

Abstract
Objective
MicroRNA (miRNA) are recognized as important regulators of a variety of fundamental biologic processes. Previously, we described increased expression of miR-155 and miR-146a in rheumatoid arthritis (RA) and showed a repressive effect of miR-155 on matrix metalloproteinase (MMP) expression in RA synovial fibroblasts (RASFs). The present study was undertaken to examine alterations in expression of miR-203 in RASFs and analyze its role in fibroblast activation.
Methods
Differentially expressed miRNA in RASFs versus osteoarthritis synovial fibroblasts (OASFs) were identified by real-time polymerase chain reaction (PCR)–based screening of 260 individual miRNA. Transfection of miR-203 precursor was used to analyze the function of miR-203 in RASFs. Levels of interleukin-6 (IL-6) and MMPs were measured by real-time PCR and enzyme-linked immunosorbent assay. RASFs were stimulated with IL-1β, tumor necrosis factor α (TNFα), lipopolysaccharide (LPS), and 5-azacytidine (5-azaC). Activity of IκB kinase 2 was inhibited with SC-514.
Results
Expression of miR-203 was higher in RASFs than in OASFs or fibroblasts from healthy donors. Levels of miR-203 did not change upon stimulation with IL-1β, TNFα, or LPS; however, DNA demethylation with 5-azaC increased the expression of miR-203. Enforced expression of miR-203 led to significantly increased levels of MMP-1 and IL-6. Induction of IL-6 by miR-203 overexpression was inhibited by blocking of the NF-κB pathway. Basal expression levels of IL-6 correlated with basal expression levels of miR-203.
Conclusion
The current results demonstrate methylation-dependent regulation of miR-203 expression in RASFs. Importantly, they also show that elevated levels of miR-203 lead to increased secretion of MMP-1 and IL-6 via the NF-κB pathway and thereby contribute to the activated phenotype of synovial fibroblasts in RA.
MicroRNA (miR) are ~21-nucleotide-long RNA molecules that regulate the expression of target genes by translational inhibition or messenger RNA (mRNA) degradation (1-3). Expression of a distinct protein can be changed by several different miRNA. In turn, one miRNA can modulate the expression of hundreds of different targets (4). In the last few years it has become clear that miRNA are indispensible for the proper functioning of the mechanisms regulating cell cycle and cellular death, cellular differentiation, and immunity (5,6). Thus, it is not surprising that dysregulated expression of miRNA is being found in an increasing number of pathologic conditions, such as cancer, cardiovascular disease, infectious disease, and autoimmunity (7,8).
Some up-regulated and down-regulated miRNA have also been identified in rheumatoid arthritis (RA). In particular, we and others have found increased expression of miR-155 in synovial fibroblasts, peripheral blood mononuclear cells (PBMCs), synovial tissue, and synovial fluid monocytes of RA patients (9,10). Of note, enforced in vitro expression of miR-155 led to decreased levels of matrix metalloproteinase 3 (MMP-3) and blocked the induction of MMP-1 and MMP-3 after stimulation with proinflammatory cytokines and Toll-like receptor (TLR) ligands (10). These data show that MMP-1 and MMP-3 are indirect targets of miR-155, but also reveal a more complex picture of miRNA regulation in RA, with increased expression of counterregulatory mechanisms. In contrast to miR-155, miR-124 was recently reported to be down-regulated in RA synovial fibroblasts (RASFs) (11). In vitro overexpression of miR-124 by transfection with its precursor (pre–miR-124) lowered the production of cyclin-dependent kinase 2 and monocyte chemoattractant protein 1. Another microRNA, miR-146, was found to be elevated in RA synovial tissue, synovial fibroblasts, and PBMCs in 3 independent studies including one from our group (9,10,12). However, functional consequences of miR-146 overexpression and direct targets of miR-146 that are affected in RA have not been identified to date.
To further elucidate the nature of miRNA dys-regulation in RA, in the current study we compared the miRNA expression profiles of synovial fibroblasts from RA patients and osteoarthritis (OA) patients and found miR-203 to be up-regulated in RA. We further studied its regulation by proinflammatory molecules and epigenetic mechanisms, and found evidence to support previous reports of the regulation of miR-203 expression via DNA methylation (13-16). Finally, we found that overexpression of miR-203 results in increased secretion of interleukin-6 (IL-6) and MMP-1 by RASFs, identifying miR-203 as a proinflammatory and joint-destructive factor in RA.
