

Abstract
Background
Pharmacological studies suggest adenosine A3AR influences motility and colitis. Aim: Functional A3−/−AR knockout mice were used to prove whether A3AR activation is involved in modulating either motility or colitis.
Methods
A3AR was probed by PCR genotyping, western blot and immunochemistry. Motility was assessed in vivo by artificial bead-expulsion, stool-frequency and FITC-Dextran transit. Colitis was induced with DSS in A3−/−AR or wild-type (WT) age, sex-matched-controls. Progression of colitis was evaluated by histopathology, changes in MPO, colon length, CD4+-cells, weight-loss, diarrhea and the Guaiac-test.
Results
Goat anti-hu-A3 antiserum identified a 66kDa immunogenic-band in colon. A3AR-immunoreactivity is expressed in SYN+-nerve varicosities, s-100+-glia and crypt cells, but not 5-HT+ (EC), CD4+ (T), tryptase+ (MC) or muscle cells. A3AR-immunoreactivity in myenteric ganglia of distal colon ≫ proximal colon by a ratio of 2:1. Intestinal transit and bead expulsion were accelerated in A3−/−AR mice compared to WT; stool retention was lower by 40%–60% and stool frequency by 67%. DSS down-regulated A3AR in epithelia. DSS histopathology scores indicated less mucosal damage in A3−/−AR mice than WT. A3−/−AR phenotype protected against DSS-induced weight-loss, neutrophil (MPO) or CD4+-T cell infiltration, colon shortening, change in splenic weight, diarrhea or occult-fecal blood.
Conclusions
Functional disruption of A3AR in A3−/−AR mice alters intestinal motility. We postulate that ongoing release of adenosine and activation of presynaptic-inhibitory A3AR can slow down transit and inhibit the defecation reflex. A3AR may be involved in gliotransmission. In separate studies, A3−/−AR protects against DSS-colitis consistent with a novel hypothesis that A3AR activation contributes to development of colitis.
Keywords: Murine DSS-Colitis, A3−/−AR mouse, Adenosine Receptors, Motility, Presynaptic inhibitory A3AR, gliotransmission, Enteric Nervous System
INTRODUCTION
Activation of adenosine A1, A2A, A2B and A3 receptors is involved in the modulation of enteric neural reflexes 1. Adenosine (ADO) A1, A2 and A3 receptor (A3AR) proteins or mRNA exist in rodent 2 and human intestinal tract3. Receptors are differentially expressed in neural and non-neural cells of the gut. Functional evidence for A1 inhibitory, non-A1 (putative A3AR) and A2 excitatory receptors exists for rodent enteric neurons1,4. Endogenous adenosine (eADO) release inhibits 5-HT release from enterochromaffin cells (EC), and suppresses neuronal excitability and synaptic transmission in the enteric nervous system (ENS) by activating high affinity A1AR, and perhaps low affinity A3AR. Low oxygen tension is a potent stimulus for eADO release to suppress peptidergic neurotransmission in networks of enteric ganglia5. In human EC or intact networks of human ENS submucous ganglia, functional inhibitory A3AR can be revealed by selective A3 agonists like N6-(3-iodobenzyl)-adenosine-5'-N-methyluronamide (IB-MECA) or 2-Cl-IB-MECA, and effects of A3AR antagonists alone suggest that eADO levels are sufficient to activate low affinity A3AR6,7.
It has been postulated that high affinity inhibitory A1AR are involved in physiological regulation whereas low affinity inhibitory A3AR are more likely to be activated by eADO in pathophysiological states because they require high levels of eADO to activate them that are more easily achieved in disease states 8–12. Recent studies are beginning to unravel possible beneficial effects of elevating extracellular levels of eADO 14–16 or activation of A3AR 17,18, A2AAR 19–22 or A2BAR 23 in models of inflammatory bowel diseases (IBD) with drug agonists or antagonists depending on the receptor. Increasing the concentration of eADO is one mechanism by which several drugs may reduce gut inflammation 14–16. Chronic administration of an adenosine kinase inhibitor 15,16 or an A3 agonist IB-MECA 17 may be beneficial in murine models of dextran sodium sulfate (DSS) – induced colitis or a rat trinitrobenzene-sulfonic acid (TNBS) – induced colitis model 18, but the mechanisms or receptors involved remain poorly understood. IB-MECA is well tolerated orally by healthy human volunteers 24 and has been in Phase IIb clinical trials for another chronic inflammatory disease, rheumatoid arthritis, and is without apparent toxicity (www.canfite.com/develop.html).
The prototypical A3AR agonist IB-MECA has been shown to have protective effects in rodent models of colitis (DSS-colitis, IL-10 knockout mice and TNBS colitis), but the effects occur at high doses ranging from 1–3 mg/Kg body weight 17,18 that may not necessarily be restricted to actions at A3AR. Furthermore, the anti-inflammatory mechanism of IB-MECA may be linked to a down-regulation of A3AR 25. In a model of cardiac ischemia, A3AR knockout (A3−/−AR) is protective 26, and not all effects at A3AR are protective 27–29. Therefore, the role of A3AR in gut inflammation needs further clarification.
Complex neural circuits in the ENS coordinate motility, secretion, vascular tone, epithelial transport and immune-responses in the gut to ensure effective digestion, absorption, transit of luminal contents, and elimination of waste products. Immune-neural communication is involved in response to pathogens, allergens in the diet, mucosal irritants, or gut inflammation, and host-defense mechanisms 30. Experimental gut inflammation causes abnormalities in A1AR or A2AR leading to alterations in purinergic modulation of enteric neurotransmission 22,31. The role of A3AR in the ENS or other cells in the gut wall of such integrated responses or reflexes in normal or disease states of the gut remains unclear. In a model of neurogenic diarrhea our recent pharmacological data suggests that eADO activation of A3AR suppresses the stereotype motor behavior of the colon in a short inter-plexus neural reflex triggered by histamine 32. Such indirect pharmacological studies do not provide definitive proof of inhibitory A3AR in modulating the motor behavior of the gut.
Thus, we used the A3AR knockout mouse model (A3−/−AR) to further evaluate whether the A3AR is involved in regulating in vivo motility and DSS – colitis. We hypothesize that functional knock-out of A3−/−AR would facilitate in vivo intestinal motility, mass movement (evacuation reflex), reduce stool retention, and also affect the development of DSS – colitis. Our data support two novel hypotheses: (1) Activation of neural A3AR at presynaptic sites of transmitter release by eADO slows-down intestinal transit, colonic emptying and mass movement (evacuation reflex) and promote stool retention. (2) Activation of A3AR by eADO may contribute to development of DSS – colitis and tissue injury. Our colitis study was not designed to test whether the A3AR−/− colitis phenotype is motility-dependent.
MATERIAL and METHODS
Breeding colonies of A3−/−AR and WT mice
Active breeding A3−/−AR homozygous trios of C57Bl/6 /mice (line 760) as well as age-matched control WT C57Bl/6 (A3AR) breeding trio-founders from Taconics (Cambridge City, IN) were provided by Marlene A. Jacobson at Merck Research Laboratory 33 and were bred in our facility at The Ohio State University. Mice 3–4 months of age (or 8–10 months of age) were used for these experiments, and separate studies of motility and DSS colitis did not provide significant age-dependent differences (duplicate data will therefore not be shown). Experiments were done using female, age, and weight-matched A3−/−AR and WT controls bred from the original founders at our facility at OSU. Heterozygotes were not available for breeding, but we took steps to insure the microbial communities were standardized. Specific pathogen – free (SPF) wild-type C57BL/6 mice and A3−/−AR knockout mice were bred under standardized conditions in cages in the same SPF room/environment in a barrier facility.
Genotyping the A3−/−AR knockout mice
Initial PCR of tail genomic DNA with specific primers for the PGKneo insert and full-length wild-type were used to identify the A3−/−AR KO mice and control A3AR founders on the basis of positive PCR analysis. Tail clips of all mice were kept to confirm the genotype of each mouse. Two sets of primers were used to genotype and confirm the identity of A3−/−AR and WT mice used in DSS-colitis or motility experiments: PCR was run using specific primers for the PGKneo plasmid insert which was used to disrupt the function of the A3AR gene. The PGKneo insert forward primer sequence used to identify the transgene was 5’-CTATGACTGGGCACAACAGACAAT-3’. The reverse primer sequence was 5’ATCAGCCATGATGGATACTTTCTC-3’. To differentiate between full-length wild-type A3AR mice and the PGKneo insert / transgene we used 5’ and 3’ end primers of the full length WT A3AR gene. The forward primer sequence was 5’-GACTGGCTGAACATCACCTACAT-3’) and the reverse primer sequence was 5’-ATAGAAGTGCATCTTGACTTGCAG-3’). Genotyping: A3−/−AR mice were identified using PCR. Genomic DNA was isolated from tail clips and incubated in 500 μl of lysis buffer overnight at 60 °C. The lysis buffer contained 50 mM Tris, pH 8, 50 mM NaCl, 25 mM EDTA, and 37.5 μl of proteinase K. The lysate was cleaned in triplicate with phenol/chloroform (1:1) and once with chloroform. The supernatant was recovered, and DNA was precipitated with 2 volumes of ethanol and spooled out in a clean tube.
