

Abstract
A successful design of peptidomimetics must come to terms with χ-space control. The incorporation of χ-space constrained amino acids into bioactive peptides renders the χ1 and χ2 torsional angles of pharmacophore amino acids critical for activity and selectivity as with other relevant structural features of the template. This review describes histidine analogues characterized by replacement of native α and/or β-hydrogen atoms with alkyl substituents as well as analogues with α, β-didehydro unsaturation or Cα-Cβ cyclopropane insertion (ACC derivatives). Attention is also dedicated to the relevant field of β-aminoacid chemistry by describing the synthesis of β2- and β3-models (β-hHis). Structural modifications leading to cyclic imino derivatives such as spinacine, aza-histidine and analogues with shortening or elongation of the native side chain (nor-histidine and homo-histidine, respectively) are also described. Examples of the use of the described analogues to replace native histidine in bioactive peptides are also given.
Keywords: amino acids, alkyl substitution, conformation, histidine, stereoselective synthesis, χ-space
1. Introduction
Design of peptidomimetics with a specific structure, conformation and topographical properties is a major issue in medicinal chemistry. Peptidic ligands elicit their bioactivity by the interaction of a portion of the three-dimensional (3D) structure with a complementary surface of the acceptor/receptor molecule or molecular complex. The peptide backbone acts as a scaffold which maintains the side chains involved in the interaction in the correct spatial position, providing at the same time the hydrogen bond network necessary to the molecular recognition and binding. The three-dimensional shape (topography) and stereoelectronic properties of the side chain moieties involved in the binding process are critical for the interaction, and provide the necessary complimentary chemical/physical environment for molecular recognition. During the past 30 years much effort has been expended to develop strategies for the design and synthesis of peptides with specific backbone conformation such as α-helix, β-sheet, extended, γ- and β-turn structures. More recently, one of the major efforts has been shifted to the design and synthesis of novel amino acids with conformationally constrained side chains so as to improve the synthesis of highly selective and potent peptide hormone and neurotransmitter analogues.
Peptide Dihedral Angles
Natural amino acids and peptides exist with a native conformational bias which starts at the backbone level. Torsional (or dihedral) angles commonly used to define the spatial orientation of the backbone peptide bonds and side chains are defined as φ, ψ, ω, and χ as illustrated in Figure 1.
Figure 1.

Conformations of peptides: definitions of the Φ, ψ, ω, χ1 and χ2 torsional angles.
The overall conformation of the peptide molecules is described by the sequence of the backbone torsion angles. The first torsion angle along the backbone is the Φ angle. This is defined as the torsional angle involving the bond sequence C(O)-N-Cα-C(O) where both C(O) atoms are carbonyl carbons. The Cα-C(O)-N-Cα torsional angle is known as the ω angle. Similarly is defined the ψ angle which involves the sequence N-Cα-C(O)-N. Finally, the χ torsional angles of the side chain groups of each amino acid are also critical. χ1 is defined by the torsional angle N-Cα-Cß-Cγ and χ2 by the torsional angle Cα-Cß-Cγ-Cδ and so forth (Figure 1) depending on the structure and extension of the side chain.
The range of the values which Φ, ψ, ω and χ torsion angles can adopt along the backbone and inside each side chain is severely restricted by steric hindrance thus leading to sets of preferred torsional angles.
The χ1 torsional angles are well illustrated by referring to the Newman projection (Figure 2). There is an energy barrier between the discrete angles, with values of +60°, −60°, and 180°having the most favorable energies.
Figure 2.

Newman projection of three staggered rotamers in l-amino acids.
It is important to note that clockwise direction on the Newman projection is defined as a (+) while a counter clockwise direction projection is defined as a (−).
Other angles in the Newman projection are less favorable due to a greater energy to overcome steric hindrance. X-ray crystallography of constrained peptides and calculations clearly show this [1,2]. The torsional angles in the rotamers reported in Figure 2 are the different values of the χ1 torsion angle. The ω angle is generally planar and trans configured with the Cα atoms of the two involved residues. Notable exception is that of the X-Pro peptide bond which can be also found as cis with deviation from the planarity higher than that of the usual peptide junctions [3,4].
Evaluation of the favored low energy conformations of the Φ and ψ angles was first examined by Ramachandran and co-workers [5], about 30 years ago, and then by many other workers. These studies show that only certain regions of the Φ, ψ space (often referred to as Ramachandran space) are actually accessible to l-amino acids. Interestingly these regions correspond to the classical secondary structures of peptides and proteins (α-helix, β-sheet, extended, etc.). An equally critical area, though much less explored and relevant to the design of new structures, is that of the 3D shape of amino acid side chains.
As illustrated in Figure 2, each χ1 torsional angle (and some of the χ2, χ3, etc. subsequent torsional angles along the chains) can adopt three low energy staggered rotameric conformations: gauche(−), gauche(+), and trans. The corresponding energy conformation is not expected to be high (about 1–2 kcal/mole for most of the simple amino acid residues).
Yet the orientation of the side chain of l-amino acid residues, with respect to the peptide or protein backbone, will be dramatically different in each of the three cases: for gauche(−) χ1: orientation towards the N-terminus; for trans: orientation towards the C-terminus; for gauche(+): orientation over the backbone. In molecular recognition processes, the side chains will adopt generally one of these three possible conformations, with preference for the gauche(−) > trans > gauche(+) for most natural l-amino acid residues. Clearly, the side chain conformations are critical to the molecular recognition and information transfer processes involving ligand-receptor/acceptor interactions [6,7].
2. Background
The incorporation of χ-space constrained amino acids into bioactive peptides renders the χ1 and χ2 torsional angles of pharmacophore amino acids critical for activity and selectivity as with other relevant structural features of the template.
A successful design of peptidomimetics in χ-space will depend upon several conditions: (1) side chains should adopt the preferred conformation found in natural amino acids; (2) the constraint should be compatible with backbone conformations; and (3) the constraint should be compatible with efficient binding kinetics. These are critical issues because the constrained amino acid residues must not significantly interfere with the conformation of the constrained template, with binding of the ligand to its receptor, or with the dynamic processes and conformational changes that accompany ligand-receptor interaction and information transduction [2].
During the design based on the topographical χ space control, authors concentrated their major efforts on aromatic amino acids. All peptide hormones and neurotransmitters have in fact one or more aromatic amino acid residues as key pharmacophore. Different structural features, chosen in order to constrain the χ space of aromatic amino acids, are shown in Figure 3.
Figure 3.

General structure of some χ-constrained aromatic amino acids: (a) β and side chain alkyl substituted amino acids; (b) imino acids; (c) α-β dehydro and α-amino cyclopropanecarboxylic acid derivatives (ACC); (d) β2/β3-homo-amino acids; (e) homo and nor-amino acids.
Histidine (His) is an important amino acid critically involved in receptor recognition and biological activity of many peptides and proteins [8].
The peculiar chemical properties of the imidazole ring are responsible for this. Histidine is often strategically located in a variety of biologically active peptides such as angiotensin [9], luteinizing hormone releasing hormone (LHRH) [10], liver cell growth factor [11], thyrotropin releasing hormone (TRH) and is preferential binding site for transition metal ions [12].
Despite the great attention dedicated to the structural modifications performed on phenylalanine, tyrosine and tryptophan aromatic residues and to the conformational and biochemical consequences of the incorporation of the obtained synthetic analogues into peptide backbones, the histidine residue has been only marginally treated. The presence of the basic and nucleophilic imidazole ring, which induces racemization processes and requires almost invariably specific temporary protection, renders the synthetic strategies more laborious than those encountered with any other aromatic amino acid. Here we summarize some relevant papers performed in the field of histidine analogues with focus on constrained models, characterized by the presence of α- and/or β-substituents, capable of limiting the χ-space available to the imidazole side chain. Results concerning other relevant analogues, such as those pertaining to the relevant field of β-amino acids are also reviewed.
3. Nomenclature
Histidine Derivatives
Since the nomenclature of imidazole ring and histidine have been subjected to several changes in the years, data reported here adopt the following nomenclature: the nitrogen atoms of imidazole ring are denoted by pros (π) and tele (τ) to show their position relative to the side chain (near and far, respectively).
The carbon atom between the two ring nitrogen atoms is numbered 2 (as in imidazole), and the carbon atom next to the τ nitrogen is numbered 5. The carbon atoms of the aliphatic chain are designated α and β [13] (Figure 4).
Figure 4.

