

Abstract
Autoantibodies that react with GRP78 expressed on the cell-surface of many tumor cell lines occur in the sera of patients with prostate cancer, melanoma, and ovarian cancer. These autoantibodies are a negative prognostic factor in prostate cancer, and when purified, stimulate tumor cell proliferation in vitro. It is unclear, however, whether these IgGs are merely a biomarker, or if they actually promote tumor growth in vivo. We immunized C57Bl/6 mice with recombinant GRP78 and then implanted the B16F1 murine melanoma cell line as flank tumors. We employed the antisera from these mice for in vitro cell signaling and proliferation assays. The immunodominant epitope in human cancer patients was well represented in the antibody repertoire of these immunized mice. We observed significantly accelerated tumor growth, as well as shortened survival in GRP78-immunized mice as compared to controls. Furthermore, antisera from these mice, as well as purified anti-GRP78 IgG from similarly immunized mice, stimulate Akt phosphorylation and proliferation in B16F1 and human DM6 melanoma cells in culture. These studies demonstrate a causal link between a humoral response to GRP78 and the progression of cancer in a murine melanoma model. They support the hypothesis that such autoantibodies are involved in the progression of human cancers and are not simply a biomarker. Because GRP78 is present on the surface of many types of cancer cells, this hypothesis has broad clinical and therapeutic implications.
Keywords: Autoantibodies to GRP78, B16 Melanoma, Cell-surface GRP78, syngeneic tumor model
Introduction
GRP78 is a highly conserved member of the HSP70 family, predominantly located in the luminal space of the endoplasmic reticulum (ER). Its primary function is to mediate the proper folding of nascent polypeptides and it is a major regulator of the unfolded protein response (UPR)[1]. The over-expression of this protein in a variety of tumors promotes cell proliferation, protects from apoptosis, and promotes tumor angiogenesis [2,3,4,5] Despite its carboxyl-terminal ER-retention signal (KDEL), GRP78 is translocated to the cell surface under certain conditions. A number of studies have demonstrated GRP78 expression on the surface of cells from a variety of lineages ranging from cancer cells and endothelial cells to activated macrophages [6,7,8,9,10]. Cell surface GRP78 plays a role in activities which include cell signaling, viral entry, and antigen presentation[11,12,13,14]. Studies of receptor-recognized α2-macroglobulin (α2M*)-mediated cell signaling demonstrated the existence of a high affinity receptor for α2M* [13]. This receptor activates signal transduction through a pertussis toxin-insensitive G-protein [15]. Cell surface GRP78 is the receptor responsible for the increase in inositol triphosphate (IP3) levels, RAC-alpha serine/threonine-protein kinase (Akt) phosphorylation, NF-κB induction, and the subsequent rise in intracellular Ca2+ when α2M* binds to the cell surface [13,16].
The effects of Akt activation are pleiotropic, but are known to promote cell survival and proliferation[17,18,19]. We have shown in activated murine peritoneal macrophages and human prostate cancer cells that cell-surface GRP78 ligation with α2M* stimulates proliferation and inhibits apoptosis, in addition to upregulating the expression of GRP78 itself [9,16].
Arap and colleagues identified GRP78 as a major autoantigen in prostate cancer patients, and presented data that anti-GRP78 autoantibody occurrence is associated with advanced disease progression and shorter patient survival [20]. An in vivo study targeting cytotoxic payloads to cancer cells by tagging them with GRP78-binding peptides further validated the significance of cell-surface GRP78 [21]. While autoantibodies are best characterized with regard to their role in the progression of autoimmune disorders such as rheumatoid arthritis and systemic lupus erythematosis, they also are markers of tumor behavior in humans [22,23]. The emergence of anti-GRP78 autoantibodies is a negative prognostic factor in prostate cancer[20]. These antibodies recognize an epitope mimicked by the phage display-derived peptide CNVSDKSC corresponding to an amino-terminal region of GRP78 where the physiologic ligand α2M* binds [24]. This, coupled with our characterization of similar prostate cancer patient-derived autoantibodies as a mitogen mimicking α2M*[24], suggests a role for a humoral response to GRP78 in promoting the growth of tumors in vivo.
In order to address this possibility, we sought to induce a humoral response to GRP78 and observe its effect on tumor growth in an animal model. Several recent studies have demonstrated a positive relationship between GRP78 expression, aggressive tumor behavior an poor prognosis in melanoma[25], gastric carcinoma[26,27,28], hepatocellular carcinoma[29], and head and neck cancer[30]. A common method in such studies is the use of siRNA-mediated knockdown of GRP78 to demonstrate the functional importance of this protein in tumor cell behaviors. This approach, however, is problematic since it causes a significant reduction in the major ER pool of GRP78 as well as its surface expression[13]. This will invariably hamper any cellular behavior requiring protein synthesis. This plus the dysregulation of ER-based Unfolded Protein Response signaling that results from GRP78 knockdown[31] make it impossible to reasonably distinguish the effects of decreased cell-membrane GRP78 from decreased intracellular protein. We have demonstrated the pro-proliferative effect of prostate cancer patient-derived anti-GRP78 autoantibodies on the human melanoma cell line DM413[24]. In the present study, we investigate the cell-surface expression and function of GRP78 in murine melanoma. Using a syngeneic mouse model of melanoma, we then demonstrate that immunization against GRP78 leads to the generation of a amino-terminal GRP78-reactive murine autoantibody as a cognate to the autoantibodies observed in human cancer patients. This is the first report demonstrating that circulating autoantibodies to GRP78 promote tumor growth in vivo.
