine infarcts, 5 days after coronary occlusion and examined whether the gap junction specific agent, Rotigaptide, could reverse it. Cx43 remodeling in the EBZ was characterized by a decrease in Cx43 protein, lateralization and increased Cx43 phosphorylation at serine (S) 368. Rotigaptide partially reversed the loss of Cx43 but did not affect the increase in S368 phosphorylation nor did it reverse Cx43 lateralization. Rotigaptide did not prevent conduction slowing in EBZ nor did it decrease the induction of sustained ventricular tachycardia (SMVT) by programmed stimulation, although it did decrease the EBZ effective refractory period (ERP).
Conclusions
We conclude that partial reversal of Cx43 remodeling in healing infarct border zone may not be sufficient to restore normal conduction or prevent arrhythmias.
Keywords: myocardial infarction, arrhythmias, gap junctions, remodeling
Introduction
The surviving rim of myocardium on the epicardial surface of healing transmural canine infarcts (epicardial border zone (EBZ)) is an arrhythmogenic substrate1. Gap junction remodeling characterized by lateralization of Cx43 contributes to genesis of reentrant arrhythmias in the EBZ2.
In addition to structural remodeling, functional remodeling of gap junctions, characterized by changes in the phosphorylation state of Cx43, is associated with pathology3. Phosphorylation at S325, S328, and S330 in the early gap junction life cycle is necessary for assembly of gap junction plaques4 and phosphorylation at S364 or S365, is important for trafficking to the plasma membrane5. Some of these sites have also been implicated in pathogenesis as their phosphorylation status is altered during acute ischemia and heart failure3,6,7 possibly leading to a decrease in Cx43. One additional site of interest is S368 which becomes phosphorylated during cellular stress that may lead to a decrease in gap junction conductance8. Phosphorylation of Cx43 has not been studied in healing infarct border zone (EBZ). In this study we further characterized Cx43 remodeling in EBZ by measuring Cx43 quantity and the involvement of phosphorylation at one site, S368.
It has been proposed that prevention or reversal of gap junction remodeling may be an effective antiarrhythmic intervention. An antiarrhythmic effect of the compound ZP123 (Rotigaptide) has been demonstrated in acutely ischemic myocardium9,10. One mechanism proposed for Rotigaptide’s effect is altering phosphorylation of Cx43 at S36811. Therefore, we determined the effects of Rotigaptide on Cx43 remodeling, phosphorylation at S368 and the associated electrophysiological changes in the EBZ.
Methods
Canine Infarct Model
Anteroseptal myocardial infarction was produced in mongrel dogs1,2,12. The electrophysiological study was done 5 days later because gap junction remodeling in the EBZ as evidenced by lateralization is well established2, sustained monomorphic ventricular tachycardia (SMVT) can be initiated by programmed electrical stimulation (PES)1 and conduction velocity and refractoriness of the remodeled regions can be determined 1,2,12,13. Electrophysiological measurements were made before and after intravenous Rotigaptide or saline infusion for 1 or 3 hours. (See Online Supplement).
Measurement of Na+ Current
Whole cell voltage clamp techniques were used to determine the effects of Rotigaptide on INa of myocytes dispersed from 5day EBZ using previously described methods14. In one series of experiments, drug was infused into the cell chamber and Na+ current measured before and after drug. In another series, Na+ current was measured after infusion of the drug in vivo for 3 hours.
Western Blot Analysis for Protein Levels
Tissue samples were obtained from EBZ of infarcted hearts with or without drug treatment, and from a remote non infarcted region on the posterior left ventricle of infarcted hearts, after the electrophysiological experiment was complete. Tissue was also taken from the left ventricular epicardium of 5 non infarcted hearts. Western blot was run as previously described 15 (See Online supplement).
Immunohistochemistry
Rapidly frozen epicardial samples were sectioned (15 microns), fixed in 4% paraformaldehyde then immunostained as previously described15 (See Online Supplement)
Statistical Methods
Data are presented as means ± SE. Effective refractory period data were tested using a repeated measures ANOVA. If significant changes occurred, means were compared using Bonferroni’s method. P < 0.05 was significant. Conduction velocity was analyzed using a paired T Test with a significance level of 0.05. Detailed statistical methods are described in the online supplement.
Results
Characteristics of Cx43 Remodeling in EBZ and Effects of Rotigaptide
Western blot analysis showed that total Cx43 levels normalized for total protein which represents mainly myocytes (see Online Supplement), were decreased in the EBZ (Figure 1A). Rotigaptide caused a significant increase in Cx43 after 1 hour, but Cx43 remained significantly less (p<0.05) than normal. Additional treatment (3 hours) did not augment this increase. Phosphorylation of Cx43 on the serine residue 368 (pS368) occurs via activation of PKC. Figure 1B shows that in normal canine epicardial tissue the amount of PKC is very low. PKC is significantly increased in the EBZ and Rotigaptide did not reverse this increase (Figure 1B). In normal epicardium, the ratio of pS368 Cx43/total Cx43 is very low (Fig 1C). In the EBZ phosphorylation at this residue was markedly increased (Fig 1C), and this increase remained significant after Rotigaptide (Figure 1C).