Adopted notations for histidine and its analogues.
4. Chemistry
4.1. α-Alkyl Substitution
The strategy based on replacement of α and β hydrogen atoms of histidine with alkyl groups started when topographical considerations, relevant to the stereochemical features required for receptor recognition and signal transduction, were still not well established.
It has been demonstrated that α substitution poorly affects in general the χ1 torsional angles. The more effective changes are found on the backbone and these analogues are in fact considered α-helix and β-turn inducers [14].
An early synthetic method (reported by Robinson and Shepherd [15]) for the α-substituted product 3 is the coupling reaction between methyl α-nitropropionate and chloromethylimidazole 1 to give 2 followed by reduction of the nitro group (Scheme 1).
Scheme 1.

Synthetic procedure for α-methyl histidine [15].
Successively (1974), Suzuki et al., found that N-tosyl-acetoxy-methylimidazole [16] 4, easily prepared from hydroxymethylimidazole [17], is a useful intermediate for the introduction of imidazole ring in the amino acid skeleton. Accordingly, α-methylhystidine was synthesized by reaction of α-isocyanoproprionate 5 with N-tosyl-acetoxymethylimidazole 4, as shown in Scheme 2.
Scheme 2.

Synthetic procedure for α-methyl histidine [16].
The coupling product 6 was obtained in 67% yield and was subsequently hydrolyzed with HCl to give α-methylhistidine dihydrochloride 7 in 80% yield.
Two years later DeGraw et al. used the route outlined in Scheme 3 to synthesize α-substituted histidines. 4-Chloromethylimidazole hydrochloride 8 [18], was added to a solution of an appropriate 2-alkyl acetoacetate 9a–d,f or 2-acetylbutyrolactone 9e in ethanol solution containing sodium ethoxide to afford the 2-alkyl 2-(4-imidazolylmethyl)acetoacetate 10. When the keto ester 10 was allowed to react with a slight excess of hydrazoic acid in sulfuric acid solution, the N-acetylhistidine esters 11a–e were obtained in 50–70% yields. Under the acidic conditions employed, the α-allyl-analogue 10f was apparently subjected to additional attack on the vinyl moiety, leading to an unidentified product rather than the anticipated product 11f. Schmidt [19] had qualitatively shown that in β-keto esters the HN3 reagent afforded selective attack on the ketone carbonyl with subsequent rearrangement occurring at the α-carbon. Synthesis of the histidines 12 and 13 were completed by acid hydrolysis of the amide and ester functions. This route seems to have been little used in the synthesis of α-substituted amino acids, but especially valuable in the histidine series.
Scheme 3.

Synthetic procedure for α-alkyl histidine [18].
An alternative synthesis (Scheme 4) of α-substituted histidine was proposed by Kelley in 1977. The synthesis was modeled after the work of Robinson and Shepherd [15] and Sletzinger and Pfister [20], as outlined in Scheme 4.
Scheme 4.

The appropriate α-nitro ester 14 [21], (Scheme 4) was alkylated with 4-chloromethylimidazole [15], in methanol or dimethylformamide. In the case of the α-phenyl analogue 14 (R1 = CH2CH3; R2 = C6H5) hexamethylphosphoramide was required to obtain the product, even if the yield remained low. Reduction of the nitro esters 15 was best accomplished by catalytic hydrogenation to give the desired product 16. Although the reduction required a high catalyst to substrate ratio and extended reaction times, in all but one case the products were obtained in high yield and purity.
In 1989, O’Donnell et al. [22] synthesized α-methyl histidine by two complementary routes using a catalytic phase-transfer (PTC) alkylation procedure (Scheme 5).
Scheme 5.

Synthetic procedure for α-methyl histidine [22].
The first route involves benzylation of the Schiff base ester 17, prepared in 90% yield from alanine ethyl ester hydrochloride and p-chlorobenzaldehyde, with the electrophile 18 [23]. In the second approach the appropriately protected histidine substrate 19 is α-methylated by using the PTC method. The intermediate 19 is easily prepared in 83% yield in one pot procedure from histidine methyl ester dihydrochloride by condensation of the free amino ester (generated in-situ) with p-chlorobenzaldehyde followed by protection of the imidazole nitrogen with tosylchloride/triethylamine. Alkylation of 17 or 19 was accomplished by catalytic solid-liquid phase transfer alkylation using powdered potassium hydroxide in acetonitrile at 15–20 °C with 10% Bu4NBr as the phase-transfer catalyst. The crude alkylated derivatives 20 (20a, R = Et, 47% crude; 20b, R = Me, 70% crude), obtained as oil, were hydrolyzed directly in a two-steps sequence. The product α-methyl histidine was obtained as its dihydrochloride 21 in overall yields of 27% and 41% from 17 and 19, respectively.
4.2. β-Alkyl Substitutions
A synthetic route for β-alkylhistidines 24 was reported by Kelley [24] and co-workers as a modification of Albertson’s classic synthesis of histidine [25], by using the appropriate 1-(4-imidazolyl)alkyl chloride 22 and the diethyl acetamidomalonate 23 (Scheme 6).
Scheme 6.

Synthetic procedure for β-alkyl histidine [24].
The synthetic route afforded the desired amino acids as diastereoisomeric mixtures. No attempt was made to separate the diastereoisomers.
A successful asymmetric synthesis of (2S,3S)-β-methylhistidine 31, by using 2-mesitylenesulfonyl (Mts-) as a protecting group at τ-N in the imidazole ring was reported by Hruby et al. [8] (Scheme 7).
Scheme 7.

Synthesis of (2S,3S)-β-methylhistidine [8].
The synthesis started from the commercially available urocanic acid 25. This was treated with 2-mesitylensulfonylchloride MtsCl in sodium hydroxide solution to furnish the derivative 26 τ-N protected at the imidazole ring. The protected urocanic acid was coupled by mixed anhydride method 27 to an optically pure chiral auxiliary according to a reported procedure [26] to give 28. The subsequent Michael addition and azide formation followed a highly enantioselective route (>99% as determined by 1H NMR). Hydrolysis of the chiral auxiliary 29 to compound 30 led to the removal of the Mts-protecting group. The final unprotected β-methylhistidine 31 was isolated, purified by ion-exchange column (Dowex 50X2-100) and recrystallized from water/ethanol [27]. According to the 1H NMR spectra some epimerization was observed in the last two steps; thus the Mts- or another protecting group should be reintroduced for this use in peptide chemistry. Although the free imidazole group can apparently catalyze the epimerization process, the adoption of the Mts protection allowed the first successful asymmetric synthesis of a (2S,3S)-β-methylhistidine derivative.
Recently, Saha et al. [28] synthesized β-amino acids based libraries on solid support, ancoring 4-formyl imidazole to 2% cross-linked PS-resin using the convenient 2-Cl trityl linker. Horner-Emmons condensation was conducted on resin bound aldehyde with an excess of tert-butylcarboxy triethyl phosphonoacetate in THF 32. Lithium amides have been utilized in conjugate Aza-Michael additions to α,β-unsatured systems for the generation of protected chiral β-amino acids 34 [29]. The amides were generated by pre-treating several amines with n-BuLi in THF at −78 °C. The resulting lithium amides were added via cannula under N2 to a pre-swelled suspension of resin 33 in THF, maintained at −78 °C. Then, the mixture was warmed to room temperature followed by work up procedure. Cleavage after Aza-Michael addition, under mild-TFA conditions produced the tert-butyl ester products 35. Cleavage with 25–50% TFA-CH2Cl2 gave the deprotected β-amino acid products 36 (Scheme 8).
Scheme 8.