Materials and Methods
Proteins and peptides
α2M isolated from human plasma was employed to prepare the receptor recognized form α2M-MeNH2 (α2M*) as previously described [32]. A recombinant murine cDNA expression construct encoding full-length (rGRP78) with a 6-His tag in pET15b was the kind gift of Dr. Sylvia Blond. This rGRP78 was expressed and purified as previously described. [33]. The GRP78 mimetic peptide CNVSDKSC identified by Mintz et al. [20] was purchased from Genemed Synthesis, Inc.(San Francisco, CA) and conjugated to KLH via its terminal cysteine residues with the heterobifunctional cross-linker sulfosuccinimidyl 4-(N-maleidomethyl)-cyclohexane-1-carboxylate (sulfo-SMMC; Pierce, Rockford, IL) as previously described [24].
Cell culture
The B16F0, B16F1, B16F10 and PC3 cell lines were obtained from the American Type Culture Collection. The DM6 human melanoma line was the kind gift of H.F. Siegler (Department of Immunology, Duke University Medical Center). The 1-LN cell line was a kind gift from Dr. Phillip Walther (Department of Urology, Duke University Medical Center). All cell lines were grown in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 2 mM glutamine, and 1% penicillin/streptomycin. Cells were maintained in a humidified 37°C incubator at 5% CO2 in logarithmic growth in 75 cm2 flasks.
Antibodies and reagents
A polyclonal antibody was rasied against the full-length recombinant GRP78 in sheep at Covance (Denver, PA). This antibody was purified by chromatography on Protein A-Sepharose, followed by immunoaffinity chromatography on recombinant GRP78 immobilized on Sepharose 4B. Non-immune sheep IgG was purchased from Sigma (Sigma-Aldrich, St. Louis, MO). We prepared normal mouse serum from exsanguinated age-matched naïve C57BL/6 mice.
On-Cell Western
Cells were grown to confluency in 24-well tissue culture plates at in DMEM with 10%FBS with penicillin and streptomycin. Media was then removed and cells were fixed with ice cold 4% using neutral-buffered formaldehyde solution for 20 min 4°C. Fixative was removed and the cells were washed five times with PBS. Fixed cells were blocked with 250 µl of Rockland IR blocking buffer for 1 h at room temperature. Primary antibody was added at 0.5µg/mL in blocking buffer and incubated overnight at 4°C. Plates were washed 5 times with PBS 0.05% Tween 20 and then secondary antibody conjugated to an IR-800nm label was added at a dilution of 1:200 in blocking buffer and incubated for 1 h. Plates were washed 5 times with PBS 0.05% Tween 20 and then dried at 37°C. IR label was protected from light at all times. Plates were read on a Li-cor Odyssey® reader, and these experiments were performed in triplicate and repeated twice.
Flow Cytometry
1 × 106 B16F1 cells were stained with 5 µg of either N20 antibody (Santa Cruz Biotechnology, Cat. No. scbt-1050) or C20 antibody (Santa Cruz Biotechnology, Cat. No. scbt-1051). The secondary antibody used was a rabbit anti-goat IgG F(Ab′)2 (Jackson Immunologicals, Cat. No. 305-096-047) at a concentration of 1 µg per 1 × 106 B16F1 cells. Cells were also stained with 7AAD as per the manufacturer’s instructions (BD Pharmingen, Cat. No. 51-68981E) in order to gate out dead cells. Samples were analyzed on a Guava EasyCyte Plus™ flow cytometer.
Murine immunization and tumor studies
Female 4–6 week-old C57BL/6 mice were used for all experiments described in this study. Prior to immunizations, we identified each mouse with a unique numbered ear tag, obtained submandibular blood samples for pre-bleeds, then randomized them into two groups of ten mice each. The each group received three identical subcutaneous inoculations comprised of either zero (adjuvant control) or 25 µg rGRP78 emulsified in 100µl of a 1:1 mixture of PBS and TiterMax Gold® adjuvant (CytRx Corporation). All mice were immunized at two-week intervals, with a final titer bleed taken two weeks following the final booster. After final titers, all mice received subcutaneous tumors in the right flank region. Briefly, we trypsinized B16F1 cells in log-phase growth and washed them thrice in DMEM (Sigma-Aldrich, St. Louis, MO)). Cells were resuspended at concentration of 5×104/ml in a 1:1 mixture of DMEM and Matrigel (BD Biosciences, San Jose, CA). Each mouse received a subcutaneous injection in the right flank of 100µl for a total of 5×103 cells. We assessed tumor growth by manual caliper measurement, and exsanguinated each animal when it reached a terminal criterion, 2 cm3 tumor volume.