Immunochemical identification of A3AR
Immunochemical identification of A3AR or other proteins was done as previously described4,7,18,34. Microdissected longitudinal muscle – myenteric plexus (LMMP) of mouse colon was used in co-labeling studies of A3AR-ir in neurons, glia, varicosities or other cells. Primary antibodies were goat anti-A3AR antibody (1:20–100 dil, Santa Cruz; R-18; sc-7510), rabbit anti-s-100 (1:80 dil from Neomarker), mouse anti-synaptophysin (1:100 dil from Sigma), mouse anti-HuC/D antibody (1:50, Molecular Probe). Co-labeling of mucosal A3AR with CD4+ cells, MC tryptase+ cells and 5-HT+ enterochromaffin cells was done in 5 μm thick transverse cut-paraffin sections from proximal, middle or distal colon of control or DSS-colitis animals; antisera were rabbit anti- MC tryptase (sc-32889, Santa Cruz), mouse anti-CD4 monoclonal antibody (ab64144, Abcam, Cambridge, MA) diluted 1:50 and mouse anti-5-HT monoclonal antibody (1:100 dilution, DAKO, M0758); secondary antibodies were FITC donkey anti-goat IgG (for visualization of A3AR), Texas red donkey anti-rabbit IgG (for s100, s100+) or Texas red donkey anti-mouse IgG (for synaptophysin, SYN+ or HuC/D+, 1:100 dilution, Jackson Immunoresearch Lab). A Zeiss LSM 410 confocal microscope 7,34 was used to image co-labeled cells. Merged images are used to show co-localization of A3AR. For IHC, CD4-ir (in mucosa) was visualized by reacting Biotin-SP-AffiniPure donkey anti-mouse IgG (712-065-150, Jackson Labs, 1:200) with a peroxidase-linked avidin-biotin complex (PK-6100, Vector Laboratories, Burlingame, CA, 1:100) and VIP substrate; methyl green (4800-30-18, Trevigen, Gaithersburg, MD) was used for counterstaining; a donkey anti-goat IgG was used for A3AR. The number of CD4+-cells was counted under a microscope at 400X magnification and expressed as CD4+-cell number / 20 crypts. The CD4 data for each mouse colon were defined as average of CD4+-cell number per 20 crypts from the 3 sections representing proximal, middle, and distal colon. Specificity was confirmed by omission of primary antibody, isotype controls for primary antibody, pre-adsorption with immunogenic peptide (according to manufacturer’s instructions, Santa Cruz) or western blots to identify a single molecular weight band of expected size. Pre-adsorption with immunogenic/control peptides was done by incubation of the antiserum with peptide overnight at 4°C.
Western blots
The proteins were isolated from colon of C57BL/6 mice. A sample of 100μg protein lysate was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Richmond, CA). Membranes were incubated at 4°C overnight withthe rabbit anti-A3 antibody (1:100, Alpha Diagnostic) or goat anti-A3 antibody (1:100, Santa Cruz) followed by incubation with goat anti-rabbit IgG-HRP (1:500, sc-2004, Santa Cruz) or donkey anti-goat IgG-HRP (1:400, sc-2020, Santa Cruz) for 2 h at room temperature. The immunogenic band for A3AR was revealed with enhanced chemiluminescence (ECL) western blot analysis detection reagent (Amersham Biosciences, Pittsburgh, PA). Immunoabsorption with immunogenic peptide was used to confirm specificity of antisera for A3AR.
Mouse dextran sulfate sodium (DSS) colitis model
Mice were group housed 5/cage maximum under a controlled temperature of 25°C and a 12:12 h light-dark cycle and they were allowed free access to standard mouse chow and water. Colitis was induced by the addition of 1.5% or 3% DSS to the drinking water for 7–8 days. The DSS-water consumption, food intake, body weight and general behavior of the animals was monitored and recorded on a daily basis. All animal experiments were approved by the Animal Care Committee of The Ohio State University (IACUC), Columbus Ohio, and followed the guidelines for the Care and Use of Laboratory Animals (published by the U.S. Public Health Service). Typically DSS experiments were carried out for 2 weeks and on the 14th day after the induction of colitis animals were euthanized by rapid cervical dislocation. The length of the colon was exposed by a midline laparotomy, the entire colon and cecum was removed, the colon length and weight (after feces removal by flushing with cold Krebs buffer) was measured and processed for histopathology or biochemical studies.
Clinical activity in DSS – colitis model
Clinical activity was evaluated using the parameters of weight loss, stool consistency, Guaiac Test, shortening of the colon:body weight ratio, increase in spleen weight:body weight ratio, neutrophil infiltration by myeloperoxidase activity. Colon length is an indicator of the severity of colitis.
Histopathological assessment of the colon in DSS colitis induced in WT or A3−/−AR mice
The colon was divided into three equal-length segments of proximal, middle and distal colon and then fixed in 4% (w/v) paraformaldehyde in PBS overnight. Paraffin sections (5μm sections, transverse orientation) were processed for histopathological scoring 35. Each slide included three sections one from each segment stained with hematoxylin and eosin (H&E) and scored blindly by an expert clinical pathologist prior to identification and statistical analysis.
Histopathological scoring was done separately for inflammation, crypt damage and ulceration. Briefly, for inflammation, score 0: rare inflammatory cells in the lamina propria; score 1: increased numbers of granulocytes in the lamina propria; score 2: confluence of inflammatory cells extending into the submucosa; score 3: transmural extension of the infiltrate. For crypt damage, score 0: an intact crypt, score 1: loss of the basal one third crypt; score 2: loss of the basal two thirds crypt; score 3: entire crypt loss; score 4: change of epithelial surface with erosion; score 5: confluent erosion. For ulceration, score 0: absence of ulcer, score 1: 1 or 2 foci of ulcerations; score 2: 3 or 4 foci of ulcerations; score 3: confluent or extensive ulceration. Aggregate scores of individual histopathology scores ranged from 0 to 11. For statistical analysis, we used SPSS 17.0 software (SPSS Inc.Chicago, IL) to average the aggregate or individual scores of the three sections for each colon. Groups were compared by using nonparametric tests, and the significance of differences between groups was assessed with the Mann–Whitney U-test; a 2-tailed p-value <0.05 was considered statistically significant.
Myeloperoxidase activity
Colonic myeloperoxidase (MPO) enzymatic activity was used as an index of neutrophils infiltration into the injured / inflamed mucosa. Briefly, MPO were measured in intestinal tissue homogenate samples lysed using a lysis buffer with composition: 200 mM NaCl, 5 mM EDTA, 10 mM tris, 10% glycerin, 1mM PMSF, 1 μg/ml leupeptin and 28 μg/ml aprotinin (pH 7.4); 200 μl lysis buffer is added to 10 mg tissue before homogenization. Samples are centrifuged twice (1500 × g at 4°C for 15 min) to avoid contamination of cell debris. Samples are stored at −80°C until used to measure MPO. MPO is measured in samples diluted at least 4× before use. A mouse MPO ELISA KIT (HK210) from Hycult Biotech (Cell Sciences Inc, Canton, MA) was used to determine the MPO levels in mouse colon lysates. Sample absorbance is measured using a spectrophotometer at A450nm (Perkin Elmer, Victor3 1420 multi-label counter) following the instructions provided by the instrument manufacturer. The MPO kit has a minimum detection level of 1.0 ng/ml and a measurable concentration range of 1.0 to 250 ng / ml.
Guaiac Test
The guaiac test (Hemoccult slides, Beckman Coulter, Inc, Fullerton, CA) is used to detect fecal occult blood in the gastrointestinal (GI) tract which is indicative of GI bleeding associated with severe intestinal inflammation and tissue damage in the DSS-colitis model. Two different approaches were used to test for occult blood in the feces and analyze the data: (1) Animals were transferred to separate cages on different days during the 2 week study and several pellets were tested for a (+) guaiac test and data was analyzed in terms of the number of animals with a (+) or (−) test at each day tested. (2) The guaiac test was applied on smears from 10 pellets obtained from each cage housing a group of 4 animals, and data analyzed in terms of (+) or (−) pellets; at least 2 separate cages housed animals for a given group and results were pooled and analyzed.
Bead Latency test
In vivo distal colonic transit and emptying was evaluated by introducing a 3 mm x 6 mm petroleum jelly-coated Teflon bead into the distal colon at 2 cm from the rectum of WT or A3−/−AR mice (n=19 WT, n=20 A3−/−AR) that were very lightly anesthetized to facilitate insertion of the bead. After bead insertion, mice were placed in individual plastic cages lined with colored paper to aid visualization of white bead expulsion. The time required for complete expulsion / evacuation of the artificial bead was recorded to the nearest second for each animal with a stop-watch. The experiment was done by two investigators working in concert to insert the bead and monitor bead expulsion time.
Stool frequency and emptying
Each mouse was placed in a clear plastic cage and monitored for 1 h. The number of stool pellets extruded per hour was used as an index of colonic emptying and reported as stool frequency. Stool weight and water content were also calculated for each animal. Pellets were collected immediately after expulsion, weighed to obtain wet weight and placed in Eppendorf tubes with a hole punched in the lid. They were then dried overnight at 65°C to obtain dry weights. The difference between wet weight and dry weight was calculated to estimate water content.