Aza-Michael reaction toward synthesis of β-amino acids on solid support [28].
4.3. β,β–Dimethyl Substitution
In 1976, De Graw et al. [30] prepared a series of substituted histidines and homo-histidines and evaluated the obtained compounds as inhibitors of specific histidine decarboxylase obtained from rat pyloric stomach.
β,β-dimethyl histidine 43 was prepared by the procedure shown in Scheme 9; 4-cyanomethylimidazole 37 was conveniently obtained in 70% yield from histidine by decarboxylation in sodium hypochloride solution [31].
Scheme 9.

Synthesis of β,β-dimethyl histidine [30].
Treatment of 37 with bis(trimethylsilyl)acetamide afforded an N-Me3Si intermediate which, when allowed to react with trityl chloride, gave the N-trityl-4-cyanomethylimidazole isomer 38 in 77% yield. Although shown as the 1-N-trityl derivative 39, the true position (Nτ or Nτ) of tritylation was not established. The blocked nitrile 38 was converted to the anion by treatment with sodium hydride in hexamethylphosphoramide (HMPA) solution. Subsequent reaction with methyl iodide at room temperature afforded a mixture of methylation products containing approximately 77% of the monomethylnitrile 39a. The dimethylnitrile 39b was obtained by exhaustive re-treatments to ensure complete methylation. Hydrolysis of the nitriles was effected by sodium hydroxide in hot 90% 2-methoxy ethanol to afford the carboxylic acids 40a and 40b in 84% and 86% yields, respectively. Reduction of the dimethyl acid 40b with diisobutylaluminum hydride in toluene gave a mixture containing about 75% of the aldehyde 41b as shown by NMR analysis. The crude product was chromatographed to remove unreacted acid 40b and to effect a separation of the aldehyde from an undesired product regarded as the alcohol 44b. The crystalline aldehyde 41 was allowed to react with KCN/(NH4)2CO3 at 110 °C in a bomb tube [32], to yield the hydantoin 42 as a crystalline product. Acid hydrolysis of 42 yielded the β,β-dimethyl histidine 43.
4.4. Constrained “Imino Acids” 1,2,3,4-Tetrahydroquinoline Derivatives (Spinacines)
The most important procedure to synthesize cyclic analogues of histidine uses the Pictet-Spengler reaction [33,34] or modifications thereof, by cyclocondensation of the amino acid His with formaldehyde in the presence of concentrated hydrochloride acid. In general these reactions proceed in good yield (70–97%) and enantiomerically pure amino acids can be obtained from enantiomerically pure precursors. If partial racemization [35] occurs fractional crystallization is necessary to obtain the desidered amino acid (yields: 90–99%) [36,37].
Among the variety of histidine analogs which provide a conformational restriction of the peptide backbone and/or of the lateral chain, the most extensively employed in structure-activity and metal-ion complexation studies have been the Nα-, Nτ- and Nπ-methyl histidine derivatives [38]. More recently, the (4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acid) (Spinacine, Spi) has also been studied [39,40].
Reaction of L-His and formaldehyde to give Spi 47 was first reported by Wellisch in 1913 [41], but only in 1991 Klutchko et al. [42] described the synthesis via Pictet-Spengler reaction on Nπ-substituted histidines, of a large number of Spi derivatives including amide, ester, 5-alkyl and acyl, and regiospecific Nπ-akyl and aralkyl derivatives.
l-Spi has been also synthesized under different experimental conditions which gave, as an intermediate, its Nπ-hydroxymethyl derivative Spi(π-MeOH) [43] 46. In this case, the starting l-His in 0.1 M sodium phosphate buffer (pH 7.0) was reacted with a 30-fold molar excess of formaldehyde at room temperature for 4 days; the precipitate was collected by filtration, washed with cold water and dried. The product, separated from the reaction medium as crystal suitable for X-ray diffraction, was obtained in 93% yield (Scheme 10). The obtained Spi(π-MeOH) 46 was dissolved in 1 M HCl and the solution concentrated at reduced pressure. The residue 45 was dissolved in water and the pH adjusted to 3.5. Absolute ethanol was added, obtaining crystals which were collected by filtration, washed with ethanol and dried (95% yield).
Scheme 10.

Synthesis of spinacine (Spi) and hydroxymethyl-spinacine Spi(π-OMe) [43].
An interesting use of the amino acid spinacine is reported by Guzman et al. [44] in their synthesis of cis/trans imidazopyridines spinacine derivatives (Scheme 11):
Scheme 11.

Synthesis of spinacine and derivatives [44].
In particular, the 1-phenyl derivative 50 was synthesized by the method of Wille [45] through the base-catalyzed Pictet-Spengler reaction of histidine 48 with benzaldehyde. Aromatization of the tetrahydroimidazopyridine derivative 49 with selenium dioxide readily afforded 50 as free base.
X-ray Studies on Spinacine Derivatives
Two tautomeric forms can be predicted for the amino acid spinacine, depending on the position of the hydrogen on the nitrogen atoms of the imidazole ring.
X-ray studies reported by Andreetti et al. [46], showed that the crystals correspond to the tautomer having the N(3) atom protonated as shown in Figure 5, in which the bond distances are also reported. Thus, the compound corresponds to the amphionic form of the 4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acid. Condensation of imidazole with tetrahydropyridine removes the π electron delocalisation on the imidazole and produces a double bond localized on C(2)-C(7) [C(2)-C(7) = 1.331(10) Å]. The other bond distances and angles are in agreement with the hybridisation state of the atoms. The imidazole ring and the C(6) and C(3) atoms lie on a plane whereas C(4) and N(1) are out of that plane by 0.208 and −0.568 Å respectively; this as a consequence of the sp3 character of the four atoms of the six-membered ring. The torsional angles around the N(1)-C(3) and C(4)-C(6) are −25.6° and 30.8° respectively. The carboxyl group is equatorial and orientated in such a way that one oxygen points to the NH2+ group to compensate the opposite electrical charge.
Figure 5.

An ORTEP view of Spi [46].
Solid state structure of spinacine analogues have been recently reported by Bertolasi et al. [43] (Figure 6) and one of these is that Spi(πMeOH) 46 which crystallizes with a water molecule and its structure displays, like that of Spi, a zwitterionic character. The carboxylate group is situated in equatorial position and is rotated by about 10° around the N1-C4 bond, the torsion angles O1-C5-C4-N1 and O2-C5-C4-N1 being −10.2(2)° and 170.3(2)°, respectively. This conformation, observed also in the structure of Spi and other amino acids [46], is favored by a short electrostatic interaction of 2.618(2) Å between the negative charged O2 atom and the positive N1. Bond distances and angles are within the normal ranges; in particular, comparing the present structure with those of imidazole [47], and l-His [48], it can be observed that the hydroxymethyl substituent at N3 and the condensation with tetrahydropyridine do not produce significant variations in the imidazole geometry. The tetrahydropyridine ring C2-C7-C6-C4-N1-C3 adopts a half-chair conformation 5H4 with puckering parameters QT = 0.483(1) Å, φ = −145.6(3)°, θ = 48.0(2)° and ΔC2(C2–C7) = 0.0182(7) Å [49,50].
Figure 6.