PATIENTS AND METHODS
Tissue specimens and cells
Synovial tissue was obtained from RA patients, OA patients, and joint trauma patients (healthy control specimens) undergoing joint replacement surgery in the Schulthess Clinic Zurich. Informed consent was obtained from all patients, and RA patients fulfilled the American College of Rheumatology criteria for classification of the disease (17). For tissue analysis, tissue was snap-frozen and stored at −80°C. For cell culture, tissue was digested and synovial fibroblasts grown as previously described (10). Synovial tissue samples from patients with very early RA were obtained from patients presenting to a rapid-access early arthritis clinic who had synovitis of at least 1 joint and a symptom duration of <3 months (n = 5). Consenting patients underwent ultrasound-guided biopsy of a knee or ankle joint at baseline, with synovial fibroblasts grown from a minimum of 8 tissue biopsy specimens per joint to account for heterogeneity. Samples were used from patients who fulfilled the American College of Rheumatology criteria for the classification of RA within the subsequent 18 months.
Reagents
Tumor necrosis factor α (TNFα; R&D Systems) was used at 10 ng/ml, interleukin-1β (IL-1β; R&D Systems) was used at 1 ng/ml, and lipopolysaccharide (LPS) from Escherichia coli (List Biological Laboratories) was used at 100 ng/ml; 5-azacytidine (5-azaC; Sigma-Aldrich) was added to cell culture medium at 1 μM every day for 1 week. Activity of IκB kinase 2 (IKK-2) was inhibited with the IKK-2 inhibitor SC-514 (Sigma-Aldrich). SC-514 was added at a concentration of 50 nM 12 hours after transfection, and supernatants were collected after a further 12 hours.
Real-time polymerase chain reaction (PCR)
For screening of miRNA expression in RASFs and OASFs, a human miRNA panel (Ambion–Applied Biosystems) consisting of 260 assays for individual miRNA was applied. For miRNA analysis, RNA was isolated from fibroblasts and synovial tissue with the mirVana Isolation Kit (Ambion–Applied Biosystems). RNA (10 ng) was reverse-transcribed using MultiScribe reverse transcriptase (RT), RT buffer, dNTPs, RNase inhibitor, and miR-specific primers in the GeneAmp 9700 PCR system (all from Applied Biosystems). The complementary DNA (cDNA) obtained was applied in real-time PCR using miR-specific TaqMan primers (Applied Biosystems). For relative quantification, expression of let-7a was used as an endogenous control. In the analysis of the expression of IL-6 and MMPs, RNA was isolated with the RNeasy Mini kit (Qiagen) including DNA digestion with RNase-free DNA. Total RNA was reverse-transcribed using MultiScribe RT, RT buffer, dNTPs, RNase inhibitor, and random hexamers (all from Applied Biosystems). TaqMan assays were used for the detection of mRNA for MMPs and the endogenous control gene 18S, and SYBR Green assays for the detection of IL-6 mRNA. For all reactions, the Ct method was used for calculations, as previously described (10).
Overexpression of miR-203
Lipofectamine 2000 was used, according to the protocol recommended by the manufacturer (Gibco Invitrogen), to transfect RASFs with 100 nM synthetic pre–miR-203 or pre-miR negative control (Pre-miR miRNA Precursor Molecules and Pre-miR miRNA Precursor Negative Controls; both from Ambion–Applied Biosystems). Pre-miR miRNA Precursor Molecules are chemically modified double-stranded RNA molecules that mimic endogenous mature miR when transfected into cells. Pre-miR miRNA Precursor Negative Controls are validated negative control, random sequence molecules that do not produce an effect on miR function.
IL-6 and MMP-1 protein measurements
Levels of IL-6 and MMP-1 were measured in cell supernatants, by enzyme-linked immunosorbent assay (ELISA). For IL-6, an OptEIA kit was used according to the instructions of the manufacturer (BD PharMingen). For MMP-1, the SensoLyte ELISA kit (AnaSpec) was used.
In silico promoter analysis and prediction algorithms
Prediction of CpG islands in miR-203 and let-7a genes was done by in silico promoter analysis with MethPrimer (18). For the prediction of miR-203 targets, algorithms from the following software packages were used: TargetScan human 5.1 (19), MicroCosm Targets version 5, and PicTar (20).