In vivo intestinal transit with FITC-dextran
Intestinal transit was evaluated in vivo by assessing the distribution of a 70 kDa FITC-conjugated dextran marker (Sigma, St Louis, MO, USA) in the small intestine and colon of A3−/−AR (n=7) and WT age-matched control mice (n=7). Each experiment consisted of a parallel study in a WT and an A3−/−AR mouse. Mice were fasted for 12 hours and 0.1 ml of 5mM FITC-dextran was orally given by gavage, and 3 hours later, mice were euthanized, and the small intestine was divided into 10 segments of equal length (S1–S10) and the colon into 4 equal segments labeled as C1–C4 from oral to distal colon and rectum. Each segment was flushed with 3 ml of 50 mM Tris buffered saline solution and samples were then centrifuged at 1200 rpm for 5min. The fluorescent activity of the supernatant was quantified using a fluorimeter at excitation 485nm and emission at 525nm. Intestinal transit was estimated according to the intestinal mean geometric center (MGC) of the distribution of FITC-dextran throughout the intestines 36. Data was analyzed according to the Mean Geometric Center (MGC).
Statistics
Mean values ± S.E.M were reported. Multiple groups were compared by a one-way analysis of variance (ANOVA) and post-hoc tests between individual groups. SPSS 17.0, GraphPad Prism 3.02 software are used for analyses. Histopathology is analyzed by a non-parametric Mann-Whitney U- test. Numbers of CD4+ T-cells in WT vs A3−/−AR is done by ANOVA (multiple comparisons) and simple t-tests between two groups. Non-linear regression analysis is used to fit a 2nd or 3rd order polynomial curve to data generated for weight-loss in DSS versus control; post-tests are used to compare pairs of curves; differences in weight-loss over time after DSS induction were analyzed by ANOVA followed by Newman-Keuls Multiple Comparison Test. For other parameters simple unpaired / 2-tail t-tests are used. Stat-View 54.51 (Abacus Concepts, Calabasas, CA) was also used for analysis. Data was analyzed from a minimum of 5 to a maximum of 20 animals as indicated in specific studies. Chi-square test was used to analyze data from the Guaiac Test for occult blood in the stools or stool consistency (diarrhea or formed pellets). Differences are considered statistically significant at p < 0.05.
RESULTS
Genotyping of A3−/−AR knockout mice and molecular expression of A3 AR
Primers for the PGKneo insert recognized the A3−/−AR functional knock-out gene but not the WT A3 AR gene (Supplement Figure 1A). Primers for the full-length wild-type A3 AR recognized both the WT and A3−/− KO genes in the mice (data not shown). Two different antibodies were used to identify the A3AR by western blot analysis. The Santa Cruz anti-A3 antibody identified a single immunogenic band at 66 kDa corresponding to the molecular size of the A3AR in lysates of whole-thick colon from A3−/−AR or WT mice (Supplement Figure 1B). There was a 1 fold up-regulation in the expression of the A3AR transgene in the A3−/−AR mice (p=0.0359) compared to WT (Supplement Figure 1D). The Alpha-Diagnostic anti-A3 antiserum recognized the same immunogenic band of 66 kDa but also revealed a number of other bands at lower molecular weight (Supplement Figure 1C).
Immunochemical identification of A3AR
In 5 animals, immunohistochemical identification was carried out in mouse colon LMMP preparations. A3AR-ir was observed in varicose nerve fibers and glial-like structures surrounding neuronal cell bodies and throughout the ganglia (Figure 1A–F). Additional co-labeling experiments were done for A3AR-ir and s-100 (glial cells), synaptophysin (varicose nerve terminals) or HuC/D (neuronal cell somas) immunoreactive structures. A3AR-ir was abundantly expressed in SYN+ varicosities (Figure 1J–L) or s-100+ Glia (Figure 1M–O) but not in HuC/D+ neurons (Figure 1G–I). In 3 additional mice, the percentage of anti-HuC/D immunoreactive ganglia in the colon with A3AR-ir was 43% in proximal colon, 77% in middle colon and 75% in distal colon (Figure 1A).
Figure 1.

Immunochemical localization of A3AR in varicose nerve terminals and glial cells. (A) Quantitative analysis of the distribution of A3AR-ir in proximal, middle and distal colon (represents positive neurons and glia; not distinguished). Data is expressed as Mean ± S.E.M; n=3 animals. ANOVA indicated a p value of 0.003 between the 3 groups of data; the number of ganglia counted in each region was 656±55.5, 742±58.1 and 695.7±66.1 respectively. (B–E) Examples of IHC - localization of A3AR immunoreactivity in myenteric varicosities and glia. (F) A3AR-ir in varicose fibers running through the circular muscle. (G–I) A3AR-ir is not co-localized in the cell body of anti-HuC/D+ myenteric neurons (J–L) A3AR-ir is co-localized with SYN+ varicose nerve terminals. (M–O) A3AR-ir is co-localized with s100+ glial cells; thin arrow, varicosities; thick arrow, glia; open arrow, varicose fiber forms basket around neuronal cell body; Secondary antibodies for anti-Huc/D, SYN, s100 conjugated to Texas Red (red color); secondary antibody to anti-A3 conjugated to FITC (green color); G–O overlay images, yellow indicates co-localization.
Bead latency – distal colonic transit and emptying
Bead latency of evacuating the artificial pellet varied from 2.5 – 8 min in WT and 1.25 – 3.75 min in the A3−/−AR KO mice. Bead latency is reduced by 50% from ~ 4 min to 2 min in A3−/−AR mice compared to WT age-matched controls in 19 WT and 20 A3−/− AR mice (p=0.0001, Figure 2).
Figure 2.

Colonic evacuation of an artificial bead occurs faster in A3−/−AR. (A, B) In vivo bead-latency (or expulsion time in min) is 50% shorter in A3−/−AR mice compared to WT controls; A is a scattergram of individual responders and B summarizes the pooled data, n=19 WT, 20 A3−/−AR mice.
Stool frequency and colonic emptying
Overall, A3−/−AR mice exhibit impaired fecal output and colonic emptying (Figure 3). Fecal-pellet output / hour (Figure 3A) and stool wet weight (Figure 3B) are reduced by more than 60% in A3−/−AR mice compared to WT. Fluid content in each pellet remains the same in the two groups (Figure 3C). Stool retention determined by weight of stool or number of pellets retained in the colon was reduced in A3−/−AR mice compared to WT (Figures 3D–E). Colon length normalized to body weight is significantly higher in A3−/−AR mice but the increase is modest (Figure 3F). A3−/−AR mice also have slightly lower body weight than WT mice (Figure 3H). Colon length alone is slightly lower (<15%) in A3−/−AR mice (Figure 3G). Food intake is not significantly different in A3−/−AR and WT mice (Figure 3I).
Figure 3.
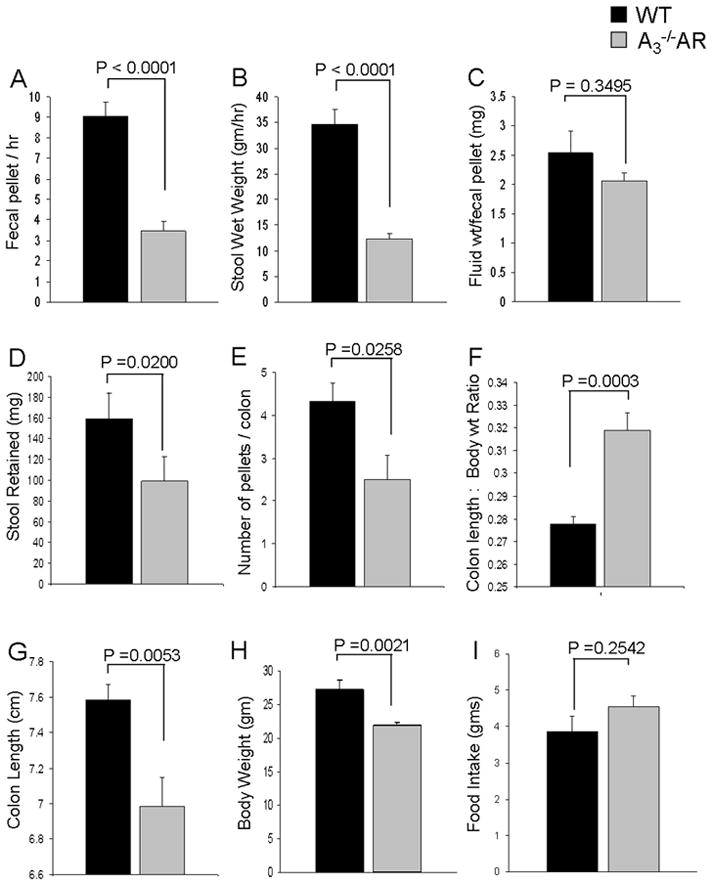
A3−/−AR mice exhibit impaired fecal output and colonic emptying (A) Fecal-pellet output and (B) Stool wet weight is reduced by more than 60% in A3−/−AR mice compared to WT. (C) Colonic water content is not altered in A3−/−AR mice. (D, E) Stool retention is significantly less in A3−/−AR mice compared to WT. (F) Colon length:body weight ratio is higher in A3−/−AR mice; (G) Colon length alone is slightly lower (<15%) in A3−/−AR mice. (H) Body weight is lower in A3−/−AR compared to WT. (I) Food intake is the same in both A3−/−AR and WT mice.