Figure 1.

Cx43 protein (A), PKC (B) and Cx43 pS368 (C) in EBZ and effects of Rotigaptide. Representative Western blots are shown at the left. Data (mean values +/− S.E. n=) are shown for normal hearts (no infarct), 5 day infarcts with saline only infusion (b), 5 day infarct with either 1 (c) or 3 (d) hour Rotigaptide infusion. Statistically significant changes indicated by horizontal lines with asterisks above each panel (N=4, p<0.05).
Cx43 was also analyzed in epimyocardial samples from non infarcted remote sites on the posterior wall from the same set of experiments. Cx43 levels were not significantly reduced (p>0.05). There was no significant change in Cx43 in the remote region after Rotigaptide (Data not shown).
A hallmark of Cx43 remodeling in EBZ is lateralization2. To determine if Rotigaptide reversed it, we did immunostaining (Figure 2) for Cx43 (red) using the scaffolding protein ZO-1 (green) as a marker of the intercalated disk. Myocytes in the EBZ exhibited lateralization of Cx43 (compare Cx43 norm and 5 day) while ZO-1 remained at the ID (compare ZO-1 norm and 5 day). The overlay image of Cx43 and ZO-1 shows that much of the Cx43 is no longer at the intercalated disk. Rotigaptide did not reverse the lateralization (compare 5 day, 1 hr and 3 hr). The overlay images indicate that there were many areas that still had lateralized Cx43 (white arrows, 1 and 3 hrs). The quantification of Cx43 lateralization shown in the bottom panel also shows a marked increase in lateralization at 5 days without reversal by Rotigaptide. Lateralization did not occur in the remote posterior left ventricle of the infarcted hearts.
Figure 2.
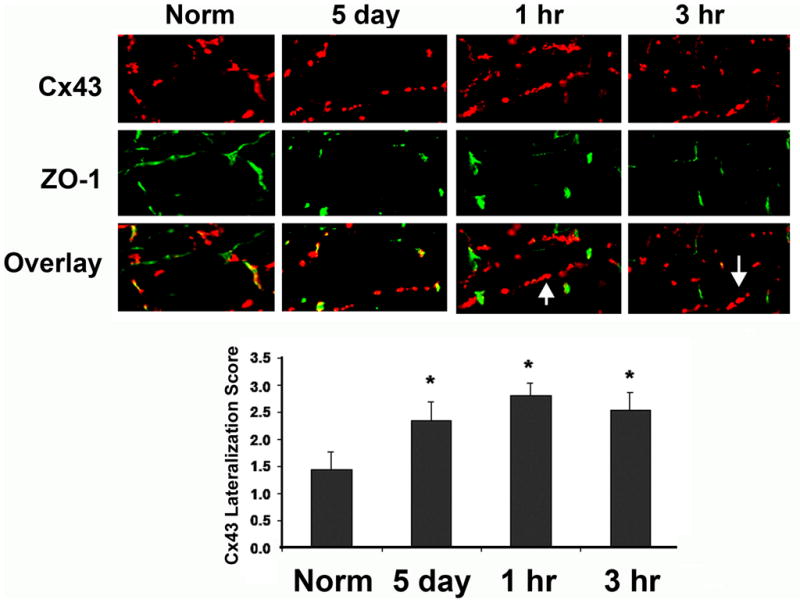
Immunofluorescent labeled myocytes for Cx43 (red) and ZO-1 (green). In normal hearts Cx43 is localized almost exclusively with ZO-1 at the intercalated disk (Norm) (overlay). After 5 day infarction lateralization of Cx43 is seen. Treatment with 1 hr or 3 hr Rotigaptide did not alter lateralization (arrows show areas of lateralized Cx43). Quantification of Cx43 on lateral membranes (not associated with ZO-1) showed no significant effect of Rotigaptide on lateralized Cx43, (N=3, Bar graphs below, mean values +/− S.E).
Effects of Rotigaptide on EBZ Conduction
Maps acquired during stimulation at the center of the large electrode array which were used to measure conduction velocities over most of the EBZ (“global” conduction velocity), were characterized by elliptical isochrones with the long axis parallel to the long axis of the myocardial fiber bundles. Figure 3 (Panels A and B) show maps from one experiment, activation pattern in control before drug in Panel A and after 3 hours of Rotigaptide infusion in Panel B. Table 1 shows the global conduction velocities calculated from the large electrode array, in the longitudinal (CVL) and transverse direction (CVT) and the anisotropic ratio for control and after 1 and 3 hours of Rotigaptide infusion. There were no significant changes in any of the measured parameters at both time periods (P>0.05). Two sets of measurements were made with the small high density electrode array (Figure 3 Panels C and D) to determine local CV at one and 3 hours. As compared with control (Figure 3C) there were no significant changes in either CVL longitudinal or CVT, or anisotropic ratios following 1 hr (Table 2) or 3 hrs of Rotigaptide infusion (Table 2, Figure 3D). Since there were only 3 experiments at 3 hours, the analysis of the 3 hour data may be underpowered (See Online Supplement). In experiments in which saline was infused for 3 hours, there were no significant changes in global CV or anisotropic ratio at 1 and 3 hours (control CVL 65±.09 cm/sec, CVT 28±.04 cm/sec, AR 2.40±0.6; 1 hour CVL 58 ±0.15 cm/sec, CVT 29 ±8 cm/sec, AR 2.11 ± 0.71; 3 hours CVL 52 ± 15 cm/sec, CVT 24 ± 9 cm/sec; AR 2.32 ± 0.84) (P>0.05 for all comparisons, n=4).