An ORTEP view of Spi(πMeOH)·H2O showing thermal ellipsoid at 30% probability [43].
4.5. 1-Amino-2-(4-imidazolyl)cyclopropanecarboxylic Acid Derivatives (ACC)
The most general route to His derivatives containing a cyclopropane ring between the Cα and the Cβ carbon atoms (ACC derivatives) involves cyclopropanation of 2-aryl-4-benzylideneoxazolones, formed by 1,3-dipolar cycloaddition of diazomethane followed by thermolysis of the intermediate pyrazoline. The cyclopropanation reaction works moderately well and the subsequent chemical transformations provide the amino acids in low to moderate overall yield (8–38%) [51].
An attractive pathway to ACC 53 appears the addition of diazomethane to 4-[(1-acetyl-4-imidazolyl)methyl-ene]-2-phenyl-2-oxazolin-5-one 51 followed by hydrolytic cleavage of the oxazolone ring 52 [52] (Scheme 12).
Scheme 12.

Synthesis of ACC derivative [52].
The choice of this route follows the findings of Awad et al. [53] and Mustafa et al. [54], according to which the carbon-to-carbon double bonds, exocyclic to certain hetero rings, including oxazolones, react with diazomethane to give cyclopropane derivatives. Moreover, Awad et al. [53], have cleaved the azlactone 1,5-diphenyl-6-oxa-4-azaspiro-[2,4]hept-4-en-7-one to 1-benzamido-2-phenylcyclopropanecarboxylic acid thus showing that the N-benzoyl group could be hydrolyzed under conditions which would not affect the cyclopropane ring.
4.6. α,β-Dehydro Amino Acids
In 1990 Easton and co-workers reported a method for the diastereoselective conversion of amino acids to their β-hydroxy derivatives by direct side chain bromination of the amino acid derivatives with NBS, followed by treatment with silver nitrate in aqueous acetone [55–57].
The side chain bromination requires an N-protecting group, such as phthaloyl or trifluoromethanesulfonyl, in order to deactivate the α-position toward hydrogen atom abstraction [58].
Crich et al. [59] reported the use of N,N-di-tert-butoxycarbonyl protected amino acids in Easton’s protocol and the advantages that this strategy affords to the synthesis of α,β-dehydrohistidine and β-hydroxyhistidine (Scheme 13).
Scheme 13.

Synthesis of α,β-dehydro-histidine [59].
The radical bromination of 54 [60] provided the threo bromide 55 and the trans oxazolidinone 56. This mixture was subsequently treated with silver nitrate in acetone to afford the trans oxazolidinones 57 and 58, which differ by the presence of τ t-butoxycarbonyl group, in 62% yield. Attempted hydrolysis of 57 or 58 under a wide variety of conditions produced the α,β-dehydrohistidine derivative 60. The free imidazole nitrogen is responsible for the elimination reaction thus, imidazole protecting group, stable under mild basic conditions, would solve the problem. Product 58 was also reacted with trityl chloride and triethylamine in dichloromethane and, after removal of the excess trityl chloride, the reaction mixture was treated with catalytic cesium carbonate in methanol leading directly to the formation of the desired (2S,3S) β-hydroxyhistidine derivative 59 in 74% yield (Scheme 13).
In 1980 Battersby et al. [61] realized a stereoselective synthesis of (αS, βS)-[β-3H1]histidine and (αS, βR)-[β-3H1]histidine, obtaining as intermediate, the 2-acetamido-3-[imidazol-4(5)yl-acrylic]acid (Scheme 14). In the first approach, NaBD4 or BT3 were added to a solution of 4(5)-formylimidazole to obtain compound 61a or 61b. These were then oxidized by MnO2 or BaMnO2 to the aldehyde 62a or 62b. Condensation of the aldehyde with N-acetylglycine in acetic anhydride afforded the oxazolinone 64 and 65 which were converted into the required acrylic acid 67 and 68 by mild basic hydrolysis. In the second way, 4(5)-formylimidazole was directly condensed with N-acetylglycine to obtain compound 63. Then 63 was worked up as above to give the imidazolylacrylic acid 66.
Scheme 14.

Synthesis of α,β-dehydro-histidine [61].
Cativiela et al. [62] published the synthesis, based on the method of Battersby [61], of α,β-dehydro-histidine with Z geometry around the double bond (ΔZ-His). The synthesis starts from 5-formylimidazole in Ac2O/AcONa which reacts with hippuric acid to give the intermediate azlactone; subsequent hydrolysis with sodium carbonate in water gave the desired Z configured compound.
Recently, an example of azlactonization of α,β-dehydro-histidine (71) starting from 5-hydroxymethyl-imidazole (69) was reported by Parker et al. [63]; as illustrated in Scheme 15, (2S, 3S)-[3-2H]histidinol, was synthesized by a stereochemically unambiguous route.
Scheme 15.

Synthesis of (Z)-α,β-dehydro-histidine (ΔZ His) [63].
Azlactone 70 was crystallized and its structure determined by X-ray crystallography. The ORTEP drawing of 70 (Figure 7) clearly indicates that the exocyclic double bond has the Z configuration, as shown in Scheme 15.
Figure 7.

An ORTEP view of azlactone 70 [63].
X-ray crystal structure of azlactone 70 is illustrated in Figure 7.
A Japanese patent [64] reported the synthesis of a series of new imidazole derivatives (Scheme 16).
Scheme 16.

Synthesis of α,β-dehydro-histidine [64].
The 5-formylimidazole 72 is treated with tert-butoxycarbonylamino-(dimethoxy-phosporyl)-acetic-acid methyl ester 73 in THF in the presence of 1,1,3,3-tetramethylguanidine (TMG) at 0 °C. The product (74) of the condensation reaction is E/Z diastereoisomeric mixture.
4.7. Homo-Histidine
The ten-steps synthesis of l-homo-histidine (Figure 8) by Bloemhoff and Kerling [65] using N-benzyloxycarbonyl-l-glutamic acid 75 as starting material, was revisited by Altman et al. [66] (Scheme 17).
Figure 8.

Series of l-homo-histidines.
Scheme 17.

Synthesis of l-homo-histidine [66].
The imidazole ring was built from the gamma-carboxyl group while the α-function was protected. The protecting group was removed in the last step. The critical step in this procedure involves imidazole ring closure with ammonia and formaldehyde forming copper salt. The reaction appears to be the most tedious step and does not exceed 50% yield. By submitting chloromethyl chetone 76 to cyclization conditions with formamidine acetate in ammonia the yield was greatly improved (71%), leading to the overall yield of 40%.
This method is not applicable to the preparation of a series of imidazole-containig amino acid derivatives by Silverman et al. [67] thus, a new enantioselective general procedure leading to 80a–e (Scheme 18) was devised by utilizing the reaction of various imidazolyl alkyl bromides 78b–e with chiral lithiated bislactim ether 79 as the key step.
Scheme 18.