Analysis of serum anti-GRP78 response
Antibodies against GRP78 in the sera of immunized mice were assayed by ELISA in 96-well culture plates coated with recombinant GRP78 (5 µg/mL) in 0.1 mol/L Na2CO3, 0.01% NaN3 (pH 9.3). Incubation of these plates with serum samples and analyses of the data were performed as previously described [34]. The epitope specificity of the anti-GRP78 IgG in the serum of prostate cancer patients was also assayed by an ELISA technique. Briefly, 96-well EIA/RIA plates were coated with the peptide CNVSDKSC conjugated to KLH (5 µg/mL) in 0.1 mol/L Na2CO3, 0.01% NaN3 (pH 9.3). All assays were done in triplicate as previously described [34]. CNVSDKSC-reactive antibody fractions were purified by affinity chromatography over columns packed with CH-Sepharose (Sigma-Aldrich) charged with CNVSDKSC peptide. We eluted bound fractions with glycine, pH 2.5.
Akt signaling assays
B16F1 or DM6 cells were grown to 90 percent confluence in 96-well plates, then washed once with serum-free DMEM, and rested for five h in fresh DMEM. We stimulated the cells for 15 min with the appropriate serum (1:200) or antibody preparation (100 pM) diluted in 200 µl DMEM before washing once with ice cold PBS and subsequent lysis. For PI3K inhibition, we pretreated cells for 30 minutes with 5µM LY29004 (Sigma-Aldrich, St. Louis, MO) before stimulation. We performed total and phospho-specific Akt ELISA assays (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. Phospho-Akt results were normalized to total Akt levels to account for any variability in cell harvesting. All conditions were in triplicate, and each experiment was repeated at least twice.
Proliferation assays
B16F1 or DM6 cells in log-phase growth were seeded at 1×104 cells per well and allowed to grow for 24 h in 96-well plates. They were washed once with serum-free DMEM, and rested for five h in fresh DMEM. The media was replaced with the appropriate serum or antibody preparation diluted in 200 µl DMEM with 0.5 µCi tritiated thymidine per well. 5 µM LY29004 was included for PI3K inhibition where appropriate. Proliferation continued for 16 h until the media was removed and the cells dissociated in 10X trypsin-EDTA and harvested them using a 12-channel microharvester (Skatron Instruments, Norway). The samples were counted in a scintillation counter. All conditions were in triplicate, and we repeated each experiment at least twice.
α2-Macroglobulin Radioligand Binding Assay
Human α2M* was labeled with Bolton-Hunter diiodo 125I according to the manufacturer’s instructions (Perkin-Elmer). Labeled α2M* used at concentrations ranging from 200 nM to 100 fM was incubated in 96 well EIA/RIA plates coated with 100 ng/well rGRP78 overnight at 4°C. Plates were washed three times with PBS and samples were read on a Wallac 1272 Clinigamma counter. For competition assays, α2M*-125I was used at a concentration of 500 pM and simultaneously incubated with pooled mouse GRP78 antiserum at dilutions from 1:1 to 1:16000. Samples were treated the same as above.
Statistical analysis
All analyses were performed employing GraphPad Prism® version 5.02. Student’s t-test, ANOVA with Tukey’s multiple comparison tests, or the Mann-Whitney U-test were used as appropriate.
Results
B16 melanoma cells express cell-surface GRP78
Nonpermeabilized monolayers of each cell line were grown in 24-well plates for these experiments (Fig. 1). These cells were incubated with 0.5µg/ml purified sheep anti-GRP78 or non-immune sheep IgG followed by an IR dye 800 DX-conjugated donkey anti-sheep secondary antibody. This cell-based ELISA analysis of the B16F0, B16F1, and B16F10 murine melanoma cell lines demonstrates substantial surface presentation of GRP78 on all three cell lines, in that relative order of increasing expression. We included known high- and low-expressing cell lines (1-LN and PC3) as positive controls [35]. Consistent with previous results, 1-LN display more GRP78 than do PC3. The signal from non-immune IgG for each cell line has been subtracted to yield the final integrated signals (Fig. 1). We confirmed this result using flow cytometry with B16F1 cells, demonstrating that both the amino and carboxyl termini of GRP78 are available on these cells (Fig. 2).
Figure 1. Cell-Surface Presentation of GRP78.