Intestinal transit
In vivo intestinal transit was monitored using FITC-dextran to evaluate its distribution in the intestinal tract (small and large intestine). A3−/−AR mice exhibit accelerated intestinal transit. Three hours after oral administration of the FITC-dextran, the peak response occurred at S8 in the small intestine of WT mice whereas in A3−/−AR mice it occurred at C1-C2 regions. A representative example of accelerated transit in A3−/−AR is shown in Figure 4A. Pooled data are summarized in Figure 4B. According to the Mean Geometric Center, A3−/−AR mice had a faster intestinal transit than WT controls.
Figure 4.

A3−/−AR mice exhibit accelerated alterations in intestinal transit via in vivo FITC-dextran GI tract distribution. (A) Raw data of intestinal transit/distribution of fluorescence in a WT and an A3−/−AR mouse calculated 3 hours after oral administration of FITC-Dextran; FITC, fluorescein isothiocyanate. Similar results obtained in 3 other pairs of animals. (B) Pooled data showing that the Mean Geometric Center (MGC) is altered in A3−/−AR compared to WT. MGC in WT = 7.03± 0.43 (n=4) and in A3−/−AR = 9.43±0.73 (n=4); p = 0.031 between the groups.
Change in Myeloperoxidase activity, spleen weight and colon length
MPO activity in WT or A3−/−AR mice is marginally detectable in the absence of DSS induction. DSS induction elevates MPO activity and there is a 50% reduction in MPO in A3−/−AR mice induced with DSS compared to WT controls induced with DSS (Figure 5A). DSS colitis caused an increase in spleen weight in both WT and A3−/−AR mice but it was significantly less in A3−/−AR knockout mice. There was no difference in spleen weight between WT and A3−/−AR control mice (Figure 5B). Mild 1.5% DSS-induced colitis causes a shortening of the colon in WT mice that was prevented in A3−/−AR mice; in fact colon length was slightly increased. At 3% DSS colitis, both WT and A3−/−AR mice had similar shortening of the colon and knockout had no effect (Figure 5C).
Figure 5.

A3−/−AR knockout inhibits myeloperoxidase (MPO) activity, change in splenic weight and shortening of the colon after DSS induction of colitis. (A) MPO enzymatic activity as an index of neutrophil infiltration in the injured colon. MPO elevation in 3% DSS-colitis is reduced 50% by A3−/−AR knockout. (B) Change in splenic weight in DSS colitis is reduced by disruption of the A3AR. (C) Shortening of the colon caused by 1.5% DSS induction of colitis is blocked by A3−/−AR knockout.
Histopathology and weight-loss
Histopathology of individual or aggregate scores for inflammation, crypt damage and ulcerations indicated less mucosal damage in A3−/−AR mice than WT mice with 1.5% or 3% DSS induced colitis. Data is summarized in Supplemental Table 1 and Figure 6. Data was analyzed a number of different ways, and significant differences were observed between A3−/−AR mice with DSS and WT mice with DSS if the average score of 3 regions (proximal, middle and distal colon) or the most severe score was used for comparison. Crypt damage or inflammation infiltrate scores were more reliable indicators of differences in histopathology between A3−/−AR mice and WT mice than ulceration. Representative examples of H&E histopathology for A3−/−AR and WT mice induced with DSS are shown in Figure 7.
Figure 6.
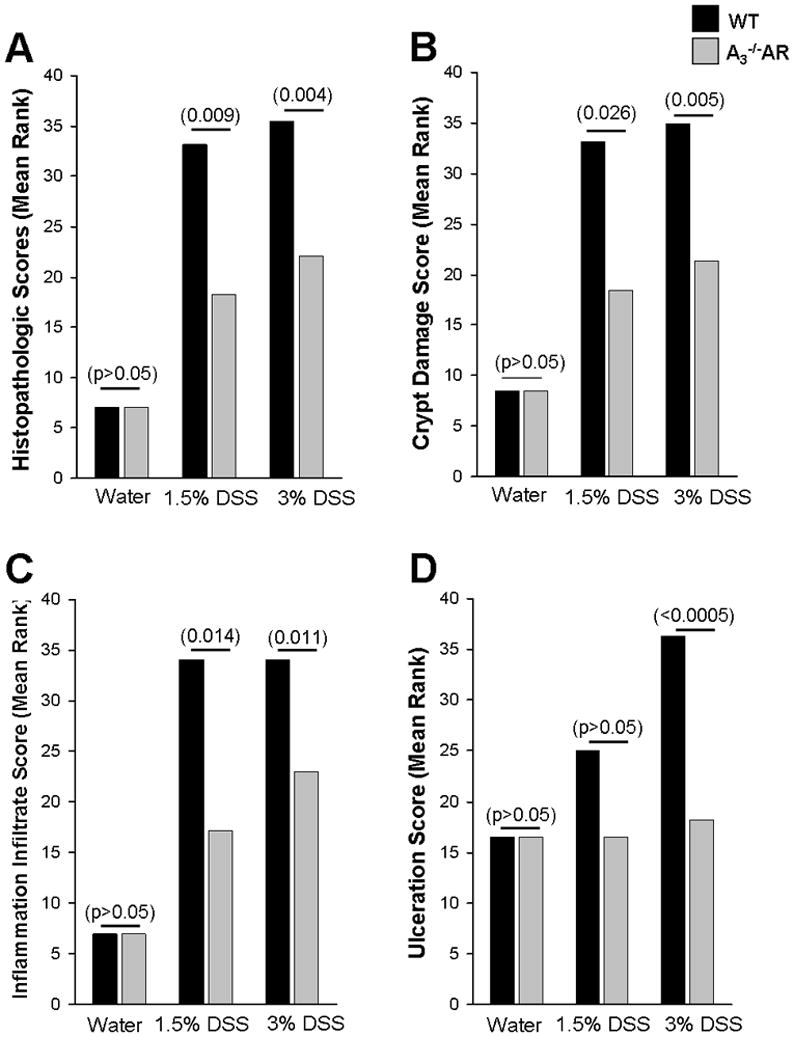
A3−/−AR knockout protects mice from gut inflammation and tissue injury. (A) Aggregate histopathologic scoring of inflammation, ulceration and crypt damage indicates protection. (B) Crypt damage score. (C) Inflammation infiltration score. (D) Ulceration score. A Mann Whitney non-parametric U-Test is used and p values are in parenthesis.
Figure 7.

Disruption of the A3AR gene reduces H&E histopathology in murine DSS colitis. (A) In WT (1, 2) or A3−/−AR (3, 4) there is no active inflammation, crypt, surface epithelial damage or ulceration. (B). WT 3% DSS with 1, transmural inflammation and crypt damage without ulceration (arrow); 2, 3, transmural active inflammation with crypt and surface epithelial damage and ulceration (arrow). (C) A3−/−AR 3% DSS with 1, focal active inflammation without crypt or surface epithelial damage or ulceration; 2, no active inflammation, crypt or surface epithelial damage or ulceration; 3, confluent active inflammation in submucosa with basal 2/3 crypt loss and epithelial damage without ulceration.
After DSS induction, WT animals lose weight over time with a maximum of 20–25% body weight lost within 8–10 days. After withdrawal of DSS from drinking water animals begin to recover and gain weight. At 3% DSS, A3−/−AR mice lost 5% body-weight compared to 20–25% in WT and recover faster than WT (Figure 8A). At 1.5% DSS, A3−/−AR knock-out prevents weight-loss and damage (Figure 8B).
Figure 8.