Synthesis of l-homo-histidines [67].
1-(N,N-dimethylsulfamoyl)imidazole 77 was protected with a tert-butyldimethylsilyl group (TBDMS) by sequential treatment with n-butylithium followed by tert-butyldimethylsilyl chloride. The lithium salt of the 1,2-diprotected imidazole was allowed to react with a series of dibromoalkanes to afford bromoalkyl substituted imidazoles 78b–e. Carbon-carbon bond formation between the alkyl bromides and lithiated bislactim ether proceeded in good yields. The homologated bislactim ether products 79a–e were hydrolyzed with 0.25 N HCl to the corresponding amino acid ethyl ester. Under these reaction conditions the TBDMS protecting group of the imidazole was cleaved. Removal of the sulfamoyl group requires relatively vigorous conditions (refluxing 30% HBr for 4 h), with consequent ethyl esters hydrolysis and formation of the desired amino acids 80b–e. The bromoethyl imidazole analogue 78a was not prepared by the route used for the other analogues, because the dibromoethane underwent elimination of HBr in the presence of n-butyllithium, and not a trace of the substitution product 79a was detected.
A preparation of racemic homo-histidine from the readily available urocanic acid has been reported by Pirrung et al. [68] (Scheme 19).
Scheme 19.

Synthesis of racemic-homo-histidine starting from urocanic acid [68].
Urocanic acid was esterified and hydrogenated to give compound 81. As reported by Browne [69], the tritylation significantly improves its solubility in ethereal solvents, permitting DIBAL-H reduction in quantity to the aldehyde 82. Strecker reaction gives an aminonitrile whose hydrolysis, accompanied by the trityl group removal, produces homo-histidine 83 in 73% overall yield.
A patent reported the synthesis of homo-histidine [70], useful for treating renin associated hypertension.
4.8. Nor-Histidine
The accidental discovery of sweet dipeptide derivative l-aspartyl-l-phenylalanine methyl ester known as aspartame, was published several years ago [71]. Since then, a variety of analogues have been prepared by different research groups seeking more stable and more potent dipeptides. The synthesis of imidazolylglycine [72] sweetener containing nor-histidine is given in Scheme 20:
Scheme 20.

Synthesis of imidazolylglycine sweetener containing nor-histidine [72].
The protected Nα,Nπ-diBoc-imidazolylglycine derivative 84 was prepared by modification of a published procedure for the Nα-Boc derivative [73,74]. Mixed anhydride coupling with (−)-α-fenchol gave Boc-amino ester in low yield (25–30%). A major side product was the ethyl ester formed by the reaction of ethanol released from the mixed anhydride upon reaction with fenchol. This severe limitation in the use of mixed anhydrides for ester formation is not a problem in amide formation where the nucleophilicity of the amine exceeds that of the ethanol released. Alternative coupling procedures also gave low yields. Acid hydrolysis gave amine 85. Coupling with N-(tert-butoxycarbony1)-β-tert-butyl-l-aspartic acid p-nitrophenyl ester to 86 followed by acid hydrolysis gave sweetener 87.
The importance of nor-histidine (Figure 9) as residue in biologically active compounds is shown in the study of Tagawa et al. [75].
Figure 9.

Structure of nor-histidine and cephalosporine containing nor-histidine (Reference 75).
Recently, a panel of amino acid analogs and conformationally-restricted amino acids bearing a sulfonic acid were synthesized and tested for their ability to preferentially inhibit the obligate cysteine glutamate transporter system xc versus the vesicular glutamate transporter (VGLUT) [76]; The target conformationally-restricted amino acids were synthesized as shown in Schemes 21; imidazolylglycine was synthesized via hydrolysis of the corresponding hydantoin intermediate 88 [77–79].
Scheme 21.

Synthesis of imidazolylglycine via the corresponding hydantoin intermediate [76].
4.9. β2-Homo-Histidine
Due to the nucleophilic character of N-atom on the (1H-imidazolyl)methyl side chain of histidine, several undesired by-products can be formed during the synthesis of β-homo-histidines. Indeed, many routes have been tested, and most were not successful. (Scheme 22) [80].
Scheme 22.

Retrosynthetic analysis for the preparation of the Fmoc-β2hHis(Tr)-OH via alkylation, Mannich-Type reaction and aldol addition of chiral acyloxazolidinones [80].
Initial attempts of diastereoselective alkylation (cf. A in Scheme 22) of homo-glycine derived acyloxazolidinones III (PG = Phth; PG = Ph2C) with (1H-imidazol-4-yl)methyl derivatives II (PG’ = Trt, X = Cl; PG’ = Ts, X = MsO) were ineffective due to the low reactivity. In a second approach (cf. B in Scheme 22), the aldol addition of the oxazolidinone derivatives III with aldehydes IV (PG’ = Tr; PG’ = Ts), followed by deoxygenation under Barton-McCombie conditions, resulted in the isolation of degradation products caused by retro-aldol reactions, occurring during the oxygenation step. Thus, the amidomethylation reaction via Ti-enolates was attempted (cf. C in Scheme 22): treatment of the acyloxazolidinone V (PG’ = Tr) with the electrophile VI resulted in a complex mixture of inseparable products; although the desired compounds had been formed, long reaction times were required for good conversion, causing the partial cleavage of the trityl protecting group. Seebach et al. envisaged the use of a more reactive electrophile, for instance, 1,3,5-trioxane (cf. D in Scheme 22), with subsequent OH/NH2 replacement. This route led eventually to the synthesis of the desired β2-homo-histidine derivatives for solid phase syntheses. For the preparation of the acyloxazolidinone 92, 1H-imidazole-4-acrylic acid (urocanic acid) was selected as the starting material. Hydrogenation of the urocanic acid, followed by esterification under acid conditions, gave the methyl ester 89, which was trityl protected to give crude 90. Product 90 was saponified to afford the acid 91 in 73% yield over four steps (Scheme 23).
Scheme 23.

Preparation of the acyloxazolidinone (R)-92 starting from urocanic acid [80].
N-acylation of the lithiated chiral auxiliary 4-isopropyl-5,5-diphenyl-1,3-oxazolidin-2-one (R)-DIOZ was accomplished by nucleophilic addition of the lithiated auxiliary (using BuLi in THF at 0 °C) to the pivaloyl mixed anhydride of 91 to give the acyloxazolidinone (R)-92, in 60% yield. The following aldol reaction was effected by reaction of the Ti-enolate of 92 with 1,3,5-trioxane to afford the hydroxymethyl derivative 93 in 70% yield. (Scheme 24).
Scheme 24.

Diastereoselective aldol reaction of (R)-92 with 1,3,5-trioxane. Further transformation towards the formation of β2-homo-histidine derivatives led to the formation of compound 95 [80].
The next step required the replacement of the OH group of 93 by a N-substituent by means of a Mitsunobu reaction. Thus, treatment of (R,S)-93 with Ph3P, DIAD and either DPPA or hydrazoic acid afforded the azide derivative (R,S)-94 in moderate to good yields (55 and 89% resp.). However attempts to cleave the auxiliary were not successful. Indeed, removal of the oxazolidinone group by BnOH/BuLi afforded the elimination product 95 (Scheme 24). To circumvent this problem, the hydroxymethyl derivative 93 was first treated with BnOH/BuLi to form the corresponding benzyl ester (S)-96, which was subsequently transformed to the azide (S)-97 under the Mitsunobu conditions in good yield (Scheme 25).
Scheme 25.