Statistical analysis
Mean ± SEM values were calculated, and Wilcoxon’s matched pairs test or the Mann-Whitney U test was used for statistical comparisons. Where noted, Bonferroni correction for multiple testing was applied. Fold changes in scores were compared to a baseline value set at 1. Spearman’s rank correlation was used for correlating miR-203 and IL-6 levels. P values less than 0.05 were considered significant. GraphPad Prism 5.01 was used for statistical analysis.
RESULTS
Increased basal expression of miR-203 in synovial fibroblasts from RA patients
To investigate for differentially expressed miR in RA and OA synovial fibroblasts, we analyzed the expression of 260 miR in synovial fibroblasts from 3 RA patients and 3 OA patients, by real-time PCR. In that analysis, miR-203 emerged among the miR that were most differentially expressed between RASFs and OASFs. Levels of miR-203 were 2.8-fold higher in cultured RASFs than in OASFs and 8.9-fold higher than in synovial fibroblasts from healthy controls (Figure 1A). Also, expression of miR-203 in OASFs was 3.2-fold higher than in healthy controls. Interestingly, the expression of miR-203 in synovial fibroblasts from patients with very early RA (<3 months since the onset of symptoms) was highly variable, with some patients expressing amounts of miR-203 similar to those expressed by patients with longstanding disease (mean ± SEM 16 ± 5 years) and others exhibiting levels similar to those in healthy controls or OA patients. Analysis of miR-203 levels in RASFs according to therapeutic regimen did not reveal any influence of medication on constitutive expression of miR-203 in cultured cells (Table 1). Measurements of miR-203 in synovial tissue showed no difference between RA and OA patients (Figure 1B).
Figure 1.
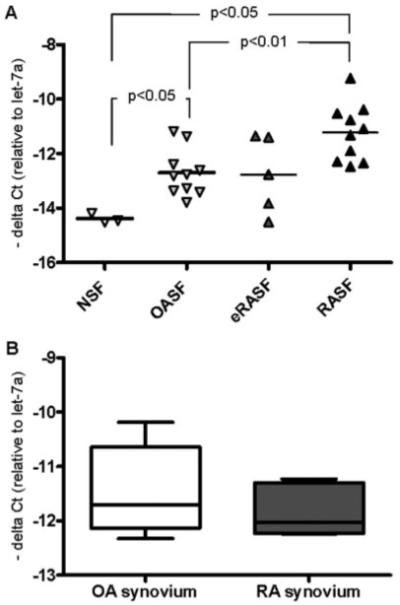
Increased basal expression of microRNA-203 (miR-203) in synovial fibroblasts from patients with established rheumatoid arthritis (RA). A, Basal expression of miR-203 in synovial fibroblasts from normal synovium (NSF), from patients with osteoarthritis (OASF), from patients with early RA (eRASF), and from patients with RA at a later stage of the disease (RASF). Bars show the mean. P values were determined by Mann-Whitney test with Bonferroni correction. B, Basal expression of miR-203 in OA synovial tissue (n = 6) and RA synovial tissue (n = 5). Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values.
Table 1.
Basal levels of microRNA-203 (miR-203) in patients with established rheumatoid arthritis, by treatment
| Patient/treatment | miR-203, ΔCt |
|---|---|
| 1/leflunomide | 9.23 |
| 2/methotrexate | 12.47 |
| 3/methotrexate | 10.77 |
| 4/methotrexate, prednisone | 11.09 |
| 5/methotrexate, prednisone | 10.53 |
| 6/methotrexate, deflazacort | 12.29 |
| 7/leflunomide, prednisone | 11.32 |
| 8/tocilizumab | 10.38 |
| 9/leflunomide, methotrexate, adalimumab, deflazacort |
11.09 |
| 10/none | 12.34 |
Regulation of miR-203 expression by epigenetic mechanisms
We next studied the regulation of miR-203 expression in synovial fibroblasts stimulated with the proinflammatory cytokines TNFα and IL-1β and the TLR-4 ligand LPS. None of these stimuli had any influence on the expression of miR-203 in RASFs (Figure 2A). Based on previous studies performed at our laboratory that showed that DNA in RASFs is generally hypomethylated (21) and published results from other groups suggesting modulation of miR-203 expression by epigenetic mechanisms (13), we hypothesized that the level of miR-203 in synovial fibroblasts might be regulated via hypomethylation of its promoter region. Additionally, in silico analysis of the upstream region of the miR-203 transcription initiation site predicted a CpG island in this region, providing evidence of transcription regulation by methylation of this gene. In contrast, the promoter of the let-7a gene showed a low CpG percentage and no CpG island (Figure 2B). To test the hypothesis, we stimulated normal synovial fibroblasts (n = 2) with the demethylating drug 5-azaC for 5 days and indeed found that expression of miR-203 was higher in demethylated than in untreated synovial fibroblasts, while the expression of let-7a remained unchanged (Figure 2C). When the fold increase of miR-203 expression relative to let-7a expression in these 2 samples was calculated using the Ct method, increases of 306-fold and 27-fold, respectively, were observed after 5-azaC treatment.