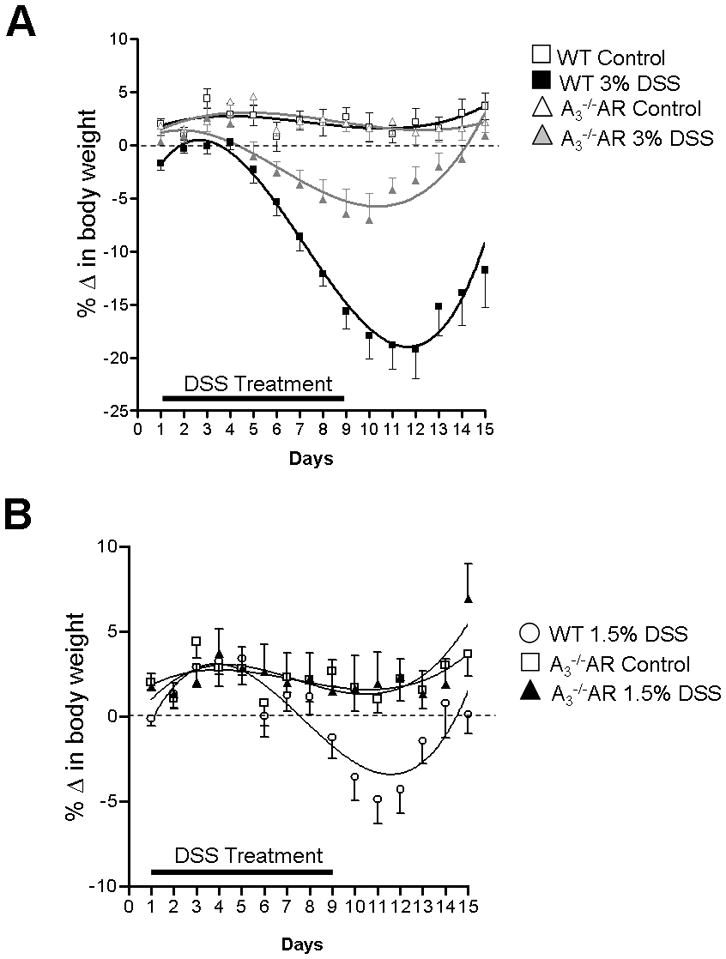
DSS-induced weight-loss is blocked by A3−/−AR knockout. (A) In animals receiving 3% DSS maximum weight loss is significantly reduced to 5% from 20% maximum weight loss. After 2 weeks animals recover completely only in A3−/−AR mice. Figure 8A, ANOVA between all groups, p<0.001; post tests, 3% DSS WT vs 3% DSS A3−/−AR, p<0.001; 3% DSS A3−/−AR vs WT control, p<0.05; 3% DSS WT vs WT control, p<0.001; 3%DSS WT vs A3−/−AR control, p<0.001; 3% DSS A3−/−AR vs A3−/−AR control, p<0.01). (B) At 1.5% DSS weight loss does not occur in A3−/−AR mice. A non-linear regression curve fit 3rd order polynomial is used to construct the curves in Prism. ANOVA, p=0.0005; 1.5% DSS WT vs 1.5% DSS A3−/−AR, p<0.01; 1.5%DSS A3−/−AR vs A3−/−AR control, p> 0.05. ANOVA followed by Newman-Keuls Multiple Comparison Test was applied to analyze different curves.
Stool consistency, diarrhea and guaiac test for occult blood
A3−/−AR knockout restores stool consistency, reduces diarrhea and occult blood. In WT 3% DSS colitis animals, diarrhea develops from days 5–11 and most animals develop diarrhea on days 7–9. By day 11 less than half of the animals have diarrhea. In contrast, A3−/−AR knockout animals induced with 3% DSS had normal stool consistency and no diarrhea (Figure 9A).
Figure 9.

A3−/−AR knockout restores stool consistency, reduces diarrhea and occult blood. (A) a, WT 3% DSS colitis animals develop diarrhea days 5–11whereas b, A3−/−AR knockout animals induced with 3% DSS have normal stool consistency (** Chi-square test comparing incidence of diarrhea between WT and A3−/−AR mice induced with DSS, p=0.001). (B) (a,b)A3−/−AR knockouts have lower incidence of occult blood than WT animals induced with 3% DSS (*, Chi-square test, WT and A3−/−AR, p=0.041). (C) Fewer pellets /day had a +Gaiac test in A3−/−AR compared to WT.
The Guaiac test is used to evaluate occult blood in stools of each animal separately. A positive Guaiac test is observed in most animals days 5–9 in WT controls induced with 3% DSS. However, A3−/−AR knockouts have lower incidence of occult blood than WT animals on day 9 after DSS induction. In WT animals 70% of animals still have a positive Guaiac test after 9 days compared to 10% of animals in A3−/−AR mice (Figure 9B). In another set of experiments, the Guaiac test was used to count the numbers of positive-pellets / cage (housing 4 animals) and used in the analysis. Group data provide similar results and show that A3−/−AR knock-out mice have lower incidence of occult blood in their stools after induction of colitis with 1.5% or 3% DSS. At 3% DSS colitis, A3−/−AR could only partially reduce the incidence of occult blood but could not prevent it. Fewer pellets /day had a positive Guaiac test in A3−/−AR compared to WT (Figure 9C).
Expression of A3AR in DSS colitis
Co-labeling experiments were done to determine whether inflammation/DSS colitis drives the expression of A3AR in murine colon – In DSS colitis there was no apparent change in expression of A3AR in s-100+, SYN+, CD4+ T-cells, mast cell tryptase+, 5-HT+ enterochromaffin or smooth muscle - cells in the gut wall. A3AR was not detected in CD4+ cells (<2%), MC tryptase+ cells or smooth muscle cells in either control or DSS animals (negative data not shown, data was analyzed by LSM confocal imaging software of pixel intensity). The expression of A3AR in crypt epithelial cells was reduced by nearly 4-fold in 3% DSS colitis (Supplemental Figure 3).
In addition, the numbers of CD4+ cells are reduced in A3−/−AR knockout in DSS colitis. Supplemental Table 2 summarizes the quantitative analysis between all groups in WT and A3−/−AR mice for numbers of CD4+ cells / 20 crypts. In WT mice DSS increases CD4+ cells in the lamina propria. In A3−/−AR mice, DSS does not elevate CD4+cells. Representative images of the distribution of CD4+-ir cells in WT and A3−/−AR mice are shown in Supplemental Figure 2.
DISCUSSION
The current study provides molecular proof for a role of A3AR in regulating in vivo motility using a functional A3−/−AR knockout mouse model. This is supported by data obtained from colonic emptying and transit studies using the in vivo bead latency test for expulsion of an artificial pellet, stool frequency, stool retention and intestinal transit of FITC-Dextran. Functional disruption of the A3−/−AR alters intestinal and colonic motility, fecal retention and evacuation. The A3−/−AR functional phenotype is characterized by a decrease in fecal retention that is associated with a reduction in frequency of fecal pellet evacuation. There is accelerated intestinal transit and in vivo artificial pellet expulsion occurred twice as fast in A3−/−AR mice compared to WT controls. Overall, disruption of A3AR facilitates transit and colonic emptying and promotes evacuation – this is consistent with the hypothesis that in the normal physiologic setting in vivo eADO release acts at low affinity A3AR to attenuate motility and the colonic evacuation reflex. A3AR activation is expected to have a constipating effect on the colon.
The expression of A3AR has been localized to enteric neurons in the guinea-pig, rat 32, human 2 or mouse colon. As shown previously in rat colon 32, the distal half of the transverse and descending mouse colon have higher expression of neural A3AR in myenteric plexus 32 – this is the functional part of the colon that is involved in mass movement and the defecation reflex. There is one important distinction however between mouse and rat A3AR distribution in the colon. In the mouse neural expression of A3 -immunoreactivity is exclusively localized to varicose nerve terminals in enteric ganglia. Presynaptic localization was confirmed by co-labeling of A3AR+ varicosities with SYN+ – immunoreactivity. It is likely that functional disruption of presynaptic inhibitory A3AR in the A3−/−AR mouse prevents eADO from acting on A3AR to attenuate motility or the defecation reflex. Our study did not test whether A3AR on SYN+ varicose terminals are indeed involved in inhibition of transmitter release. Pharmacological data with a selective A3 antagonist indicates that eADO can activate A3AR to suppress excitatory synaptic transmission 7 in human ENS. A potential caveat is that species differences may exist in A3AR distribution in the ENS between mouse (current study), rat, guinea-pig and human 1,7,32.
Experiments in A3−/−AR knockout mice in this study indicate that disruption of the A3AR protects mice from developing experimental DSS-colitis. That is, A3−/−AR transgenic mice (TG) are less sensitive than wild-type (WT) age-matched controls in developing DSS colitis. Weight-loss and recovery is a reliable and sensitive indicator of DSS-induced inflammation and injury. A3−/−AR mice are resistant to developing DSS-colitis and they recover faster than WT mice. A3AR mice can still develop inflammation but are more resistant to it. At 1.5% DSS, TG mice do not develop colitis or lose weight compared to WT mice with DSS.
A3−/−AR mice develop less histopathology than WT mice induced with DSS. The 3 parameters scored individually or in aggregate fashion were crypt damage, inflammation infiltrate and ulceration – according to these criteria, there was less mucosal / crypt damage and inflammatory infiltration in A3−/−AR than WT. Protection was also associated with a reduction in neutrophil infiltration as indicated by a decrease in MPO activity in A3−/−AR. Quantitative analysis indicates that in A3−/−AR knockout mice induced with DSS there are relatively few or no CD4+ T-cells in the lamina propria compared to WT-DSS mice. It has been suggested that unrestrained activation of CD4+ T cells is a mechanism of protraction of IBD 37. A3−/−AR mice induced with DSS have less occult blood in their stools and they do not have any diarrhea unlike their WT counterparts. Transgenic mice are also partially protected against shortening of the colon and change in splenic weight that occur in DSS colitis.