Removal of the auxiliary with BnOLi followed by Mitsunobu Reaction and functional group modifications for the preparation of Fmoc-(S)-β2hHis(Tr)-OH 98 [80].
Simultaneous benzyl ester cleavage and azide reduction, by catalytic hydrogenation followed by Fmoc-protection, afforded the histidine derivative (S)-98 in moderate yield. It should be noted that the Tr protection group is stable under the hydrogenation conditions only to a certain extent; prolonged reaction times cause complete cleavage of that group.
In this way, the synthesis of (S)-Fmoc-β2hHis(Tr)-OH was achieved with an overall yield of 11% over ten steps from 4-isopropyl-5,5-diphenyl-1,3-oxazolidin-2-one (DIOZ).
4.10. β3-Homo-Histidine
First attempts at synthesizing the homo-histidine derivatives by Seeback et al. [80] using the Arndt-Eistert homologation from Fmoc-His(τ-BOM)-OH, Fmoc-His(τ-Tr)-OH, Boc-His(τ-Bn)-OH, Boc-His(π-Bn)-OH, and Boc-His(τ-BOM)-OH were unsuccessful. Only Ts-protected histidines reacted with CH2N2 to form the corresponding diazo ketones, probably due to the strong electron-withdrawing effect of the tosylate, which renders the 1H-imidazolyl-N-atom less nucleophilic. Thus, commercially available Fmoc-His(Ts)-OH and Boc-His(Ts)-OH were converted via their mixed anhydrides (NMM/ClCO2Et or NEt3/ClCO2iBu) to the diazo ketones 99a and 99b in 56 and 86% yields respectively (Scheme 26). Attempts at converting diazo ketone 99a to a β3-homo-histidine derivative were shown to be ineffective, due to its insolubility in most solvents suitable for the Wolff rearrangement (e.g., BnOH, H2O, THF, dioxane, etc.). On the other hand, decomposition of diazo ketone 99b in the presence of the corresponding alcohol (MeOH, BnOH), by reaction with catalytic amounts of Ag+ (CF3CO2Ag dissolved in Et3N) gave the Boc-protected methyl or benzyl ester 100 as a mixture of three products in a 1:1:1 ratio. It was found that the Ag+ ion interacts with the 1H-imidazole ring inducing the partial displacement of the Ts protecting group from the τ-N to the π-N, as well as the complete removal of this protecting group. With this result at hand, they decided to use Boc-His(πTs)-OH as starting material for conversion to the β3-amino acid derivative Fmoc-β3hHis(πTr)-OH. Since the use of histidine derivatives with unprotected 1H-imidazolyl side chains for peptide couplings in solution or on solid support are known to cause side reactions, Tr-protected 1H-imidazolyl group of the ester 100 was used: treatment with TrCl and Et3N afforded compound 101 in quantitative yield. Hydrolysis of the methyl ester with LiOH in MeOH/H2O gave the acid 102 as a single product (Scheme 26). The Boc-β3hHis(Tr)-OH 102 was thus prepared from α-Boc-His(Ts)-OH in ca. 75% yield over four steps.
Scheme 26.

Preparation of the β3-homo-histidine derivative 102 by Arndt-Eistert homologation of Boc-His(Ts)-OH, followed by Tr-protection and saponification of the methyl ester group in 101 [80].
The successful preparation of Fmoc-β3hHis(Tr)-OH was accomplished by using the Boc-protected homo-histidine esters 100 as starting materials. As such, Boc deprotection, followed by saponification or hydrogenolysis of the ester groups, gave the completely unprotected β3-homo-histidine, which was then phthaloyl(Phth)-protected and acidified to yield the HCl salt 103 in 67% yield. Subsequent tritylation and N-Phth/N-Fmoc protecting-group exchange afforded the acid 104 in 61% yield over the three steps (Scheme 27).
Scheme 27.

Preparation of Fmoc-β3hHis(Tr)-OH 104 starting from 100 [80].
In this way, the synthesis of Fmoc-β3hHis(Tr)-OH was achieved in eight steps with an overall yield of 32%, starting from commercial Boc-His(Ts)-OH.
Recently, Wyatt and co-workers [81] accomplished the synthesis of Boc-β3hHis-(Boc)-OH 110 via the Kolbe reaction, i.e., (Scheme 28), by reducing α-Mts-His(Mts)-OMe 105 to the corresponding amino alcohol 106 in 58% yield, the OH group of which was then activated as its methanesulfonate 107 (81%) and replaced by CN, to give compound 109; treatment of the methanesulfonate 107 with NaCN (1 equiv.) in DMF at room temperature gave mainly the aziridine 108, which could be ring-opened by excess cyanide to give the desired nitrile 109; by using two or more equivalents of NaCN in DMF at room temperature the methanesulfonate 107 could be converted into the nitrile 109 (63%) in a single step. The CN group was, in turn, hydrolyzed to the MeNH group, which was Boc-protected to give the final product 110 in 7% yield over five steps. The authors state that this derivative is suitable for direct use in the synthesis of peptides, but so far no applications are known.
Scheme 28.

Preparation of Boc-β3hHis(Boc)-OH 110 via Kolbe reaction [81].
4.11. Aza-Histidine
The preparation of aza-histidine (Figure 10) has been already described [82] and the synthetic routes published to date to obtain the fully deprotected amino acid are usually rather long (seven steps) [83].
Figure 10.
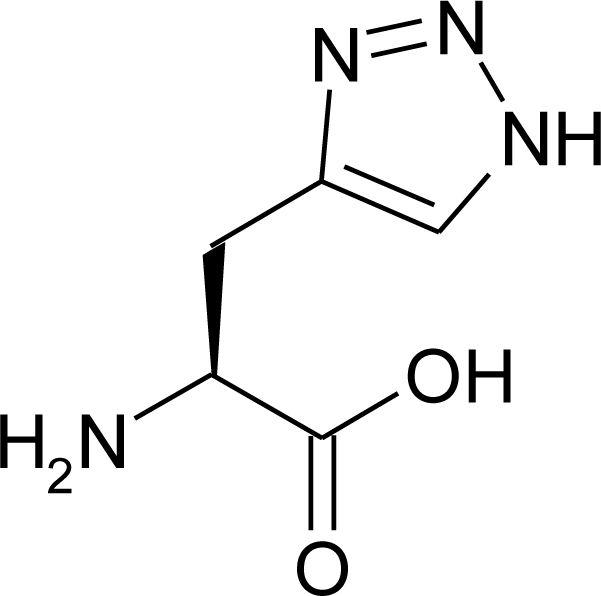
Triazole analogue of histidine.
The recent development of the copper catalysed version of Huisgen cycloaddition allows the rapid access to 1,2,3-triazoles then aza-histidine derivatives have been obtained with this method, but no real attention has been paid to the removal of the protecting groups.
Cintrat et al. [82] described the click chemistry based access to fully protected aza-histidine, suitable for solid phase peptide synthesis (Scheme 29).
Scheme 29.

Aza-histidine analogues via [3 + 2] Huisgen cycloaddition [82].
Aza-histidine were obtained using either copper-catalysed click chemistry or the more recently described ruthenium catalyzed cycloaddition, affording the 1,5-disubstituted triazole. Nevertheless, the latter requires the fully protected propargylglycine as starting material, since the catalyst does not allow the use of free carboxylic acids. Using either condition, rapid access to protected aza-histidine was demonstrated and the expected regioisomers were obtained with fair to satisfactory yields.
Robinson et al. [84] proposed the synthesis of dl-α-Amino-l,2,3-triazole-4-propionic acid 112, prepared by two independent routes. The catalytic hydrogenation of the oximino acid was extremely slow and the yield of the amino acid was small (17%). As an unambiguous method, the synthesis was also accomplished through the azlactone. From 1,2,3-triazole-4-carboxaldehyde 111, the crude azlactone was obtained (52%) as a mixture of a ring-acetylated azlactone and a small amount of the non-acetylated form which was separated through its insolubility in chloroform. The latter compound was readily converted into the former by acetylation. Purified samples of both forms gave α-benzamido-1,2,3-triazole-4-acrylic acid dihydrate on hydrolysis (86–88%). The yield of the acrylic acid dihydrate from the crude mixture of azlactones was 85%. α-Benzamido-l,2,3-triazole-4-propionic acid was obtained in 61% yield by hydrogenation of the acrylic acid in glacial acetic acid using platinum oxide. Hydrolysis of this benzamido acid gave analytically pure dl-amino-l,2,3-triazole-4-propionic acid 112 in 51% yield (Scheme 30).
Scheme 30.