Figure 2.
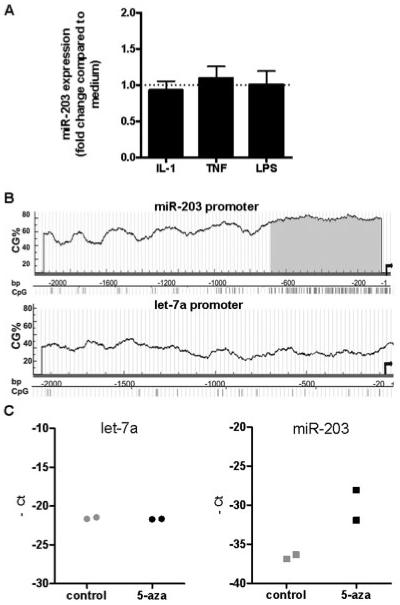
Epigenetic regulation of the expression of miR-203. A, Changes in the expression of miR-203 by RASFs (n = 6) after stimulation for 12 hours with interleukin-1 (IL-1), tumor necrosis factor (TNF), and lipopolysaccharide (LPS). Expression was measured by real-time polymerase chain reaction (PCR). Values are the mean ± SEM. B, CpG content (CG%) in 2,000 bp of miR-203 promoter and let-7a promoter. Gray shading shows particularly high density of CpG sites in the 680 bp adjacent to the miR-203 gene transcription initiation site (arrows). C, Changes in the expression of let-7a and miR-203 in healthy synovial fibroblasts (n = 2) after incubation for 1 week with 1 μM 5-azacytidine (5-aza). Expression was measured by real-time PCR. See Figure 1 for other definitions.
Increased expression of miR-203 results in higher levels of MMP-1 in RASFs
To elucidate the functional consequences of increased miR-203 expression in RASFs, we used miR-203–specific precursors for gain-of-function experiments. To ascertain whether overexpression of miR-203 has any effect on the expression of MMPs, we measured mRNA for MMP-1, MMP-3, MMP-9, and MMP-13 in pre–miR-203– and control pre-miR–transfected RASFs. After 24 and 48 hours, MMP-1 transcripts were increased by 2-fold (Figure 3A). RASFs transfected with pre–miR-203 also produced significantly more MMP-1 protein than control pre-miR–transfected RASFs (Figure 3B). Specifically, mean ± SEM levels of MMP-1 increased from 2,851 ± 714 pg/ml to 4,982 ± 1,027 pg/ml (69 ± 10%) after pre-miR–203 transfection. Levels of MMP-3, MMP-9, and MMP-13 transcripts were not significantly influenced by pre–miR-203 transfection (Figure 3C). Even though a trend toward higher MMP-13 levels was seen after pre–miR-203 transfection, the changes were not consistent and therefore not statistically significant.
Figure 3.