All together, it is concluded that A3−/−AR KO mice are resistant to developing DSS-induced mucosal inflammation and tissue injury. In A3−/−AR KO mice eADO can no longer activate A3 AR and hence mice are protected from developing colitis. A3AR activation in WT mice contributes to the development of gut inflammation, mucosal damage and disease progression. This is an unexpected finding and one that could not easily be predicted from previous studies using oral administration of the A3 agonist IB-MECA.17,18 If eADO release and chronic activation of low affinity A3AR (or A2BAR in other studies) contributes to the development of DSS colitis as suggested by our findings, then the mechanism of A3 protection is likely to be more complicated than a simple interaction and activation of the A3AR.17,18
Overall, the prototypical A3AR agonist IB-MECA has some protective effects in rodent models of colitis, and this at first glance does not fit with our findings in A3−/−AR mice in the current study. Data on A3−/−AR mice and in vivo administration of IB-MECA seem to be diametrically opposed. A number of possible reasons are forwarded to explain these results. First of all, effects with in vivo IB-MECA in previous studies occurred at doses of 1–3 mg/Kg/day that are likely not restricted to actions at A3AR and both high affinity (A1, A2A) and low affinity (A2B and A3AR) receptors may be activated by oral A3AR drugs. It is plausible that a non-A3 receptor mediates effects of IB-MECA in vivo. The pharmacology is complex since all 4 adenosine receptors are potential therapeutic targets in experimental models of IBD and agonist drugs (A1,A2A,A3), antagonist drugs (A2B) or drugs that modulate eADO levels (e.g. ADO KINASE or ADO DEAMINASE inhibitors, methotrexate or sulfasalazine) may have protective or therapeutic effects in colitis models or other inflammatory diseases. 2,18,21,23,25,38,39
Secondly, there may be differences in sensitivity to IB-MECA in different models of IBD. In this regard, oral administration of 1.5mg/Kg IB-MECA (b.i.d.) for 7 days protects animals from developing TNBS-induced colitis that has features of CD18. To a lesser extent, IB-MECA was also reported to have protective effects in DSS–induced colitis, a model that more closely resembles UC, as well as in spontaneous colitis induced in IL-10 knockout mice 17 . IB-MECA was more effective in preventing weight-loss, histopathology and other abnormalities induced by TNBS colitis compared to DSS colitis suggesting a differential sensitivity to IB-MECA in these models of IBD; in the DSS model, IB-MECA was marginally effective in reducing weight-loss at 1.5mg/Kg (b.i.d) and had no effect at 0.5mg/Kg (b.i.d) although it was effective in reducing other more sensitive indicators of inflammation (i.e. MPO, cytokine production). Therefore, its effects in DSS colitis are modest in protecting against damage and our findings in A3−/−AR mice suggest that a straight forward A3AR activation mechanism does not explain the protection. In the current study, A3−/−AR knockout was very effective in protecting the gut against mucosal damage and clinical pathology in contrast to high dose IB-MECA treatment. Furthermore, activation of the high affinity A2AAR by high doses of IB-MECA is conceivable in previous studies that have protective effects 19–22; such protective effects could also be offset by activation of low affinity A2BAR 23 by IB-MECA.
Third, not all effects at A3AR are protective 27–29. In some studies effects of A3AR activation are pro-inflammatory and are detrimental rather than protective. For instance, intense activation of the A3AR is pro-apoptotic 40 and adenosine A3R stimulation exacerbates global ischemia in gerbils 27 or seizures 41. A later study by the same group however, showed that cerebroprotection or damage (i.e. increase in infarct size) may be dependent on when IB-MECA is administered (i.e. before or after ischemia) 28.
Fourth, A3AR activation can contribute to pro-inflammatory/immune responses (mast cells) 33- and if eADO during inflammation is high enough, it could cause A3AR activation on mast cells, and degranulation of mast cells to release various products 42,43 and cause a net pro-inflammatory effect. This may not be the case since MC tryptase+ mast cells in the mouse do not appear to express A3AR.
Fifth, alterations in the PI3K-NF-kB signal transduction pathway has been shown to be involved in the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis 25 – Extended life-span of inflammatory cells is believed to involve inhibition of apoptosis due to stimulation of the PI3K pathway that leads to activation of PKB/Akt. This in turn phosphorylates proteins that cause failure to induce apoptosis. Giving IB-MECA in that study caused down regulation of A3AR and the initiation of a molecular mechanism that involves de-regulation of the PI3K-NF-kB signaling pathway – this resulted in a potent anti-inflammatory effect that resulted in protection. Analogous to this, A3−/−AR knockout mimics the consequences of chronic A3AR activation by IB-MECA to down-regulate the A3AR on inflammatory cells, leading to a similar mechanism of protection and amelioration of the inflammatory process. Such a mechanism would protect against DSS colitis. If so, molecular changes in the pathway of A3−/−AR mice with DSS afford protection. It remains unknown whether chronic A3AR in vivo agonist administration leads to A3AR down-regulation and protection. It cannot be ruled out whether knockout of A3R leads to compensations in A2AAR (protect against colitis) or A2BAR (exacerbate colitis) or enzymes involved in eADO metabolism 23,44–46. A potential caveat is that heterozygote A3 knockouts were not available for breeding, and although we took steps to insure the microbial communities were essentially the same, studies in littermate controls bred from heterozygotes in the same cages would be better.
A2BAR activation by eADO contributes to development of gut colitis and A2B−/−AR knockout mice protect against colitis 23. Therefore, knockout of either of the low affinity adenosine receptors (i.e. A3−/−AR and A2B−/−AR) protects mice from developing colitis. However, these two low affinity receptors can be clearly distinguished by the opposite effect they mediate on motility. A2B−/−AR knockout has a constipating effect 23,44 in contrast to the effect of A3−/−AR knockout that accelerates motility/transit and evacuation (our study). It has been suggested that stimulation of A2BAR could represent a novel strategy to treat constipation. In contrast, one would expect that blockade or down-regulation of A3AR in the ENS 7,32 could offer yet another novel strategy to treat constipation. These low affinity adenosine receptors, A2B and A3AR are potential novel targets in motility disorders such as IBS, IBD, and perhaps various developmental motility disorders in children.
ADO A3 ARs have a wide range of physiological and disease-related effects with promise for treatment of colorectal cancer and inflammation 15,47–49. IB-MECA is in Phase IIb clinical trials for rheumatoid arthritis. Compounds like ADO kinase inhibitors show promise in mouse models of colitis 16,17 but whether blockade of conversion of ADO to AMP is the mechanism remains unknown. Our study in A3−/−AR KO mice raises the question of the role of eADO in gut inflammation. Studies in A2B−/− KO mice indicate protection further raising the question of the detrimental effects of eADO under certain circumstances. Available data suggest that eADO release from inflamed gut cells is likely to be sufficient to activate low affinity A3AR and A2BAR 6,7,9–13,32 that could cause detrimental effects. A3AR is expressed in glial cells in both the murine and rat ENS. The significance of this novel finding is not yet clear, but the expression of the A3 AR in glia suggests a role in glial function. Neural-glial communication occurs in both rodent 51 and human ENS 52 and involves purine nucleotides. This extends those findings to adenosine, a metabolite of purine nucleotide breakdown (i.e. ATP, ADP or AMP). Glial cells also serve an important role in immune-neural communication in inflammatory conditions of the gut ENS and CNS 53, 54 and glial expression of the A3AR suggests a novel role for the receptor in ENS.
Our colitis study was not designed or intended to test whether the A3AR−/− colitis/protection phenotype is motility-dependent. Definitive data on the inter-relationship between motility and colitis in animals or humans is lacking. In situations of an infection like cholera toxin or c.dificile (TxA) increase in motility would serve to enhance transport of fluid in an aboral direction in an effort to expel a noxious stimulus and alleviate the problem2. Abnormal motility55 and diarrheal states do occur56 in both IBD and IBS (diarrhea - predominant IBS) but the inter-relationship between the two is not clear. Diarrhea is the most debilitating symptom of UC, and is blocked by A3−/−AR knockout in DSS colitis. Its pathogenesis is presumed to involve changes in colonic motility and transit, leakage of fluid across the inflamed mucosa and abnormal ion transport mechanisms.57
Inflammation can drive A3AR expression and cause up-regulation in peripheral blood mononuclear cells (PBMCs) in rheumatoid arthritis, psoriasis and Crohn’s Disease (CD)58; other studies don’t show this39 A3AR is upregulated in colonic extracts from TNBS colitis animals18 and in mucosa of patients with human colorectal cancer59.Up-regulation also occurs in lung-tissue from patients with asthma or COPD60,61. A3AR up-regulation is involved in the anti-inflammatory mechanism of glucocorticoids62. Co-labeling experiments tested whether inflammation/DSS colitis drives the expression of A3AR in murine colon – In DSS colitis there was no apparent change in expression of A3AR in s-100+, SYN+, CD4+ T-cells, mast cell tryptase+, 5-HT+ enterochromaffin or smooth muscle - cells in the gut wall. Therefore inflammation does not seem to drive A3AR expression in these cells. However, the expression of A3AR in crypt epithelial cells was reduced by nearly 4-fold in DSS colitis. This may be of clinical relevance since the A3AR transcript is down-regulated in mucosal biopsies from patients with Crohn’s Disease (Rybaczyk et al, 2009). The function of A3AR in secretion is unknown, but further studies are warranted on A3AR down-regulation in the pathophysiology of colitis.
CONCLUSIONS
In conclusion, our study provides evidence to support the hypotheses that disruption of the A3AR has two important consequences (1) to alter intestinal motility and (2) protect against DSS–colitis. Therefore A3AR is involved in these actions. Whether the A3AR−/− colitis / protection phenotype is motility-dependent is left to future studies. Our findings may be important in a number of ways: First of all, data suggest that activation of presynaptic A3AR by eADO attenuates the evacuation reflex and slows-down intestinal transit, colonic emptying and mass movement in the mouse colon. It is postulated that the A3AR is a novel target for motility disorders, where A3AR antagonists could be tested as potential drugs for constipation. Secondly, glial A3AR expression suggests an important new role for adenosine in gliotransmission and neuroglial communication. Thirdly, our findings challenge the current paradigm on use of an A3AR agonist as a therapeutic target in colitis – and are consistent with the possibility that activation of A3AR by eADO may contribute to development of DSS-colitis. Further studies on the molecular mechanism of action of A3AR and effects of A3 AR agonists or antagonists can further test these novel hypotheses.