Figure 3.

Activation maps from stimulation of EBZ at the center of the large electrode array in A and B. 10 msec interval isochrones are represented by colors progressing from earliest activation at the site of stimulation (red) to latest activation (dark blue). Panel A shows a control map at a basic cycle length (S1-S1) of 300ms (CVL = 48 cm/s, CVT = 33 cm/s, AR = 1.39) and panel B after 3 hour infusion of Rotigaptide (CVL = 40 cm/s, CVT = 25 cm/s, AR = 1.68). Panels C and D show activation maps with high density array during stimulation from the center at a CL of 300ms. Color code is the same as in Panels A and B, small numbers indicate activation times. The long axes of the elliptical isochrones indicate longitudinal activation (CVL) while the short axes indicate transverse activation (CVT). Panel C shows activation map during control (CVL = 35 cm/s, CVT = 15 cm/s, AR = 2.19), Panel D shows activation map after 3 hours of Rotigaptide (CVL = 33 cm/s, CVT = 16 cm/s, AR = 2.18).
Table 1.
Effect of Rotigaptide on Global Conduction Velocity
| Control | 1 Hour | |
|---|---|---|
| Number | 6 | 6 |
| CVL(cm/s) | 59.6±4.0 | 56.3±3.0 |
| CVT(cm/s) | 32.8±4.0 | 32.9±4.3 |
| AR | 1.92± 0.21 | 1.8±0.18 |
| Control | 3 Hour | |
|---|---|---|
| Number | 4 | 4 |
| CVL(cm/s) | 51.1±5.89 | 54.3±6.79 |
| CVT(cm/s) | 21.4±3.26 | 21.2±2.8 |
| AR | 2.73±0.79 | 2.84±0.74 |
Table 2.
Effect of Rotigaptide on Local Conduction Velocity
| Control | 1 Hour | |
|---|---|---|
| Number | 4 | 4 |
| CVL(cm/s) | 42.74±3.12 | 46.09±2.51 |
| CVT(cm/s) | 26.08±1.94 | 29.42±2.86 |
| AR | 1.65± 0.10 | 1.59±0.08 |
| Control | 3 Hour | |
|---|---|---|
| Number | 3 | 3 |
| CVL(cm/s) | 38.64±1.74 | 41.90±2.83 |
| CVT(cm/s) | 21.41±5.52 | 19.84±5.05 |
| AR | 2.00±0.39 | 2.29±0.35 |
Effects of Rotigaptide on EBZ Effective Refractory Period
Rotigaptide significantly shortened ERP in the EBZ after 3 hours of perfusion (Figure 4A) (control 237.5 ± 4.8 msec; 1 hr 230.0 ± 4.1 msec; 2 hr 217.5 ± 2.5 msec, 3 hr 207.5 ± 4.8 msec (p<0.05 3hr vs control ERP and 1hr)). In the experiments in which saline was infused for 3 hours there was no significant change in ERP in the EBZ (control 165.0±22 msec; 2 hr 156.67±10.8 msec p>0.05; 3 hr 171.6 ±14.2 msec (p>0.05). Rotigaptide infusion also did not significantly alter the ERP measured at the right ventricular stimulation site (n=3) either at 1 hour (control 152± 5.8 msec, 1 hour 152±7.3 msec (P>0.05)) or 3 hours (control 155 ±7.6 msec, 3 hour 167 ± 15 msec; P>0.05) of infusion. Likewise there was no significant change in the ERP at the non infarcted right ventricular site in the saline experiments over a 3 hour infusion period (n=5, control ERP 165±7.6 msec; 3 hour infusion 183±12 msec; p>0.05).
Figure 4.

Panel A. Panel A. Effective refractory periods (EBZ-RP) in control before Rotigaptide infusion (Ctr circles;N=4), and 1 hour (1hr squares; N=4), 2 (2hr diamonds; N=4) and 3 (3hr triangles; N=4) after infusion. Data from each individual experiment are represented by a different color (black, red, yellow and green). Mean±SE at each time is represented by the unfilled symbols with error bars. *P<0.05 3hr vs Ctr and #P<0.05 3hr vs 1hr determined by repeated measures ANOVA/Bonferroni’s. Panel B shows activation of the earliest propagated premature impulse in control (S1–S2= 260 ms) and Panel C shows the activation of the earliest propagated premature impulse after 3 hours Rotigaptide infusion (S1–S2 = 220 ms), when ERP was decreased. Longitudinal activation (arrows) is similar before and after Rotigaptide.