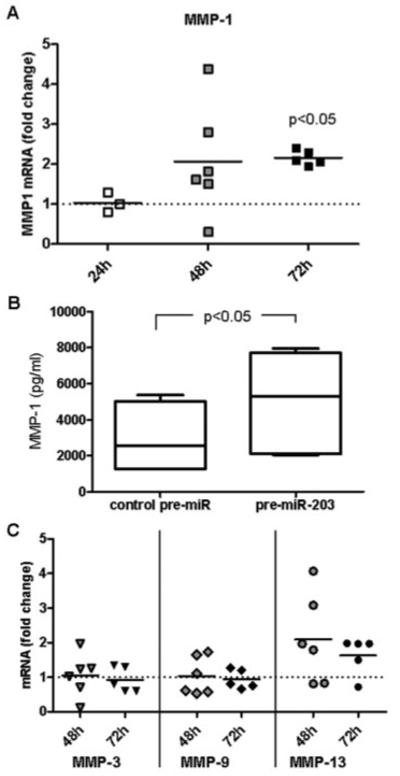
Increased levels of matrix metalloproteinase 1 (MMP-1) associated with increased expression of microRNA-203 (miR-203) in rheumatoid arthritis synovial fibroblasts (RASFs). A, RASFs were transfected with miR-203 precursor (pre–miR-203) or control pre-miR. MMP-1 transcripts were measured by real-time polymerase chain reaction after 24, 48, and 72 hours. Bars show the mean. P values (versus levels in control pre-miR–transfected cells [reference; set at 1]) were determined by Wilcoxon’s matched pairs test. B, MMP-1 levels in the supernatants of control pre-miR–transfected and pre–miR-203–transfected RASFs (both n = 5) were measured by enzyme-linked immunosorbent assay after 72 hours. Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values. P value was determined by Wilcoxon’s matched pairs test. C, MMP-3, MMP-9, and MMP-13 transcripts were measured 48 and 72 hours after transfection of RASFs with control pre-miR or pre–miR-203. Levels in control pre-miR–transfected cells (set at 1) were used as the reference.
IL-6 regulation by miR-203 via the NF-κB pathway
After transfection with pre–miR-203, a gradual increase in IL-6 mRNA levels was observed over time, and 48 hours after transfection, IL-6 mRNA levels were significantly increased in pre–miR-203–transfected RASFs compared with negative control–transfected cells (mean ± SEM 2.4 ± 0.3–fold increase) (Figure 4A). In addition, IL-6 protein levels were significantly increased, from 617 ± 132 pg/ml in control-transfected to 901 ± 162 pg/ml in pre-miR-203–transfected RASFs (58 ± 13% increase) (Figure 4B). Consistent with these findings, basal expression levels of IL-6 mRNA in synovial fibroblasts strongly correlated with basal miR-203 levels (rs = 0.76, P = 0.007) (Figure 4C).
Figure 4.
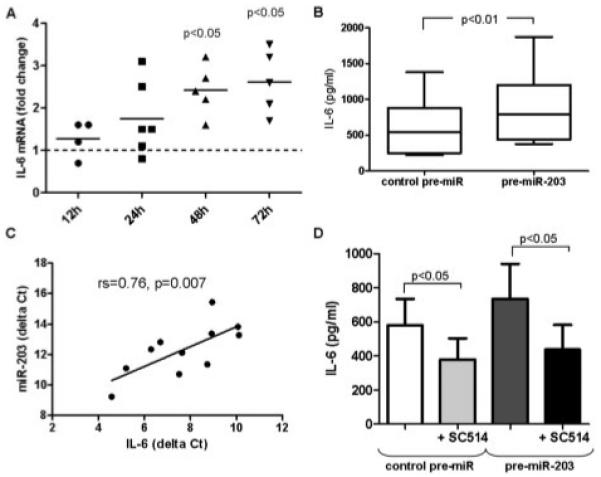
Increased levels of interleukin-6 (IL-6) associated with increased expression of miR-203 in RASFs. A, RASFs were transfected with pre–miR-203 or control pre-miR. IL-6 transcripts were measured by real-time polymerase chain reaction after 12, 24, 48, and 72 hours. Bars show the mean. P values (versus levels in control pre-miR–transfected cells [reference; set at 1]) were determined by Wilcoxon’s matched pairs test. B, IL-6 levels in the supernatants of control pre-miR–transfected and pre–miR-203–transfected RASFs (both n = 9) were measured by enzyme-linked immunosorbent assay (ELISA) after 48 hours. Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the minimum and maximum values. P value was determined by Wilcoxon’s matched pairs test. C, The correlation between basal levels of IL-6 and basal levels of miR-203 in synovial fibroblasts was determined by Spearman’s rank correlation test. D, RASFs were transfected with control pre-miR or pre–miR-203. Twelve hours after transfection, RASFs were left without further treatment or were treated for a further 12 hours with the IκB kinase 2 inhibitor SC-514. IL-6 levels in the supernatants (n = 6) were measured by ELISA. Values are the mean ± SEM. P values were determined by Wilcoxon’s matched pairs test. See Figure 3 for other definitions.