Synthesis of dl-α-Amino-1,2,3-triazole-4-propionic acid [84].
Finally, Boyd et al. [85] explored variations in the imidazole portion of α-adrenoceptor agonists, synthesizing a series of compounds, among them substituted (+,−)-aza-histidine, shown in Scheme 31. The anion of ketoamide 113 was alkylated with a benzyl-protected heteroarylmethyl chloride to furnish the protected amidoketone 114. Debenzylation of these compounds was accomplished by catalytic hydrogenation with Pearlman’s catalyst [Pd(OH)2]. Finally, the target compounds were obtained by cyclization of these intermediate to thiazole.
Scheme 31.

Synthesis of substituted aza-histidine [85].
5. Conclusions
Topographical considerations are an important approach for exploring the stereochemical requirements for receptor recognition and for signal transduction [2,86].
In this approach, side chain constrained novel amino acids are designed and incorporated into peptide templates. The use of pure chiral α- or β-substituted amino acids in bioactive peptide ligands as key pharmacophore residues has proven to be a powerful tool for understanding ligand-receptors binding interaction and in peptidomimetics design [7].
One of the major goals of medicinal chemistry has been the design and synthesis of novel amino acids with conformationally constrained side chains in order to obtain highly selective and potent peptide hormone and neurotransmitter analogues. Incorporation of different β-substituted amino acids (Figure 11), e.g., β-methylphenylalanine 115, β-methyltyrosine 116, β-methyl-2’,6’-dimethyltyrosine 117, and β-methyltryptophan 118, provided new insights into the stereochemical requirements of peptide-receptor interactions [87–90]. Beta-methylhistidine (119 in Figure 11) is an important amino acid involved in signal transduction mechanism necessary to express biological activities [91] and in several key role receptor interactions, one of which being glucagone with its receptor.
Figure 11.

Topographical changes can greatly affect the potency and selectivity of peptidic ligands. Though this approach to ligand design is embryonal, significant progress has been made and some very impressive peptide-based ligands have been discovered.
The major difficulties to synthesize substituted asymmetric histidine derivatives arise from the imidazole ring, which is a general base and a nucleophile. Thus, the imidazole group can cause epimerization and may participate in different reactions during the synthesis of asymmetric amino acid derivatives [8].
There remains a need for a deeper understanding of the conformational properties of many of these amino acids as well as for the design, synthesis and conformational analysis of novel amino acids with well-defined χ1 and χ2 angles.
The Importance of Histidine Amino Acid as Residue in Bioactive Molecules and in the Synthesis of Biologically Active Compounds
An example of the use of l-histidine in the synthesis of biologically active compounds is reported by Gonzales et al. [92]: They prepared the cyclic carbamate analogs of 120–122 as a part of a novel series of muscarinic agonists containing the 2-oxazolidinone ring system, and the conformationally restricted derivatives 123–125 (Figure 12), the latter synthesized by the Pictet-Spengler reaction.
Figure 12.

(+) Pilocarpine analogs [92].
It is of interest that no rigid analog of (+)-pilocarpine has been reported to have more than approximately 1% of the agonist activity of the naturally occurring alkaloid.
Tourwè et al. [93] replaced the histidine residue in angiotensin IV by various conformationally constrained amino acids (Figure 13).
Figure 13.