As a result of ERP shortening, conduction of premature impulses was facilitated (Figure 4; Panels B and C). Panel B shows the conduction pattern of a control premature impulse with a coupling interval of 260 msec, the shortest coupled impulse that propagated in this experiment. Panel C shows conduction of a premature impulse at a coupling interval of 220 msec, the earliest propagated premature impulse after the 3 hour Rotigaptide infusion, indicating a decrease in the ERP of 40 msec. At this shorter coupling interval, conduction velocities in both longitudinal and transverse directions are similar to control velocities, at the 260 msec coupling interval. Similar effects were found in all other experiments.
Effects of Rotigaptide on SMVT
Rotigaptide did not have a consistent antiarrhythmic effect. In the 8 experiments in which Rotigaptide was infused for either 1 or 3 hours, SMVT was induced in 4 either from the EBZ or the RV stimulation site (Figure 5). SMVT was caused by reentrant excitation that was mapped in the EBZ (Fig 6A). Cycle length ranged from 170–300 msec. In 3 of these experiments in which there was a 3 hour infusion period, SMVT was still inducible at the end of the 3 hours (Figures 5) although the pattern of excitation of the EBZ sometimes changed (Figure 6). There were no or only small changes in the PES coupling values at which SMVT was induced after drug. In experiment 1, SMVT was induced in control with rapid pacing from the center site at a cycle length of 260 msec. After Rotigaptide 2 hours it was still induced at a pacing cycle length of 260 msec and after 3 hours at a pacing cycle length of 240 msec. In experiment 2, SMVT was induced in control from the right ventricular site with double extrastimuli; 280 BCL/170S2/165S3, and after 3 hours Rotigaptide at 280BCL/180S2/165S3 (ERP at the normal stimulation site increased by about 8 ms). In experiment 3, SMVT was induced from the right ventricular site by single premature stimuli; control 350 BCL/220-200S2, after 2 hr Rotigaptide 350BCL/220S2 and after 3 hours Rotigaptide 350BCL/190S2.
Figure 5.
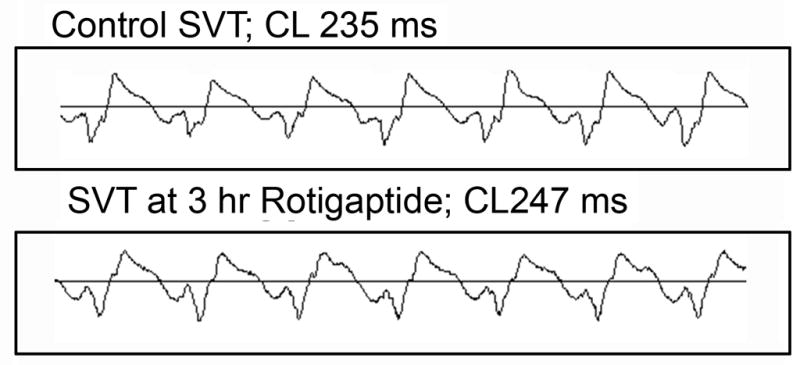
ECG recorded during control SMVT (top panel) and after 3 hour Rotigaptide infusion.
Figure 6.

Effects of Rotigaptide on a reentrant circuit causing SMVT. Each panel shows an activation map of the EBZ, isochrones at 10 msec intervals, arrows indicate direction of activation. Dark lines indicate regions of block. Panel A shows activation pattern in control. The pattern is a figure of eight reentrant circuit with a central common pathway between the two lines of block. In this central region of the EBZ there is no indication of transmural break through although at the periphery where the EBZ abuts non infarcted myocardium (within 100 msec isochrones) there is likely transmural activation. Panel B shows the activation pattern after 1 hour Rotigaptide infusion. The lines of block have shifted and the reentrant cycle length is prolonged because the direction of activation in the central common pathway has shifted from longitudinal, to transverse. Panel C shows the pattern of activation during the SVT induced after 2 hours of Rotigaptide infusion. The pattern is similar to control with minor shifts in block lines. Panel D shows activation of SVT after 3 hours of Rotigaptide infusion. A reentrant circuit is no longer mapped in the EBZ. The border zone is activated nearly simultaneously by multiple wave fronts moving from the margins toward the center. There is transmural breakthrough around the entire margin of the electrode array which overlies non infarcted myocardium.
SMVT cycle length decreased in two of these experiments (170 ms control to 160 ms Rotigaptide; 300 ms control to 230 ms Rotigaptide) and increased in one (235 ms control to 247 ms Rotigaptide). In the experiment shown in Figure 5, the increased cycle length despite a shortening of the ERP was related to a change in the activation pathway during reentry.
In the other (4th) experiment in which SMVT was induced in control, it could not be induced at one hour despite testing with the exact same PES protocol. Drug infusion was not continued beyond this time period. In two experiments in which SMVT could not be initiated in control, it was initiated after 1 and 3 hours of Rotigaptide infusion. SMVT was initiated in 2 of 4 saline experiments. In 1 experiment it could be re induced periodically for the 3 hours of saline infusion while in the other it could not be induced after 1–3 hours of saline infusion. In the other 2 experiments in which SMVT was not induced in control, it was also not induced after 3 hr saline infusion.