Formalin-fixed non-permeabilized B16F0, B16F1, B16F10, 1-LN, and PC3 cells were labeled with either 0.5µg/ml sheep anti-GRP78 polyclonal antibody or with non-immune sheep IgG. The secondary antibody was labeled with a near-IR 800nm fluor and read on a Li-Cor Odyssey® detector. The upper panel shows a histogram of the integrated signal from scans of 24-well plates after subtraction of the background non-immune signal. Below is an example of this cell-based ELISA.
Figure 2. Flow Cytometry.
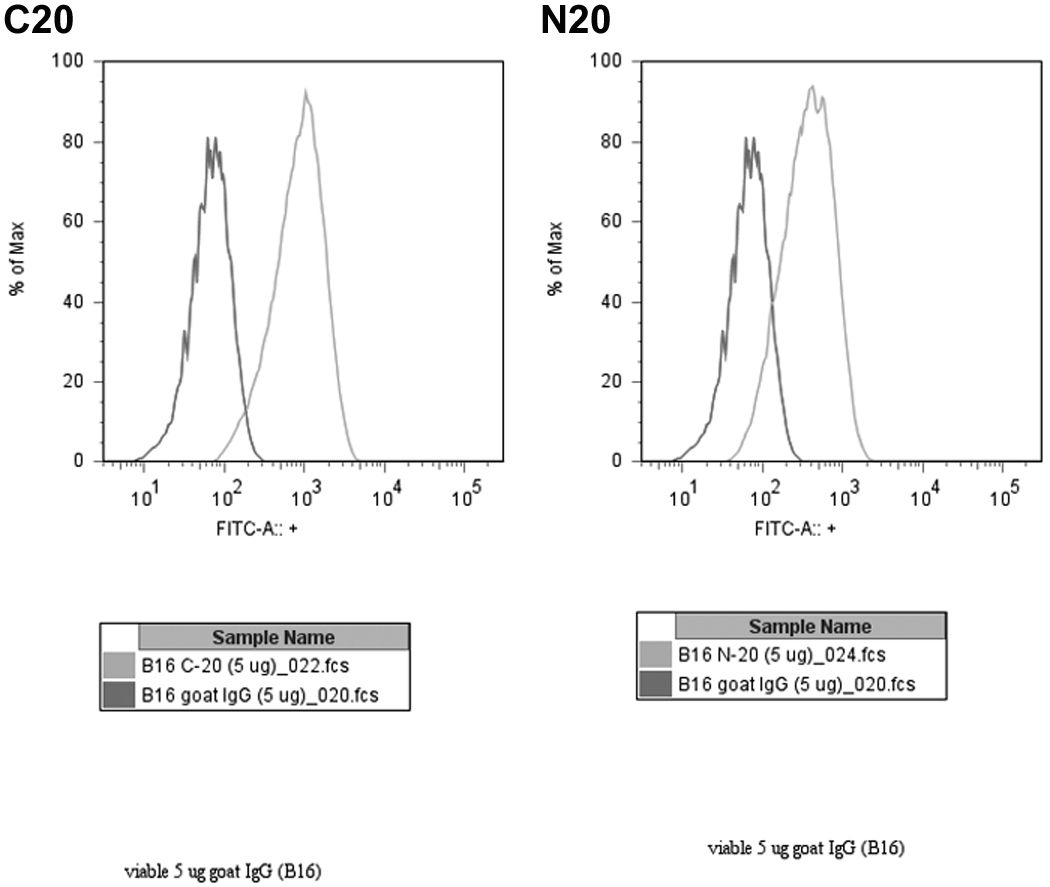
Live, non-permeabilized cells were labeled with 5µg/106 cells of N20 or C20 antibody, or else normal goat IgG. Data shown are representative of several such experiments, and are gated only on live cells.
GRP78 immunization induces autoantibodies
The average endpoint titers of sera raised against recombinant murine GRP78 (1.8×106, N=10) and against TiterMax Gold® adjuvant alone (1×103, N=9) determined by ELISA against rGRP78 are shown on in Figure 3A. Because the peptide CNVSDKSC contains a motif recognized by anti-GRP78 autoantibodies produced by prostate cancer patients (23), we developed an ELISA assay with this peptide conjugated to KLH and immobilized on 96-well culture plates. We show significant (Mann-Whitney U test, P<0.0001) reactivity of these sera against this dominant epitope observed in human cancer in Figure 3B.
Figure 3. Murine Anti-GRP78 Autoantibodies.

Panel A. Endpoint titers of mouse sera taken before tumor implantation. Samples were diluted and then assayed on plates coated with rGRP78. Endpoints were defined as the highest dilution yielding absorbance threefold higher than background. Panel B. ELISA data from adjuvant-only (N=9) and GRP78-immunized (N=10) mice. EIA/RIA plates were coated with CNVSDKSC peptide conjugated to KLH. Sera were diluted 1:100 in 1%BSA and incubated overnight at 4°C. Data are presented as absorbance units at 450nm and P value here is derived from the nonparametric Mann-Whitney test.