Since IL-6 is obviously not a direct target of miR-203, we sought upstream pathways that are affected by miR-203 overexpression. Previous studies have shown that RASFs spontaneously produce more IL-6 than OASFs (22,23). This constitutive transcription of IL-6 has been ascribed to the autonomous activation of NF-κB (22,24). Based on this knowledge, we used an IKK-2 inhibitor to determine whether up-regulation of IL-6 via miR-203 was dependent on the NF-κB pathway. Figure 4D shows that IKK-2 inhibition totally blocked pre–miR-203–induced IL-6 production. In accordance with the above-mentioned findings, inhibition of NF-κB also significantly blocked the spontaneous production of IL-6 in RASFs.
Expression of NF-κB pathway modulator genes after overexpression of miR-203
Based on the above data, we speculated that the direct target of miR-203 might be an inhibitor of the NF-κB pathway. Using different miRNA target gene prediction algorithms (Pic-Tar, TargetScan, and MicroCosm Targets), we browsed the predicted targets of miR-203 for modulators of the NF-κB pathway. In this list we found 9 genes whose down-regulation by miR-203 might lead to increased activity of NF-κB. The predicted targets included the members of the IκB family nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor and NF-κB p100, as well as NF-κB–repressing factor, and Tax1-binding protein 1, TNFα-induced protein 3, and ring finger protein 11, which form an NF-κB inhibitory complex. We also analyzed suppressor of cytokine signaling 3 (SOCS-3) and SOCS-6 and methyl CpG binding protein 2. Transcripts of each of these genes were measured (n = 3) 12 hours, 24 hours, 48 hours, and 72 hours after pre–miR-203 transfection. However, no down-regulation of the expression of any of the measured predicted targets was seen (Figure 5).
Figure 5.
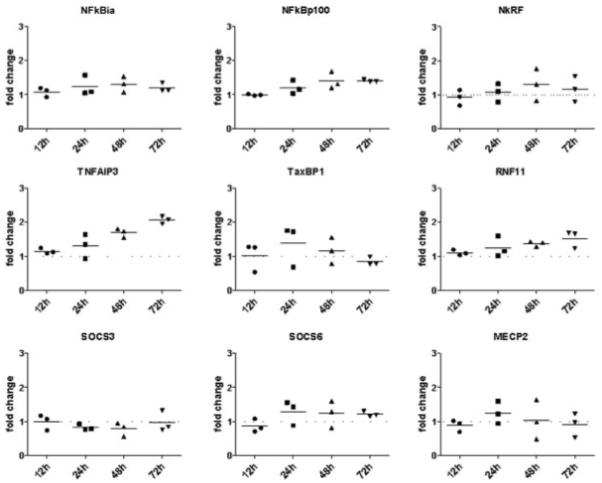
Expression of predicted targets of miR-203 after miR-203 overexpression. RASFs were transfected with pre–miR-203 or control pre-miR. Transcripts of the genes for nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor (NFkBia), NF-κB p100, NF-κB–repressing factor (NkRF), tumor necrosis factor α–induced protein 3 (TNFAIP3), Tax1-binding protein 1 (TaxBP1), ring finger protein 11 (RNF11), suppressor of cytokine signaling 3 (SOCS-3), SOCS-6, and methyl CpG binding protein 2 (MECP2) were measured by real-time polymerase chain reaction after 12, 24, 48, and 72 hours. Levels in control pre-miR–transfected cells (set at 1) were used as the reference. Bars show the mean. See Figure 3 for other definitions.
DISCUSSION
In the current study we showed that the basal expression of miR-203 is increased in RASFs compared to OASFs and that overexpression of miR-203 in these cells leads to increased production of MMP-1 and IL-6. By demonstrating this, we identified miR-203 as another miRNA with altered expression in RASFs.
Pathologic expression of miR-203 has mainly been identified in various malignant tumors, where its expression levels are mostly found to be higher than in normal tissue (25-28). In some malignancies, however, miR-203 levels are down-regulated (14,16,29), indicating that it can also function as a tumor suppressor. In particular in hematopoietic malignancies, restoration of miR-203 leads to decreased expression of the oncoprotein BCR-ABL1 (13). In accordance with our data, studies of cancer cells have also shown that the expression of miR-203 is regulated by CpG methylation and can be modulated by demethylating agents (13-16).
Apart from tumor pathology, miR-203 has been shown to play an important role in keratinocyte differentiation in the skin and was found to be highly expressed in psoriatic skin plaques (30,31). Most interestingly, cultured dermal cell suspensions from lesional psoriatic skin spontaneously produce more IL-6 protein than do nonlesional psoriatic or healthy cells (32). Our findings, together with reports of increased miR-203 expression in psoriasis (32), suggest that the link between IL-6 and miR-203 might also apply in other chronic inflammatory diseases.