Effects of Rotigaptide on Remodeled Na+ currents of EBZ Myocytes
To determine if Rotigaptide might decrease Na+ current to offset possible effects of improved gap junction coupling on conduction, or alter recovery from inactivation to influence ERP, myocytes isolated from the EBZ were voltage clamped, peak INa was recorded14 and then the cells exposed to Rotigaptide (80nM) for 30 mins. There was no significant effect of drug on peak INa (n=4, predrug 4.01±0.63 pA/pF, +30 mins, 3.48±0.50pA/pF, Vh=−100mV) or recovery from inactivation (n=4) (data not shown). In an additional three myocytes dispersed from the EBZ which had been exposed to 3 hours of Rotigaptide infusion, we found peak INa (5.54±1.84 pA/pF) to be similar to previously published INa data for EBZ myocytes14. There was no evidence for a direct depressant effect of Rotigaptide on peak sodium current function.
Discussion
We investigated gap junction remodeling in EBZ and the possibility of reversing it to improve conduction and prevent arrhythmias using Rotigaptide as a pharmacological tool based on it increasing gap junction coupling during metabolic stress and acute ischemia9,10,16. We found a significant reduction of total Cx43 in EBZ with an increase in phosphorylation at residue S368, which has been associated with a decrease in gap junctional conductance17. In contrast to previous studies on acute ischemia9,10, Rotigaptide partially reversed Cx43 remodeling in EBZ by increasing Cx43. However, Rotigaptide did not affect conduction velocity nor prevent induction of SMVT although it did decrease the ERP.
Gap Junction Remodeling in Infarct Border Zones
Serine and tyrosine phosphorylation sites in the Cx43 molecule participate in functional regulation18. Tyrosine phosphorylation is associated with channel closure18. Acute ischemia leads to loss of Cx43 phosphorylation at S325, S328 and S330, accompanied by a decrease in Cx43 protein and lateralization of Cx434,6,7. Dephosphorylation at those sites and the decrease in protein, all contribute to decreased cell-to-cell coupling, manifested as an increase in resistivity within 15–20 min of acute ischemia19. Dephosphorylation of Cx43 at S364/365 also occurs rapidly within 5 min. Other sites that are dephosphorylated during acute ischemia are S297 and S30611,20 which may contribute to decreased coupling. In contrast, serine residue S368, becomes phosphorylated during acute ischemia as a result of activation of protein kinase C (PKC)5,17,21–23. This occurs only after dephosphorylation of a “gatekeeper” serine, S36524 which in its normal phosphorylated state inhibits phosphorylation of S368. Once S365 is dephosphorylated, S368 is accessible for PKC phosphorylation which causes decreased channel conductance23. This regulation is important for insuring that gap junctional conductance is not decreased by other signaling pathways that may activate PKC.
The above changes occur in acutely ischemic myocardium, much of which dies and forms the infarct core. Gap junction remodeling also occurs in infarct border zones which form arrhythmogenic regions1,2. During the healing phase of infarction, Cx43 lateralization persists in the EBZ2. Within several hours of coronary occlusion, the separation of Cx43 from ZO-1 that is caused by the decreased intracellular pH, is a primary step in the movement of Cx43 to the lateral membranes, probably leading to its eventual internalization and destruction15. Lateralization continues to be present at 5 days2,13. The increase in PKC that we measured in the EBZ at this time might lead to compromised Cx43 targeting/retention at intercalated disks resulting in lateralization and the marked decrease in Cx4322,25. The increased phosphorylation of the remaining Cx43 at S368 which has not been previously described in EBZ, may contribute to decreased gap junctional coupling26.
Relationship of Gap Junction Remodeling to EBZ Arrhythmogenic Electrophysiology
Cx43 remodeling in EBZ contributes, along with ion channel remodeling, to altered electrophysiology that leads to this site being an arrhythmogenic source. The EBZ in healing infarcts is characterized by non uniform anisotropic conduction1. Control conduction velocities in Tables 1 and 2 are in agreement with our previous studies12,13. Gap junction conductance in the transverse direction is decreased in EBZ myocytes despite an increase in lateralized Cx43 suggesting that lateralized Cx43 does not form functional gap junctions26. The decrease in conductance may be related to the decreased Cx43 and increased S368 phosphorylation that we show in this study. The 76% decrease in Cx43 quantity has been shown to slow conduction27. The precise relationship of the reduction in Cx43 protein to slowed conduction is not straight forward owing to a contribution from decreased Na+ current14 and structural changes (separation of myocardial fiber bundles by edema) that may disrupt gap junction connections28. The EBZ is also characterized by a prolonged ERP that facilitates block of premature impulses that initiate SMVT29. Computer modeling has shown that gap junction coupling can influence time course for repolarization in regions where intrinsic repolarization of myocytes is heterogeneous30–32, making repolarization and ERP more homogeneous33. Uncoupling of myocytes prolongs repolarization in these models and has been proposed to underlie ERP prolongation in the epicardium in heart failure where Cx43 is reduced34. Such uncoupling may contribute to prolongation of the ERP in the EBZ although remodeling of reactivation kinetics of Na+ currents also contributes to post repolarization refractoriness14.