GRP78 immunization affects B16F1 flank tumor growth rate
The growth kinetics of B16F1 flank tumors in control (N=9) and rGRP78-immunized (N=10) mice are shown in Figure 4A. Tumors in immunized mice were significantly (Student’s t-test, P<0.05) larger by day 14 post-implantation and remained so throughout the study. Kaplan-Meier analysis of mouse survival in this study is shown in Figure 4B. GRP78 immunization significantly shortened median mouse survival to 26 days from the adjuvant control group’s median survival of 29 days (log-rank test, P<0.05). One mouse in the control group died unexpectedly within one week of tumor implantation. That mouse was censored from analysis here.
Figure 4. Effects of GRP78 Immunization on B16F1 Flank Tumor Growth.

Panel A. Growth of 5×103 B16F1 cells implanted in the flanks of C57BL/6 mice. Average tumor volumes in GRP78-immunized mice (N=10) are represented by squares while those of adjuvant-only mice (N=9) are represented by circles. Data shown represent mean tumor volume in cm3 ± SEM versus day post-implantation. Asterisks represent statistically significant differences in mean tumor volume at a given timepoint (P<0.05, Student’s t-test). Panel B. Kaplan-Meier analysis of mouse survival compares GRP78-immunized mice to controls. The median survival for the two groups is 26 and 29 days, respectively (P<0.05, log-rank test). The GRP78-immunized group is represented by a solid line, while the adjuvant control group is represented by a dashed line.
Mouse antisera against GRP78 stimulate Akt phosphorylation
Serum-starved B16F1 or human DM6 melanoma cells were stimulated with a variety of preparations of mouse sera and we show the resulting phosphorylation of Akt in Figure 5. Application of normal mouse serum (N=5) or adjuvant-group, tumor-bearing mouse serum at a dilution of 1:200 (N=9) caused only negligible Akt activation. Sera from the rGRP78-immunized, tumor-bearing mice (N=10) stimulated a significant (P<0.05), roughly three-fold increase in phospho-Akt as compared to naïve mouse serum and adjuvant-group serum. Pooled sera from rGRP78-immunized mice that were not exposed to B16 tumors (N=20) evoked a similar response to that of the tumor-bearing mice. We purified the anti-CNVSDKSC fraction of this last pool of sera by affinity chromatography and subsequently stimulated B16F1 cells. Anti-CNVSDKSC-specific IgG at 100pM elicited a response similar (no significant difference) to whole antiserum, while CNVSDKSC depletion of the antiserum almost completely abrogated its signaling potential (P<0.05). We repeated these experiments with the human DM6 cell line (Fig. 5B). Whole murine sera raised against rGRP78 stimulated Akt phosphorylation in DM6, as did CNVSDKSC-specific IgG. Moreover, CNVSDKSC depletion of antisera also abrogated signaling in DM6 as it did in B16F1. In all of the above experiments, 5µM LY29004 completely abolished Akt activation by GRP78 antibodies or antisera.
Figure 5. Murine Anti-GRP78 Autoantibodies Stimulate Akt Activation.

Panel A. ELISA-based analysis of Akt signaling in murine B16F1 cells. Cells were grown to 90% confluency, deprived of serum for five hours, and and then stimulated for 15 minutes with a variety of reagents. Data shown represent mean ±SD of experiments performed in triplicate on two different days. Phospho-Akt results were normalized to the amount of total Akt in each sample. Asterisks represent significant increases in phospho-Akt over baseline, normal sera, and adjuvant-control sera levels (P<0.05) Panel B. ELISA-based analysis of Akt signaling in human DM6 cells. This experiment was performed exactly as in 4A.
Murine GRP78 antisera stimulate melanoma proliferation
B16F1 or DM6 cells (1×104 per well) were incubated with either 1:200 dilutions of mouse antisera, 100 pM purified anti-CNVSDKSC IgG, or 100 pM α2M* (positive control) in serum-free medium for 24 h, followed by a further 16 h in the presence of 0.5 µCi tritiated thymidine per well. Differential proliferation was assayed by scintillation counting of incorporated labeled thymidine. In keeping with previous experiments, α2M* stimulated a dramatic ~threefold increase in proliferation (P<0.05)(Fig.6A). Both whole pooled (N=20) antiserum and purified anti-CNVSDKSC stimulated a similar proliferative response as compared α2M*. CNVSDKSC-depleted antiserum treatment was indistinguishable from normal sera. These responses were closely recapitulated in DM6 cells (Fig.6B). LY29004, 5µM, completely abrogated GRP78 antisera- or antibody-induced cellular proliferation in all of these experiments.
Figure 6. Murine Anti-GRP78 Autoantibodies Stimulate Tumor Cell Proliferation.