In earlier studies we demonstrated for the first time that expression of miR-155 and miR-146a is altered in RA synovium and synovial fibroblasts and provided evidence of a functional role of miR-155 as a modulator of MMP-3 and MMP-1 expression in RASFs (10). Recently, published data by another group revealed that miR-124a is down-regulated in RASFs and contributes to the regulation of cell proliferation and the production of monocyte chemoattractant protein 1 (11). Based on these reports a complex picture of altered expression of miRNA in RASFs emerges, and its functional relevance to the development of the disease phenotype becomes evident.
Previously, our group suggested that the longstanding, inflammatory milieu in RA leads to epigenetic changes in synovial fibroblasts and showed that DNA in RASFs is globally hypomethylated (21). In the present study, we demonstrated that demethylating treatment of synovial fibroblasts leads to increased expression of miR-203. Therefore, we propose that the constitutively high production of miR-203 in RASFs may be due to changes in epigenetic modifications in the promoter of the miR-203 gene. The variable levels of miR-203 that we measured in RASFs from patients with symptoms of very recent onset (<3 months) support the hypothesis that this epigenetic switch is turned on during the development of the disease, which may eventually lead to the imprinted activated phenotype of synovial fibroblasts seen in RA (33). Interestingly, the fact that 2 of the patients with early RA already exhibited levels of miR-203 as high as those in patients with established RA suggests that miR-203 is involved in the pathogenesis of the disease in its very early stage. It must be stressed, however, that further in vivo experiments are needed to confirm this hypothesis.
The finding that expression of miR-203 was not increased in the synovial tissue of RA patients can be explained by the pronounced heterogeneity and vast interindividual differences in the cellular composition of inflamed synovial tissue. If increased miR-203 levels are a specific feature of hypomethylated synovial fibroblasts, the high numbers of inflammatory and vascular cells in synovial tissue lysates might conceal differences between the fibroblast populations.
IL-6 is one of the major proinflammatory cytokines present in abundance in rheumatoid joints, and recently, therapeutic regimens targeting IL-6 have been successfully introduced into clinical practice (34,35). Of interest, resident cells of the joint, i.e., synovial fibroblasts, are recognized as a major source of this cytokine, and even more interestingly, RASFs in vitro are characterized by higher spontaneous production of IL-6 as compared to OASFs (22,23). To better understand the contribution of miR-203 to the modulation of IL-6 levels in RASFs, knowing that NF-κB is the major transcription factor driving basal expression of IL-6 in synovial fibroblasts, we analyzed the influence of miR-203 on the NF-κB signaling pathway and were able to clearly show that miR-203 is dependent on NF-κB activity to modulate IL-6 levels. Therefore, our study identifies miR-203 as a novel endogenously altered modulator of IL-6 expression in RASFs, which is upstream of NF-κB. However, using miR target prediction algorithms, we could not identify the direct miR-203 target involved in this mechanism.
In recent years it has become clear that epigenetic modifications play an important role in the regulation of microRNA and that dysregulated expression of microRNA occurs in a variety of pathologic conditions (36-38). Herein we have shown that the high expression of miR-203 in RASFs leads to increased secretion of IL-6 and MMP-1, two molecules that are highly associated with RA pathogenesis and contribute importantly to chronic inflammation and joint destruction in the disease. Given recent reports of successful approaches to targeting miRNA in vivo (39-41), our data suggest that miR-203 may be a new therapeutic target in RA.
ACKNOWLEDGMENTS
We thank Ferenc Pataky and Katherine Howlett for excellent assistance with cell cultures.
Supported by the European Union Sixth Framework Programme (project AutoCure) and Seventh Framework Programme (project Masterswitch) and by the Institute for Arthritis Research, Lausanne, Switzerland. Dr. Stanczyk’s work was supported by the Foundation for the Medical Research University of Zurich/Abbott Rheumatology grant. Dr. Kyburz’ work was supported by the Swiss National Foundation.
Footnotes
Dr. Raza has received consulting fees and honoraria from Wyeth (less than $10,000). Dr. Buckley has received consulting fees, speaking fees, and/or honoraria from Pfizer (less than $10,000).