Reversal or Prevention of Cx43 Remodeling as an Antiarrhythmic Intervention
Since gap junction remodeling contributes to arrhythmogenesis3, preventing the reduction in gap junction coupling that accompanies it, may have antiarrhythmic effects. Rotigaptide has been touted to do this based on experiments showing that it prevents the decrease in conduction velocity caused by acute ischemia and metabolic stress35–37 without affecting sarcolemmal ion channels16,37–39. This effect has been associated with the prevention of reentrant ventricular and atrial arrhythmias associated with acute ischemia9,10,40. It has been proposed that Rotigaptide exerts its effects by PKC activation to prevent dephosphorylation (or induce rephosphorylation) of Cx43 during acute ischemia39–41.
Emphasis has been placed on Rotigaptide-mediated phosphorylation at S368 which was found to be dephosphorylated in an acute global ischemia rat model11. These findings are contrary to studies cited above which showed enhanced phosphorylation of S368 in acute ischemia and the present study (Figure 1). We believe this discrepancy is due to the fact that total Cx43 is dramatically reduced in ischemia, thus overall levels of phosphoCx43 are reduced. The increase in phosphorylation at this residue then can only be discussed in terms of the percent of Cx43 that is phosphorylated at S368. In normal heart the percent of pS368/total Cx43 is quite low. In contrast, in the EBZ there is a dramatic increase in the percent of Cx43 that is phosphorylated at S368. This indicates that not only is total Cx43 decreased, the remainder of the Cx43 is primarily phosphorylated at S368.
Since Rotigaptide increases gap junction conductance42 and prevents acute ischemic arrhythmias9, we investigated whether the drug would have similar effects in healing infarction. Rotigaptide partially reversed Cx43 remodeling in EBZ as evidenced by increased Cx43 quantity at one hour (Figure 1), an effect not previously shown in models of acute ischemia or metabolic stress. This was unrelated to any effect on PKC or phosphorylation at S368 which remained elevated and unchanged. However, it may have been related to phosphorylation at other sites that control Cx43 life cycle, which we did not measure, or increases in transcription/translation. Since lateralization of Cx43 is related to degradation15 and was not affected, it is unlikely that reduction of degradation applies to Rotigaptide’s effects. At 3 hours the increase in Cx43 was not significant compared to control (but also not significantly different than at one hour), indicating that either sample size was too small, variability was too large or the effects at one hour wanes with time.
Conduction velocity in EBZ did not increase despite the increased Cx43. It is uncertain whether the increased Cx43 formed functional gap junctions. Since Cx43 remained highly phosphorylated at S368, if gap junctions were formed, they still may have reduced conductance characteristic of pS368 Cx43 gap junctions. If some of the increased Cx43 was lateralized, it also would not be expected to be functional although we did not detect an increased lateralized Cx43. In addition, conduction slowing in EBZ is also a result of remodeled Na+ current which was not affected by Rotigaptide. Failure to improve conduction velocity was not a result of depression of Na+ current by Rotigaptide to negate an effect of increasing gap junction conductance. Our results are consistent with other studies showing that Rotigaptide is not effective when structural remodeling and/or ion channel remodeling of the arrhythmogenic substrate has occurred37,40,43. Nevertheless, Rotigaptide reduced the ERP which was specific for the EBZ since it did not occur in normal ventricle. This effect which has also been described in chronic human heart failure43 may have been an indicator of some restoration of gap junction coupling, reversing ERP prolongation caused by uncoupling (see above discussion on gap junction uncoupling and repolarization). Rotigaptide did not affect reactivation kinetics of the Na+ channel but, we cannot rule out an effect on repolarizing ionic currents in EBZ myocytes although it has not been seen in normal myocytes.
Rotigaptide did not prevent SMVT in 3 of 4 experiments. There were small changes in coupling intervals of stimulated impulses inducing SVT. Whether that had any effect on inducibility is uncertain but we interpret these results to indicate that the drug did not have a robust effect to prevent SMVT. It is possible that in a larger series of experiments it would exhibit some antiarrhythmic activity. The failure to prevent induction of SMVT may be related to persistence of conduction abnormalities in the EBZ. Whether the changes in SMVT cycle lengths or patterns of reentry were related to an effect of Rotigaptide is uncertain. In 2 experiments, SMVT was inducible after Rotigaptide. More experiments are necessary to determine if it has a significant proarrhythmic effect.
Conclusion
Improving myocyte coupling is an attractive idea for a new approach to antiarrhythmic therapy. The results of our study indicate that interventions that are effective in restoring cell to cell coupling under conditions of acute ischemia may not be effective in a substrate that has already undergone a prolonged period of gap junction remodeling such as the border zone of healing infarcts. Drugs with properties that would restore coupling in this environment might still be effective.
Supplementary Material
Acknowledgments
Funding Sources: Supported by HL066140 (PAB) and HL083205 (HSD) from the National Heart Lung and Blood Institute Bethesda, a grant from Wyeth Pharmaceuticals (ALW) and a Medtronic fellowship (EM).
Footnotes
Conflict of Interest Disclosures: This work was supported in part by a grant from Wyeth Pharmaceuticals who supplied the Rotigaptide for these studies (ALW).