Panel A. Effect of murine GRP78 antisera on B16F1 proliferation. Cells in 96-well plates were deprived of serum for five hours and then stimulated for 16 hours in the presence of various stimuli. Data shown represent mean ±SD of experiments performed in triplicate on two different days. Asterisks represent significant increases in proliferation over baseline and normal sera stimulation (P<0.05). Panel B. Effect murine GRP78 antisera on proliferation in human DM6 cells. This experiment was performed exactly as in 5A.
GRP78 antisera compete with α2M* for GRP78 binding
Activated α2M labeled with I125 was incubated with pooled serum from GRP78-immunized mice in 96-well plates coated with rGRP78. The Kd-apparent for α2M*-125I derived from initial binding assays was 47pM, so we used roughly 10-fold higher, 500pM α2M*-125I in our competition assays. GRP78-immunized mouse antiserum completely abrogated α2M*-125I binding to rGRP78 at dilutions out to 1:16000, while naïve mouse serum showed no competition.
Discussion
A humoral response against GRP78 is correlated with a poor prognosis in prostate cancer[20], and we have previously demonstrated the in vitro mitogenic potential of prostate cancer patient-derived anti-GRP78 IgG on the 1-LN and DU145 prostate cancer cell lines, as well as the DM413 melanoma cell line[24]. Circulating antibodies to GRP78 are also associated with ovarian carcinoma [36] and have been employed experimentally to distinguish women with benign masses from those with frank ovarian carcinomas[37]. We have detected anti-GRP78 antibodies in melanoma patients, and their titers increase with time after diagnosis with metastatic melanoma (unpublished observations, Dr. Mario Gonzalez-Gronow). Our current results taken together with previous studies suggest that a humoral response against GRP78 is not only a marker of cancer progression, but also a contributor to tumor cell proliferation and malignant behavior in vivo.
Here we present the first experimental evidence that humoral immunity against GRP78 can hasten the growth of tumor in vivo. In order to actively immunize animals against GRP78 and then observe an effect on a tumor challenge, a syngeneic tumor model is necessary, and the tumor cells employed must express cell-surface GRP78. Our observations that a human melanoma line responds pro-proliferatively to anti-GRP78 IgG [24] and that similar antibodies arise in melanoma patients led us to the B16 melanoma model.
The B16F1 line is frequently used in flank tumor models, however, there is no published characterization of B16 cell surface presentation of GRP78. Our cell-based ELISA analysis of surface expression on B16F0, B16F1 and B16F10 revealed significant amounts of GRP78 on all three cell lines. Interestingly, their expression (Fig1) increases in order of increasing metastatic potential[38,39]. We have observed the same relationship with PC3 and 1-LN prostate cancer cells; namely, the more aggressive 1-LN subclone of PC3 also expresses more surface GRP78 [13,16]. We confirmed the surface expression of GRP78 on B16F1 cells by flow cytometry (Fig. 2). Polyclonal antibodies directed at either the amino or carboxyl terminal of GRP78 positively labeled these cells, confirming the presence of both termini extracellularly.
The robust anti-GRP78 responses reported here (Fig. 3A) are consistent with previous observations of the immunogenicity of this protein in studies of rheumatoid arthritis[34,40,41]. In the mouse melanoma model employed here GRP78 also is an autoantigen, since we employed recombinant murine protein for immunization. We were able to mimic the humoral anti-GRP78 response seen in human cancers, since the dominant human epitope was very well represented in our experimental antisera (Fig.3B). In our study, GRP78 immunity caused B16F1 flank tumors to grow substantially faster than in PBS-adjuvant control mice (Fig. 4A) or naïve mice (data not shown). Moreover, the GRP78-immunized mice experienced significantly shorter survival than the controls (Fig. 4B). Given the very fast growth of the B16F1 tumor, achieving significance with relatively small groups of animals indicates a very robust effect of GRP78 immunity on tumor growth.
The PI3K/Akt axis signaling downstream of GRP78 ligation by its ligand α2M* has been extensively characterized [8,15,16]. Because the dominant GRP78 epitope in human cancer corresponds to the α2M*-binding region, we hypothesized that cell-surface GRP78 ligation by agonist autoantibodies would drive tumor growth. We applied antisera derived from terminal bleeds of the tumor-challenged mice in this study to melanoma cells in vitro to test this mechanistic hypothesis. Antisera raised against GRP78 strongly induced Akt phosphorylation in both the B16F1 cell line and the human DM6 cell line (Fig. 5A–B), as did purified anti-CNVSDKSC IgG from such antisera. This finding indicates the cross-species biological significance of an immunologic response to GRP78 and validates the relevance of our murine model to human cancer. Moreover, we have demonstrated the significance of the dominant human epitope by depleting the mitogenic potential of GRP78 antiserum by affinity chromatography against CNVSDKSC peptide. Similarly, we confirmed the downstream effect of anti-GRP78 antiserum Akt activation on proliferation in both murine and human cell lines (Fig. 6A–B). Both Akt activation and proliferative responses were completely compromised by the inhibitor LY29004, which indicates the PI3K dependence now canonical for GRP78-mediated signaling. This potentiation of cellular proliferation alone provides intriguing evidence for the pathogenic role of anti-GRP78 autoantibodies in tumor growth, but other downstream effects of Akt activation beg examination for their contributions as well.