Supplementary Material
Molecular identification of the A3−/−AR transgene and expression of A3AR. (A) The transgene was only identified in the A3−/−AR transgenic founders (T1–T6) and was absent in wild-type (WT) age matched control mouse founders. PCR was run using specific 5’ and 3’ primers for the PGKneo plasmid insert which was used to disrupt the function of the A3AR gene. (B) Western blot analysis with a goat anti-A3AR antibody from Santa Cruz identified a 66 kDa immunogenic band for the A3AR. (C) A 66 kDa band was revealed by a rabbit anti-A3 antibody from Alpha Diagnostics but additional lower molecular weight bands were also evident. (D) The expression of A3AR in the A3−/−AR mouse was 1 fold greater than in WT mice.
CD4+ T-cells are reduced by A3−/−AR knockout in DSS colitis. (A) Representative immunohistochemical image of WT control. CD4+-ir cells are rare in the lamina propria (arrows). (B) CD4+-ir cells in A3−/−AR. (C) Abundance of CD4+ cells in 1.5% DSS colitis. (D) A3−/−AR almost prevents infiltration of CD4+ cells after induction of DSS colitis. (E). Pooled results for CD4+ cell infiltration. A3−/−AR reduces CD4+ cell infiltration.
Inflammation causes down-regulation of A3AR expression in mucosal crypt cells. Expression of A3AR in crypt epithelial cells is reduced by nearly 4-fold in DSS colitis. The crypt number was not significantly different between WT and DSS (p>0.05); crypt number counted in WT: 212.8±66.1; WT+1.5% DSS: 181.8±38.2; WT+3%DSS: 256.8±83.0. The crypt number represents average crypt number of three 1 cm2 sections / colon (proximal, middle, distal colon).
Acknowledgments
This work is supported by National Institutes of Health (NIH) Grant RO1 DK44179-15 and NIH ARRA supplement and NCRR 1S10RR11434 to F.L. Christofi.
References
- 1.Christofi FL. Unlocking mysteries of gut sensory transmission: is adenosine the key? News Physiol Sci. 2001;16:201–207. doi: 10.1152/physiologyonline.2001.16.5.201. [DOI] [PubMed] [Google Scholar]
- 2.Christofi FL. Purinergic recepors and gastrointestinal secretomotor function. Purinergic Signalling. 2008;4:213–236. doi: 10.1007/s11302-008-9104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dixon AK, Gubitz AK, Sirinathsinghji DJ, et al. Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol. 1996;118(6):1461–1468. doi: 10.1111/j.1476-5381.1996.tb15561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christofi FL, Zhang H, Yu JG, Guzman J, et al. Differential gene expression of adenosine A1, A2a, A2b, and A3 receptors in the human enteric nervous system. J Comp Neurol. 2001;439(1):46–64. doi: 10.1002/cne.1334. [DOI] [PubMed] [Google Scholar]
- 5.Deshpande NA, McDonald TJ, Cooke MA. Endogenous interstitial adenosine in isolated myenteric neural networks varies inversely with prevailing P02. Am J Physiol Gastrointest Liver Physiol. 1999;276:G875–G885. doi: 10.1152/ajpgi.1999.276.4.G875. [DOI] [PubMed] [Google Scholar]
- 6.Christofi FL, Kim M, Wunderlich JE, et al. Endogenous adenosine differentially modulates 5-hydroxytryptamine from a human enterochromaffin cell model. Gastroenterology. 2004;127:188–202. doi: 10.1053/j.gastro.2004.04.070. [DOI] [PubMed] [Google Scholar]
- 7.Wunderlich JE, Needleman BJ, Chen Z, et al. Dual purinergic synaptic transmission in the human enteric nervous system. Am J Physiol Gastrointest Liver Physiol. 2008;294(2):G554–G566. doi: 10.1152/ajpgi.00500.2007. [DOI] [PubMed] [Google Scholar]
- 8.Baggott JE, Morgan SL, Ha TS, et al. Antifolates in rheumatoid arthritis: a hypothetical mechanism of action. Clin Exp Rheumatol. 1993;11:S101–S105. [PubMed] [Google Scholar]
- 9.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50(3):413–492. [PubMed] [Google Scholar]
- 10.Martin C, Leone M, Viviand X, et al. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med. 2000;28(9):3198–3202. doi: 10.1097/00003246-200009000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Sperlagh B, Doda M, Baranyi M, et al. Ischemic-like condition releases norepinephrine and purines from different sources in superfused rat spleen strips. J Neuroimmunol. 2000;111(1–2):45–54. doi: 10.1016/s0165-5728(00)00365-9. [DOI] [PubMed] [Google Scholar]
- 12.Sottofattori E, Anzaldi M, Ottonello L. HPLC determination of adenosine in human synovial fluid. J Pharm Biomed Anal. 2001;24(5–6):1143–1146. doi: 10.1016/s0731-7085(00)00574-4. [DOI] [PubMed] [Google Scholar]
- 13.Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25(1):33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Cronstein BN, Naime D, Ostad E. The anti-inflammatory mechanism of methotrexate: increased adenosine release at inflamed sites diminished leukocyte accumulation in an in vivo model of inflammation. J Clin Invest. 1993;92:2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cronstein B, Naime D, Firestein G. The anti-inflammatory effects of an adenosine kinase inhibitor are mediated by adenosine. Arthritis & Rheumatism. 1995;38:1040–1045. doi: 10.1002/art.1780380804. [DOI] [PubMed] [Google Scholar]
- 16.Siegmund B, Rieder F, Albrich S, et al. Adenosine kinase inhibitor GP515 improves experimental colitis in mice. JPET. 2001;296:99–105. [PubMed] [Google Scholar]
- 17.Mabley J, Soriano F, Pacher P, et al. The adenosine A3 receptor agonist, N6-(3-iodobenyl)-adenosine-5’-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–329. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 18.Guzman J, Yu JG, Suntres Z, et al. ADOA3R as a therapeutic target in experimental colitis: proof by validated high-density oligonucleotide microarray analysis. Inflamm Bowel Dis. 2006;12(8):766–789. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 19.Antonioli L, Fornai M, Colucci R, et al. Pharmacological modulation of adenosine system: novel options for treatment of inflammatory bowel diseases. Inflamm Bowel Dis. 2008;14:566–574. doi: 10.1002/ibd.20316. [DOI] [PubMed] [Google Scholar]
- 20.Cavalcante IC, Castro MV, Barreto AR, et al. Effect of novel A2A adenosine receptor agonist ATL 313 on Clostridium difficile toxin A-induced murine ileal enteritis. Infect Immun. 2006;74(5):2606–2612. doi: 10.1128/IAI.74.5.2606-2612.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naganuma M, Wiznerowicz EB, Lappas CM, et al. Cutting edge: critical role for A2A adenosine receptors in the T-cell- mediated regulation of colitis. J Immunol. 2006;177(5):2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 22.Odashima M, Bamias G, Rivera-Nieves J, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129 (1):26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 23.Kolachala VL, Vijay-Kumar M, Dalmasso G, et al. A2B adenosine receptor gene deletion attenuates murine colitis. Gastroenterology. 2008;135(3):861–870. doi: 10.1053/j.gastro.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Troostenburg AR, Clark EV, Carey WD, et al. Tolerability, pharmacokinetics and concentration–dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int J Clin Pharmacol Ther. 2004;42(10):434–442. doi: 10.5414/cpp42534. [DOI] [PubMed] [Google Scholar]
- 25.Fishman P, Bar-Yehuda S, Madi L, et al. The PI3K-NF-kappaB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther. 2006;8(1):R33. doi: 10.1186/ar1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo Y, Bolli R, Bao W, et al. Targeted deletion of the A3 adenosine receptor confers resistance to myocardial ischemic injury and does not prevent early preconditioning. J Mol Cell Cardiol. 2001;33(4):825–830. doi: 10.1006/jmcc.2001.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Von Lubitz DK, Lin RC, Popik P, et al. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263(1–2):59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Von Lubitz DK, Simpson KL, Lin RC. Right thing at a wrong time? Adenosine A3 receptors and cerebroprotection in stroke. Ann N Y Acad Sci. 2001;939:85–96. doi: 10.1111/j.1749-6632.2001.tb03615.x. [DOI] [PubMed] [Google Scholar]
- 29.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. Protective roles of adenosine A1, A2A, and A3 receptors in skeletal muscle ischemia and reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;293(6):H3685–6891. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooke HJ, Javed N, Christofi FL. Enteric neural reflexes and secretion. In: Goetzl EJ, Blennerhassett MG, Bienestock J, editors. Autonomic Nervous System, Autonomic Neuroimmunology. Vol. 16. Reading, UK: Hardwood Academic; 2003. pp. 35–59. [Google Scholar]