References
- 1.Dillon S, Allessie M, Ursell PC, Wit AL. Influence of anisotropic tissue structure on reentrant circuits in the subepicardial border zone of subacute canine infarcts. Circulation Research. 1988;63:182–206. doi: 10.1161/01.res.63.1.182. [DOI] [PubMed] [Google Scholar]
- 2.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 3.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodeling of gap junctions and connexin expression in diseased myocardium. Cardiovascular Research. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–3442. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solan JL, Lampe PD. Key Connexin 43 Phosphorylation Events Regulate the Gap Junction Life Cycle. J Membrane Biol. 2007;217:35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kle’ber AG, Schuessler RB, Saffitz JE. Dephosphorylation and Intracellular Redistribution of Ventricular Connexin43 during Electrical Uncoupling Induced by Ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 7.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol Rev. 2003:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 9.Xing D, Kjølbye AL, Nielsen MS, Petersen JS, Harlow KW, Holstein-Rathlou NH, Martins JB. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol. 2003;14:510–520. doi: 10.1046/j.1540-8167.2003.02329.x. [DOI] [PubMed] [Google Scholar]
- 10.Hennan JK, Swillo RE, Morgan GA, Keith JC, Schaub RG, Smith RP, Feldman HS, Haugan K, Kantrowitz J, Wang PJ, Abu-Qare A, Butera J, Larsen BD, Crandall DL. Rotigaptide (ZP123) Prevents Spontaneous Ventricular Arrhythmias and Reduces Infarct Size during Myocardial Ischemia/Reperfusion Injury in Open-Chest Dogs. JPET. 2006;317:236–243. doi: 10.1124/jpet.105.096933. [DOI] [PubMed] [Google Scholar]
- 11.Axelsen LN, Stahlhut M, Mohammed S, Larsen BD, Nielsen MS, Holstein-Rathlou N-H, Andersen S, Jensen ON, Hennan JK, Kjølbye AL. Identification of ischemia-regulated phosphorylation sites in connexin43: A possible target for the antiarrhythmic peptide analogue rotigaptide (ZP123) Journal of Molecular and Cellular Cardiology. 2006;40:790–798. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Coromilas J, Saltman AE, Waldecker B, Dillon SM, Wit AL. Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation. 1995;91:2245–2263. doi: 10.1161/01.cir.91.8.2245. [DOI] [PubMed] [Google Scholar]
- 13.Cabo C, Yao J, Boyden PA, Chen S, Hussain W, Duffy HS, Ciaccio EJ, Peters NS, Wit AL. Heterogeneous gap junction remodeling stabilizes reentrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72:242–249. doi: 10.1016/j.cardiores.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Pu J, Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997–81:110–9. doi: 10.1161/01.res.81.1.110. [DOI] [PubMed] [Google Scholar]
- 15.Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, Hirst-Jensen BJ, Duffy HS, Sorgen PL. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1100–1112. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haugan K, Olsen KB, Hartvig L, Petersen JS, Holstein-Rathlou NH, Hennan JK, Nielsen MS. The antiarrhythmic peptide analog ZP123 prevents atrial conduction slowing during metabolic stress. J Cardiovasc Electrophysiol. 2005;16:537–545. doi: 10.1111/j.1540-8167.2005.40687.x. [DOI] [PubMed] [Google Scholar]
- 17.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;126:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36:1171–86. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleber AG, Riegger CB, Janse MJ. Electrical Uncoupling and Increase of Extracellular Resistance After Induction of Ischemia in Isolated, Arterially Perfused Rabbit Papillary Muscle. Circulation Research. 1987;61:271–279. doi: 10.1161/01.res.61.2.271. [DOI] [PubMed] [Google Scholar]
- 20.Procida K, Jørgensen L, Schmitt N, Delmar M, Taffet SM, Holstein-Rathlou N-H, Nielsen MS, Braunstein TH. Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm. 2009;6:1632–1638. doi: 10.1016/j.hrthm.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao X, Reuss L, Altenberg GA. Regulation of purified and reconstituted connexin 43 hemichannels by protein kinase C-mediated phosphorylation of serine 368. J Biol Chem. 2004;279:20058–66. doi: 10.1074/jbc.M311137200. [DOI] [PubMed] [Google Scholar]
- 22.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of Connexin 43 Channels Is Regulated Through Protein Kinase C–Dependent Phosphorylation. Circ Res. 2006;98:1498–1505. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Yao J-A, Hussain W, Patel P, Peters NS, Boyden P, Wit AL. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circulation Research. 2003;92:437–443. doi: 10.1161/01.RES.0000059301.81035.06. [DOI] [PubMed] [Google Scholar]