In summary, we have demonstrated the utility of syngeneic melanoma modeling of the humoral response to GRP78 observed in human cancer. Considering that autoantibodies that react with cell-surface GRP78 are also observed in prostate[20] and ovarian carcinomas[36,37], future studies employing the syngeneic TRAMP murine prostate tumor line and ID8 murine ovarian tumor line may broaden the significance of our findings. The ultimate goal of such studies should be therapeutic interference with cell-surface GRP78 ligation and/or signaling, whether driven by autoantibodies or the physiological ligand α2M*. While anti-GRP78 autoantibodies arise in patients with several types of cancer, all cancer patients will have high local concentrations around tumors of activated α2M. This is because α2M is a broad-range protease inhibitor, and tumor cells as well as tumor-associated leukocytes tend to release significant amounts of proteases. Therefore, any tumor with functional cell-surface GRP78 may be inhibited by agents interfering with α2M* or anti-GRP78 IgG signaling.
Acknowledgments
We sincerely thank Marie Thomas and Steven Conlon of the Duke University Department of Pathology for their assistance in preparing this manuscript.
Grant Support
These studies were supported in part by an NCI Grant No. CA 131235.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Quinones QJ, de Ridder GG, Pizzo SV. GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histol Histopathol. 2008;23:1409–1416. doi: 10.14670/HH-23.1409. [DOI] [PubMed] [Google Scholar]
- 2.Cai B, Tomida A, Mikami K, Nagata K, Tsuruo T. Down-regulation of epidermal growth factor receptor-signaling pathway by binding of GRP78/BiP to the receptor under glucose-starved stress conditions. J Cell Physiol. 1998;177:282–288. doi: 10.1002/(SICI)1097-4652(199811)177:2<282::AID-JCP10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 4.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–3499. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 5.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 6.Xiao G, Chung TF, Pyun HY, Fine RE, Johnson RJ. KDEL proteins are found on the surface of NG108-15 cells. Brain Res Mol Brain Res. 1999;72:121–128. doi: 10.1016/s0169-328x(99)00188-6. [DOI] [PubMed] [Google Scholar]
- 7.Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005;65:4663–4672. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 8.Misra UK, Deedwania R, Pizzo SV. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J Biol Chem. 2005;280:26278–26286. doi: 10.1074/jbc.M414467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misra UK, Pizzo SV. Up-regulation of GRP78 and antiapoptotic signaling in murine peritoneal macrophages exposed to insulin. J Leukoc Biol. 2005;78:187–194. doi: 10.1189/jlb.1104685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C, Bhattacharjee G, Boisvert W, Dilley R, Edgington T. In vivo interrogation of the molecular display of atherosclerotic lesion surfaces. Am J Pathol. 2003;163:1859–1871. doi: 10.1016/S0002-9440(10)63545-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Triantafilou K, Fradelizi D, Wilson K, Triantafilou M. GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J Virol. 2002;76:633–643. doi: 10.1128/JVI.76.2.633-643.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Triantafilou M, Fradelizi D, Triantafilou K. Major histocompatibility class one molecule associates with glucose regulated protein (GRP) 78 on the cell surface. Hum Immunol. 2001;62:764–770. doi: 10.1016/s0198-8859(01)00269-5. [DOI] [PubMed] [Google Scholar]
- 13.Misra UK, Gonzalez-Gronow M, Gawdi G, Hart JP, Johnson CE, Pizzo SV. The role of Grp 78 in alpha 2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J Biol Chem. 2002;277:42082–42087. doi: 10.1074/jbc.M206174200. [DOI] [PubMed] [Google Scholar]
- 14.Jindadamrongwech S, Thepparit C, Smith DR. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch Virol. 2004;149:915–927. doi: 10.1007/s00705-003-0263-x. [DOI] [PubMed] [Google Scholar]
- 15.Misra UK, Chu CT, Gawdi G, Pizzo SV. The relationship between low density lipoprotein-related protein/alpha 2-macroglobulin (alpha 2M) receptors and the newly described alpha 2M signaling receptor. J Biol Chem. 1994;269:18303–18306. [PubMed] [Google Scholar]