- 31.De Man JG, Seerden TC, De Winter BY, et al. Alteration of the purinergic modulation of enteric neurotransmission in the mouse ileum during chronic intestinal inflammation. Br J Pharmacol. 2003;139:172–184. doi: 10.1038/sj.bjp.0705218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bozarov A, Wang YZ, Yu JG, et al. Activation of adenosine low-affinity A3 receptors inhibits the enteric short interplexus neural circuit triggered by histamine. Am J Physiol Gastrointest Liver Physiol. 2009;297(6):G1147–G1162. doi: 10.1152/ajpgi.00295.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salvatore CA, Tilley SL, Latour AM, et al. Disruption of the A(3) adenosine receptor gene in mice and its effect on stimulated inflammatory cells. J Biol Chem. 2000;275(6):4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 34.Cooke HJ, Xue J, Yu JG, et al. Mechanical stimulation releases nucleotides that activate P2Y1 Rs to trigger neural reflex chloride secretion in guinea pig distal colon. J Comp Neurol. 2004;469:1–15. doi: 10.1002/cne.10960. [DOI] [PubMed] [Google Scholar]
- 35.Castaneda FE, Walia B, Vijay-Kumar M, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129(6):1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 36.Miller MS, Galligan JJ, Burks TF. Accurate measurement of intestinal transit in the rat. J Pharmacol Methods. 1981;6(3):211–217. doi: 10.1016/0160-5402(81)90110-8. [DOI] [PubMed] [Google Scholar]
- 37.Kabashima K, Saji T, Murata T, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109(7):883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cremon C, Gargano L, Morselli-Labate AM, et al. Mucosal immune activation in irritable bowel syndrome: gender-dependence and association with digestive symptoms. Am J Gastroenterol. 2009;104(2):392–400. doi: 10.1038/ajg.2008.94. [DOI] [PubMed] [Google Scholar]
- 39.Rybaczyk L, Rozmiarek A, Circle K, et al. A new bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm Bowel Dis. 2009;15(7):971–984. doi: 10.1002/ibd.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Appel E, Kazimirsky G, Ashkenazi E, et al. Roles of BCL-2 and caspase 3 in the adenosine A3 receptor-induced apoptosis. J Mol Neurosci. 2001;7(3):285–292. doi: 10.1385/JMN:17:3:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Von Lubitz DK, Carter MF, Deutsch SI, et al. The effects of adenosine A3 receptor stimulation on seizures in mice. Eur J Pharmacol. 1995;275(1):23–29. doi: 10.1016/0014-2999(94)00734-o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith SR, Denhardt G, Terminelli C. A role for histamine in cytokine modulation by the adenosine A(3) receptor agonist, 2-Cl-IB-MECA. Eur J Pharmacol. 2002;457(1):57–69. doi: 10.1016/s0014-2999(02)02645-6. [DOI] [PubMed] [Google Scholar]
- 43.Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem. 1993 Aug 15;268(23):16887–16890. [PubMed] [Google Scholar]
- 44.Chandrasekharan BP, Kolachala VL, Dalmasso G, et al. Adenosine 2B receptors (A(2B)AR) on enteric neurons regulate murine distal colonic motility. FASEB J. 2009;23(8):2727–2734. doi: 10.1096/fj.09-129544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart ML, Much C, Gorzolla IC, et al. Extracellular adenosine production by ecto-5'-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology. 2008;135(5):1739–1750. doi: 10.1053/j.gastro.2008.07.064. [DOI] [PubMed] [Google Scholar]
- 46.Beldi G, Wu Y, Sun X, et al. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology. 2008;135(5):1751–1760. doi: 10.1053/j.gastro.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Firestein G, Boyle D, Bullough D, et al. Protective effect of an adenosine kinase inhibitor in septic shock. J Immunol. 1994;152:5853–5859. [PubMed] [Google Scholar]
- 48.Marak GE, Jr, de Kozak Y, Faure JP, et al. Pharmacologic modulation of acute ocular inflammation. I. Adenosine. Ophthalmic Res. 1988;20:220–226. doi: 10.1159/000266646. [DOI] [PubMed] [Google Scholar]
- 49.Rosengren S, Bong G, Firestein GS. Anti-inflammatory effects of an adenosine kinase inhibitor: decreased neutrophil accumulation and vascular leakage. J Immunol. 1995;154:5444–5451. [PubMed] [Google Scholar]
- 50.Cronstein BN, Van de Strouwe M, Druska L, et al. Nonsteroidal anti-inflammatory agents inhibit stimulated neutrophil adhesion to endothelium: adenosine dependent and independent mechanisms. Inflammation. 1994;18:323–335. doi: 10.1007/BF01534273. [DOI] [PubMed] [Google Scholar]
- 51.Sharkey KA, Kroese A. Consequences of intestinal inflammation on the enteric nervous system: Neuronal activation induced by inflammatory mediators. The Anat Rec. 2001;262:79–90. doi: 10.1002/1097-0185(20010101)262:1<79::AID-AR1013>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 52.Wunderlich JE, Ren T, Needleman BJ, et al. Novel Purinergic P1, P2X and P2Y Receptor Signaling Targets in the Human Enteric Nervous System and Glia. Gastroenterology. 2009;136(Suppl 1):A-3. [Google Scholar]
- 53.Abbracchio MP, Burnstock G, Verkhratsky A, et al. Purinergic Signalling in the Nervous System: An Overview. Trends in Neuroscience. 2008;32(1):19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Rühl A, Nasser Y, Sharkey KA. Enteric glia. Neurogastroenterol Motil. 2004;16(Suppl 1):44–49. doi: 10.1111/j.1743-3150.2004.00474.x. [DOI] [PubMed] [Google Scholar]
- 55.Camilleri M. Motor function in irritable bowel syndrome. Can J Gastroenterol. 1999;13 (Suppl A):8A–11A. doi: 10.1155/1999/240329. [DOI] [PubMed] [Google Scholar]
- 56.Hansen MB. The enteric nervous system II: gastrointestinal functions. Pharmacol Toxicol. 2003;92(6):249–257. doi: 10.1034/j.1600-0773.2003.920601.x. [DOI] [PubMed] [Google Scholar]
- 57.Seidler U, Lenzen H, Cinar A, et al. Molecular mechanisms of disturbed electrolyte transport in intestinal inflammation. Ann N Y Acad Sci. 2006;1072:262–275. doi: 10.1196/annals.1326.024. [DOI] [PubMed] [Google Scholar]
- 58.Ochaion A, Bar-Yehuda S, Cohen S, et al. The anti-inflammatory target A(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn's disease. Cell Immunol. 2009;258(2):115–122. doi: 10.1016/j.cellimm.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 59.Gessi S, Cattabriga E, Avitabile A, et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin Cancer Res. 2004;10(17):5895–5901. doi: 10.1158/1078-0432.CCR-1134-03. [DOI] [PubMed] [Google Scholar]
- 60.Spruntulis LM, Broadley KJ. A3 receptors mediate rapid inflammatory cell influx into the lungs of sensitized guinea-pigs. Clin Exp Allergy. 2001;31:943–951. doi: 10.1046/j.1365-2222.2001.01087.x. [DOI] [PubMed] [Google Scholar]
- 61.Walker BAM, Jacobson MA, Knight DA, et al. Adenosine A3 receptor expression and function in eosinophils. Am J Respir Cell Mol Biol. 1997;16:531–537. doi: 10.1165/ajrcmb.16.5.9160835. [DOI] [PubMed] [Google Scholar]
- 62.Barczyk K, Ehrchen J, Tenbrock K, et al. Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood. 2010;116(3):446–455. doi: 10.1182/blood-2009-10-247106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Molecular identification of the A3−/−AR transgene and expression of A3AR. (A) The transgene was only identified in the A3−/−AR transgenic founders (T1–T6) and was absent in wild-type (WT) age matched control mouse founders. PCR was run using specific 5’ and 3’ primers for the PGKneo plasmid insert which was used to disrupt the function of the A3AR gene. (B) Western blot analysis with a goat anti-A3AR antibody from Santa Cruz identified a 66 kDa immunogenic band for the A3AR. (C) A 66 kDa band was revealed by a rabbit anti-A3 antibody from Alpha Diagnostics but additional lower molecular weight bands were also evident. (D) The expression of A3AR in the A3−/−AR mouse was 1 fold greater than in WT mice.
CD4+ T-cells are reduced by A3−/−AR knockout in DSS colitis. (A) Representative immunohistochemical image of WT control. CD4+-ir cells are rare in the lamina propria (arrows). (B) CD4+-ir cells in A3−/−AR. (C) Abundance of CD4+ cells in 1.5% DSS colitis. (D) A3−/−AR almost prevents infiltration of CD4+ cells after induction of DSS colitis. (E). Pooled results for CD4+ cell infiltration. A3−/−AR reduces CD4+ cell infiltration.
Inflammation causes down-regulation of A3AR expression in mucosal crypt cells. Expression of A3AR in crypt epithelial cells is reduced by nearly 4-fold in DSS colitis. The crypt number was not significantly different between WT and DSS (p>0.05); crypt number counted in WT: 212.8±66.1; WT+1.5% DSS: 181.8±38.2; WT+3%DSS: 256.8±83.0. The crypt number represents average crypt number of three 1 cm2 sections / colon (proximal, middle, distal colon).