- 27.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, Gutstein DE. Modulation of Cardiac Gap Junction Expression and Arrhythmic Susceptibility. Circ Res. 2004;95:1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner PI, Ursell PC, Fenoglio JJ, Jr, Wit AL. Electrophysiologic and anatomic basis for fractionated electrograms recorded from chronically ischemic and infarcted regions of the heart. Circulation. 1985;72:596–611. doi: 10.1161/01.cir.72.3.596. [DOI] [PubMed] [Google Scholar]
- 29.Gough WB, Mehra R, Restivo M, Zeiler RH, El-Sherif N. Reentrant ventricular arrhythmias in the late myocardial infarction period in the dog. Correlation of activation and refractory maps. Circ Res. 1985;57:432–442. doi: 10.1161/01.res.57.3.432. [DOI] [PubMed] [Google Scholar]
- 30.Lesh MD, Pring M, Spear JF. Cellular Uncoupling Can Unmask Dispersion of Action Potential Duration in Ventricular Myocardium: A Computer Modeling Study. Circulation Research. 1989;65:1426–1440. doi: 10.1161/01.res.65.5.1426. [DOI] [PubMed] [Google Scholar]
- 31.Tan RC, Joyner RW. Electrotonic influences on action potentials from isolated ventricular cells. Circ Res. 1990;67:1071–1081. doi: 10.1161/01.res.67.5.1071. [DOI] [PubMed] [Google Scholar]
- 32.Spitzer KW, Pollard AE, Yang L, Zaniboni M, Cordeiro JM, Huelsing DJ. Cell-to-Cell Electrical Interactions During Early and Late Repolarization. J Cardiovasc Electrophysiol. 2006;17:S8–S14. doi: 10.1111/j.1540-8167.2006.00379.x. [DOI] [PubMed] [Google Scholar]
- 33.Anyukhovsky EP, Sosunov EA, Rosen MR. Regional Differences in Electrophysiological Properties of Epicardium, Midmyocardium, and Endocardium: In Vitro and In Vivo Correlations. Circulation. 1996;94:1981–1988. doi: 10.1161/01.cir.94.8.1981. [DOI] [PubMed] [Google Scholar]
- 34.Akar FG, Rosenbaum DS. Transmural Electrophysiological Heterogeneities Underlying Arrhythmogenesis in Heart Failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- 35.Eloff BC, Gilat E, Wan X, Rosenbaum DS. Pharmacological modulation of cardiac gap junctions to enhance cardiac conduction: evidence supporting a novel target for antiarrhythmic therapy. Circulation. 2003;108:3157–3163. doi: 10.1161/01.CIR.0000101926.43759.10. [DOI] [PubMed] [Google Scholar]
- 36.Kjølbye AL, Dikshteyn M, Eloff BC, Desche nes I, Rosenbaum DS. Mantenance of intercellular coupling by the antiarrhythmic peptide rotigaptide suppresses arrhythmogenic discordant alternans. Am J Physiol Heart Circ Physiol. 2008;294:H41–H49. doi: 10.1152/ajpheart.01089.2006. [DOI] [PubMed] [Google Scholar]
- 37.Guerra JM, Everett TH, 4th, Lee KW, Wilson E, Olgin JE. Effects of the gap junction modifier rotigaptide (ZP123) on atrial conduction and vulnerability to atrial fibrillation. Circulation. 2006;114:110–118. doi: 10.1161/CIRCULATIONAHA.105.606251. [DOI] [PubMed] [Google Scholar]
- 38.Kjølbye AL, Knudsen CB, Jepsen T, Larsen BD, Petersen JS. Pharmacological characterization of the new stable antiarrhythmic peptide analog Ac-D-Tyr-DPro-D-Hyp-Gly-D-Ala-Gly-NH2 (ZP123): in vivo and in vitro studies. J Pharmacol Exp Ther. 2003;306:1191–1199. doi: 10.1124/jpet.103.052258. [DOI] [PubMed] [Google Scholar]
- 39.Dhein S, Larsen BD, Petersen JS, Mohr FW. Effects of the new antiarrhythmic peptide ZP123 on epicardial activation and repolarization pattern. Cell Commun Adhes. 2003;10:371–378. doi: 10.1080/cac.10.4-6.371.378. [DOI] [PubMed] [Google Scholar]
- 40.Shiroshita-Takeshita A, Sakabe M, Haugan K, Hennan JK, Nattel S. Model-dependent effects of the gap junction conduction-enhancing antiarrhytmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation. 2007;115:310–318. doi: 10.1161/CIRCULATIONAHA.106.665547. [DOI] [PubMed] [Google Scholar]
- 41.Weng S, Lauven M, Schaefer T, Polontchouk L, Grover R, Dhein S. Pharmacological modification of gap junction coupling by an antiarrhythmic peptide via protein kinase C activation. FASEB J. 2002;16:1114–1116. doi: 10.1096/fj.01-0918fje. [DOI] [PubMed] [Google Scholar]
- 42.Lin X, Zemlin C, Hennan JK, Petersen JS, Veenstra RD. Enhancement of ventricular gap-junction coupling by Rotigaptide. Cardiovascular Research. 2008;79:416–426. doi: 10.1093/cvr/cvn100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiegerinck RF, de Bakker JMT, Opthof T, de Jonge N, Kirkels H, Wilms-Schopman FJG, Coronel R. The effect of enhanced gap junctional conductance on ventricular conduction in explanted hearts from patients with heart failure. Bas Res Cardiol. 2009;104:321–332. doi: 10.1007/s00395-008-0771-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.