- 16.Misra UK, Pizzo SV. Potentiation of signal transduction mitogenesis and cellular proliferation upon binding of receptor-recognized forms of alpha2-macroglobulin to 1-LN prostate cancer cells. Cell Signal. 2004;16:487–496. doi: 10.1016/j.cellsig.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 18.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 19.Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- 20.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 21.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Tan EM, Zhang J. Autoantibodies to tumor-associated antigens: reporters from the immune system. Immunol Rev. 2008;222:328–340. doi: 10.1111/j.1600-065X.2008.00611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu H, Goodell V, Disis ML. Humoral immunity directed against tumor-associated antigens as potential biomarkers for the early diagnosis of cancer. J Proteome Res. 2008;7:1388–1394. doi: 10.1021/pr700818f. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Gronow M, Cuchacovich M, Llanos C, Urzua C, Gawdi G, Pizzo SV. Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 2006;66:11424–11431. doi: 10.1158/0008-5472.CAN-06-1721. [DOI] [PubMed] [Google Scholar]
- 25.Papalas JA, Vollmer RT, Gonzalez-Gronow M, Pizzo SV, Burchette J, Youens KE, et al. Patterns of GRP78 and MTJ1 expression in primary cutaneous malignant melanoma. Mod Pathol. 2010;23:134–143. doi: 10.1038/modpathol.2009.152. [DOI] [PubMed] [Google Scholar]
- 26.Zheng HC, Zheng YS, Xia P, Xu XY, Xing YN, Takahashi H, et al. The pathobiological behaviors and prognosis associated with Japanese gastric adenocarcinomas of pure WHO histological subtypes. Histol Histopathol. 2010;25:445–452. doi: 10.14670/HH-25.445. [DOI] [PubMed] [Google Scholar]
- 27.Zheng HC, Takahashi H, Li XH, Hara T, Masuda S, Guan YF, et al. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum Pathol. 2008;39:1042–1049. doi: 10.1016/j.humpath.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L, et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastasis. 2006;23:401–410. doi: 10.1007/s10585-006-9051-9. [DOI] [PubMed] [Google Scholar]
- 29.Su R, Li Z, Li H, Song H, Bao C, Wei J, et al. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer. 2010;10:20. doi: 10.1186/1471-2407-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu CC, Lin CY, Lee LY, Chen YJ, Kuo TF, Chang JT, et al. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol Cancer Ther. 2008;7:2788–2797. doi: 10.1158/1535-7163.MCT-08-0172. [DOI] [PubMed] [Google Scholar]
- 31.Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 32.Imber MJ, Pizzo SV. Clearance and binding of two electrophoretic "fast" forms of human alpha 2-macroglobulin. J Biol Chem. 1981;256:8134–8139. [PubMed] [Google Scholar]
- 33.King LS, Berg M, Chevalier M, Carey A, Elguindi EC, Blond SY. Isolation, expression, and characterization of fully functional nontoxic BiP/GRP78 mutants. Protein Expr Purif. 2001;22:148–158. doi: 10.1006/prep.2001.1424. [DOI] [PubMed] [Google Scholar]
- 34.Cuchacovich M, Gatica H, Pizzo SV, Gonzalez-Gronow M. Characterization of human serum dipeptidyl peptidase IV (CD26) and analysis of its autoantibodies in patients with rheumatoid arthritis and other autoimmune diseases. Clin Exp Rheumatol. 2001;19:673–680. [PubMed] [Google Scholar]
- 35.Asplin IR, Misra UK, Gawdi G, Gonzalez-Gronow M, Pizzo SV. Selective upregulated expression of the alpha2-macroglobulin signaling receptor in highly metastatic 1-LN prostate carcinoma cells. Arch Biochem Biophys. 2000;383:135–141. doi: 10.1006/abbi.2000.2052. [DOI] [PubMed] [Google Scholar]
- 36.Chinni SR, Falchetto R, Gercel-Taylor C, Shabanowitz J, Hunt DF, Taylor DD. Humoral immune responses to cathepsin D and glucose-regulated protein 78 in ovarian cancer patients. Clin Cancer Res. 1997;3:1557–1564. [PubMed] [Google Scholar]
- 37.Taylor DD, Gercel-Taylor C, Parker LP. Patient-derived tumor-reactive antibodies as diagnostic markers for ovarian cancer. Gynecol Oncol. 2009;115:112–120. doi: 10.1016/j.ygyno.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 38.Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Characterization of mouse melanoma cell lines by their mortal malignancy using an experimental metastatic model. Life Sci. 2002;70:791–798. doi: 10.1016/s0024-3205(01)01454-0. [DOI] [PubMed] [Google Scholar]
- 40.Blass S, Union A, Raymackers J, Schumann F, Ungethum U, Muller-Steinbach S, et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001;44:761–771. doi: 10.1002/1529-0131(200104)44:4<761::AID-ANR132>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 41.Purcell AW, Todd A, Kinoshita G, Lynch TA, Keech CL, Gething MJ, et al. Association of stress proteins with autoantigens: a possible mechanism for triggering autoimmunity? Clin Exp Immunol. 2003;132:193–200. doi: 10.1046/j.1365-2249.2003.02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]