REFERENCES
- 1.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 2.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 3.Liu J. Control of protein synthesis and mRNA degradation by microRNAs. Curr Opin Cell Biol. 2008;20:214–21. doi: 10.1016/j.ceb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839–45. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 6.Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, et al. Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008;181:1055–63. doi: 10.1083/jcb.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tili E, Michaille JJ, Costinean S, Croce CM. MicroRNAs, the immune system and rheumatic disease. Nat Clin Pract Rheumatol. 2008;4:534–41. doi: 10.1038/ncprheum0885. [DOI] [PubMed] [Google Scholar]
- 8.Visone R, Croce CM. MiRNAs and cancer. Am J Pathol. 2009;174:1131–8. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10:R101. doi: 10.1186/ar2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–9. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 11.Nakamachi Y, Kawano S, Takenokuchi M, Nishimura K, Sakai Y, Chin T, et al. MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 2009;60:1294–304. doi: 10.1002/art.24475. [DOI] [PubMed] [Google Scholar]
- 12.Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58:1284–92. doi: 10.1002/art.23429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bueno MJ, Perez de Castro I, Gomez de Cedron M, Santos J, Calin GA, Cigudosa JC, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–76. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- 15.Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 16.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 17.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 18.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 19.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 20.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 21.Karouzakis E, Gay RE, Michel BA, Gay S, Neidhart M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:3613–22. doi: 10.1002/art.25018. [DOI] [PubMed] [Google Scholar]
- 22.Miyazawa K, Mori A, Yamamoto K, Okudaira H. Constitutive transcription of the human interleukin-6 gene by rheumatoid synoviocytes: spontaneous activation of NF-κB and CBF1. Am J Pathol. 1998;152:793–803. [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazawa K, Mori A, Okudaira H. IL-6 synthesis by rheumatoid synoviocytes is autonomously upregulated at the transcriptional level. J Allergy Clin Immunol. 1999;103:S437–44. doi: 10.1016/s0091-6749(99)70159-4. [DOI] [PubMed] [Google Scholar]
- 24.Bondeson J, Brennan F, Foxwell B, Feldmann M. Effective adenoviral transfer of IκBα into human fibroblasts and chondro-sarcoma cells reveals that the induction of matrix metalloproteinases and proinflammatory cytokines is nuclear factor-κB dependent. J Rheumatol. 2000;27:2078–89. [PubMed] [Google Scholar]
- 25.Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P, et al. Micro-RNA profiling in kidney and bladder cancers. Urol Oncol. 2007;25:387–92. doi: 10.1016/j.urolonc.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 26.Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 126:73–80. doi: 10.1002/ijc.24687. [DOI] [PubMed] [Google Scholar]
- 27.Petillo D, Kort EJ, Anema J, Furge KA, Yang XJ, Teh BT. MicroRNA profiling of human kidney cancer subtypes. Int J Oncol. 2009;35:109–14. doi: 10.3892/ijo_00000318. [DOI] [PubMed] [Google Scholar]
- 28.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–36. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feber A, Xi L, Luketich JD, Pennathur A, Landreneau RJ, Wu M, et al. MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg. 2008;135:255–60. doi: 10.1016/j.jtcvs.2007.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonkoly E, Wei T, Janson PC, Saaf A, Lundeberg L, Tengvall-Linder M, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS One. 2007;2:e610. doi: 10.1371/journal.pone.0000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi R, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing ‘stemness.’. Nature. 2008;452:225–9. doi: 10.1038/nature06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. 2009;183:3170–6. doi: 10.4049/jimmunol.0803721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ospelt C, Gay S. The role of resident synovial cells in destructive arthritis. Best Pract Res Clin Rheumatol. 2008;22:239–52. doi: 10.1016/j.berh.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, et al. CHARISMA Study Group Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–29. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- 35.Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, et al. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. 1997;40:217–25. doi: 10.1002/art.1780400206. [DOI] [PubMed] [Google Scholar]
- 36.Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle. 2007;6:1001–5. doi: 10.4161/cc.6.9.4209. [DOI] [PubMed] [Google Scholar]
- 37.Sheedy FJ, O’Neill LA. Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann Rheum Dis. 2008;67(Suppl III):iii50–5. doi: 10.1136/ard.2008.100289. [DOI] [PubMed] [Google Scholar]
- 38.Trang P, Weidhaas JB, Slack FJ. MicroRNAs as potential cancer therapeutics. Oncogene. 2008;27(Suppl 2):S52–7. doi: 10.1038/onc.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 40.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–17. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]