Conformationally constrained aromatic residues used as His replacements in Ang IV [93].
The substitution of the His4-Pro5 dipeptide sequence by the constrained Trp analogue Aia-Gly, in combination with β2hVal substitution at the N-terminus, provided a new stable analogue H-(R)-β2hVal-Tyr-Ile-Aia-Gly-Phe-OH (AL-40) that is a potent ligand for the Ang IV receptor IRAP and selective versus AP-N and the AT1 receptor.
References
- 1.Deschamps JR, Flippen-Anderson JL, George C. X-ray studies on ligands. Biopolymers. 2003;66:287–293. doi: 10.1002/bip.10308. [DOI] [PubMed] [Google Scholar]
- 2.Hruby VJ, Li G, Haskell-Luevano C, Shenderovich MD. Design of peptides, proteins, and peptidomimetics in χ space. Biopolymers. 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Pinnen F, Zanotti G, Lucente G, Cerrini S, Fedeli W, Gavuzzo E. Cyclodepsitripeptides. Synthesis, Crystal Structure and Molecular Conformations of cyclo(-l-2-Hydroxyisovaleryl-l-prolyl-l-prolyl-) and cyclo(-d-2-Hydroxyisovaleryl-l-prolyl-l-prolyl-) J. Chem. Soc. Perkin Trans. II. 1985;12:1931–1937. [Google Scholar]
- 4.Cerrini S, Gavuzzo E, Lucente G, Luisi G, Pinnen F, Radics L. Ten-membered cyclotripeptides. Int. J. Peptide Prot. Res. 1991;38:289–297. [PubMed] [Google Scholar]
- 5.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv. Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 6.Hruby VJ. Conformational and topographical considerations in the design of biologically active peptides. Biopolymers. 1993;33:1073–1082. doi: 10.1002/bip.360330709. [DOI] [PubMed] [Google Scholar]
- 7.Hruby VJ. In: Peptides: Chemistry, Structure and Biology. Hodges RS, Wieland T, Karger S, editors. Karger; Basel, Switzerland: 1995. pp. 207–220. [Google Scholar]
- 8.Wang S, Tang X, Hruby VJ. First stereoselective synthesis of an optically pure β-substituted histidine: (2S,3S)-β-methylhistidine. Tetrahedron Lett. 2000;41:1307–1310. [Google Scholar]
- 9.Schiavone MT, Santos RA, Brosnihan KB, Khosla MC, Ferrario CM. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1–7) heptapeptide. Proc. Natl. Acad. Sci. USA. 1988;85:4095–4098. doi: 10.1073/pnas.85.11.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeburg PH, Adelman JP. Characterization of cDNA for precursore of human luteinizing hormone releasing hormone. Nature. 1984;311:666–668. doi: 10.1038/311666a0. [DOI] [PubMed] [Google Scholar]
- 11.Pickart L, Freedman JH, Loker WJ, Peisach J, Perkins CM, Stenkamp RE, Weinstein B. Growth-modulating plasma trypeptide may function by facilitating cooper uptake into cells. Nature. 1980;288:715–717. doi: 10.1038/288715a0. [DOI] [PubMed] [Google Scholar]
- 12.Sundberg RJ, Martin RB. Interactions of histidine and other imidazole derivatives with transition metal ions in chemical and biological system. Chem. Rev. 1974;74:471–517. [Google Scholar]
- 13.McNaught AD, Wilkinson A. Compendium of Chemical Terminology: IUPAC Reccomandations. 2nd ed. Blackwell Scientific Publications; Oxford, UK: 1997. p. 43. [Google Scholar]
- 14.Morera E, Nalli M, Mollica A, Paradisi MP, Aschi M, Gavuzzo E, Mazza F, Lucente G. Peptides containing 4-amino-1,2-dithiolane-4-carboxylic acid (Adt): Conformation of Boc-Adt-Adt-NHMe and NH...S interactions. J. Pept. Sci. 2005;11:104–112. doi: 10.1002/psc.602. [DOI] [PubMed] [Google Scholar]
- 15.Robinson B, Shepherd DM. The preparation of dl-α-methylhistidine dihydrochloride. J Chem Soc. 1961:503–508. [Google Scholar]
- 16.Snyder HR, Smith CW, Curtius W, Stewart JM. C-Alkylation with quaternary ammonium salts. A new approach to the synthesis of compounds containing the β-indolemethylene group. J. Am. Chem. Soc. 1944;66:200–204. [Google Scholar]
- 17.Weidenhagen R, Harrmann R. A new synthesis of imidazole derivatives. Chem. Ber. 1935;68:1953–1961. [Google Scholar]
- 18.Turner RA, Huebner CF, Scholz CR. Studies on imidazole compounds. I. 4-Methylimidazole and related compounds. J. Am. Chem. Soc. 1949;71:2801–2803. [Google Scholar]
- 19.Schmidt KF. The imine residue. Chem. Ber. 1924;57:704–706. [Google Scholar]
- 20.Sletzinger M, Pfister K. α-alkyl histidines. 1965 US Patent 3,169,971. [Google Scholar]
- 21.Kornblum N, Blackwood RK. Ethyl-α-nitrobutyrate. Org. Synth. 1957;37:44–46. [Google Scholar]
- 22.O’Donnell MJ, Rusterholz DB. Synthesis of α-methylhistidine by catalytic phase-transfer alkylations. Synt. Commun. 1989;19:1157–1165. [Google Scholar]
- 23.Matsumoto K, Miyahara T, Suzuki M, Miyoshi M. Synthesis of amino acids and related compounds. 8. Improved synthesis of dl-histidine. Agric. Biol. Chem. 1974;38:1097–1115. [Google Scholar]
- 24.Kelley JL, Miller CA, McLean EW. Attempted inhibition of histidine decarboxylase with β-alkyl analogs of histidine. J. Med. Chem. 1977;20:721–723. doi: 10.1021/jm00215a021. [DOI] [PubMed] [Google Scholar]
- 25.Albertson NF, Archer S. Use of Et acetamidomalonate in the synthesis of amino acids. Preparation of dl-histidine, dl-phenilalanine and dl-leucine. J. Am. Chem. Soc. 1945;67:308–310. [Google Scholar]
- 26.Evans DA, Weber AE. Asymmetric glycine enolate aldol reactions: Synthesis of cyclosporin’s unusual amino acid, MeBmt. J. Am. Chem. Soc. 1986;108:6757–6761. [Google Scholar]
- 27.Pyman FL. Derivatives of glyoxaline-4(or 5)-formaldehyde and glyoxaline-4(or 5)-carboxylic acid. A new synthesis of histidine. J. Chem. Soc. 1916;109:186–202. [Google Scholar]
- 28.Saha AK, End DW. Novel β-(imidazol-4-yl)-β-amino acids: Solid-phase synthesis and study of their inhibitory activity against geranylgeranyl protein transferase type I. Biorg. Med. Chem. Lett. 2005;15:1713–1719. doi: 10.1016/j.bmcl.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 29.Davies SG, Ichihara O. Asymmetric synthesis of R-β-amino butanoic acid and S-β-tyrosine: Homochiral lithium amide equivalents for Michael additions to α,β-unsaturated esters. Tetrahedron: Asimmetry. 1991;2:183–186. [Google Scholar]
- 30.De Graw JI, Engstrom J, Ellis M, Johnson HL. Potential histidine decarboxylase inhibitors. I. α- and β-substituted histidine analogues. J. Med. Chem. 1977;20:1671–1674. doi: 10.1021/jm00222a027. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch A, Richardson K. Reactions of Histidine. J. Appl. Chem. 1969;19:83–85. [Google Scholar]
- 32.Goodson LH, Honigberg IL, Lehman JJ, Burton WH. Potential growth antagonists. I. Hydantoins and di-substituted glycine. J. Org. Chem. 1960;25:1920–1924. [Google Scholar]
- 33.Pictet A, Spengler T. Formation of isoquinoline derivatives by the action of methylal on phenylethylamine, phenylalanine and tyrosine. Ber. Dtsch. Chem. Ges. 1911;44:2030–2036. [Google Scholar]
- 34.Whaley WM, Govindachari TR. The Pictet-Spengler of tetrahydroisoquinolines and related compounds. Org. React. 1951;6:151–190. [Google Scholar]
- 35.Verschueren K, Toth G, Tourwe D, Lelb M, van Binst G, Hruby VJ. A facile synthesis of 1,2,3,4-tetrahydro-7-hydroxyisoquinoline-3-carboxyilic acid, a conformationally constrained tyrosine analog. Synthesis. 1992:458–460. [Google Scholar]
- 36.Shinkai H, Toi K, Kumashiro I, Seto Y, Fukuma M, Dan K, Toyoshima S. N-acylphenylalanines and related compounds. A new class of oral hypoglycemic agents. J. Med. Chem. 1988;31:2092–2097. doi: 10.1021/jm00119a007. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi K, Ozaki Y, Nunami K, Uchida T, Kato J, Kinashi K, Yoneda N. Studies on angiotensin-converting enzyme inhibitors. I. Syntheses and angiotensin-converting enzyme inhibitory activity of 2-(3-mercaptopropionyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives. Chem. Pharm. Bull. 1983;31:570–576. doi: 10.1248/cpb.31.570. [DOI] [PubMed] [Google Scholar]
- 38.Remelli M, Munerato C, Pulidori F. Binary and ternary copper (II) complexes of Nτ- and Nπ- methyl-l-histidine in aqueous solution. J. Chem. Soc. Dalton Trans. 1994;14:2049–2056. [Google Scholar]
- 39.Remelli M, Rossi S, Guerrini R, Pulidori F. Binary and ternary copper (II) complexes of l-spinacine in aqueous solution. Ann. Chim. 1995;85:503–518. [Google Scholar]
- 40.Salvadori S, Guerrini R, Forlani V, Bryant SD, Attila M, Lazarus LH. Prerequisite for His4 in deltorphin A for high δ-opioid receptor selectivity. Amino Acids. 1994;7:291–304. doi: 10.1007/BF00807704. [DOI] [PubMed] [Google Scholar]
- 41.Wellisch J. Synthetic alkaloids from tyrosine, tryptophane and histidine. Biochem. Z. 1913;49:173–194. [Google Scholar]
- 42.Klutchko S, Hodges JC, Blankley CJ, Colbry NL. 4,5,6,7-Tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acids (Spinacines) J. Heterocycl. Chem. 1991;28:97–108. [Google Scholar]
- 43.Remelli M, Pulidori F, Guerrini R, Bertolasi V. Synthesis of spinacine and spinacine derivatives: Crystal and molecular structures of Nπ-hydroxy-methyl spinacine and Nα-methyl spinaceamine. J. Chem. Crystallogr. 1997;27:507–513. [Google Scholar]
- 44.Guzman F, Cain M, Larscheid P, Hagen T, Cook JM, Schweri M, Skolnick P, Paul SM. Biomimetic approach to potential benzodiazepine. Receptor agonists and antagonists. J. Med. Chem. 1984;27:564–570. doi: 10.1021/jm00371a002. [DOI] [PubMed] [Google Scholar]
- 45.Wille MA. The base-catalyzed Pictet-Spengler reaction Synthesis of nitrogen heterocyclic compounds from the reaction of histamine and tryptamine analogs with aromatic aldehydes in alkaline medium. 1969. PhD Thesis. University of Pennsylvania, Philadelphia, PA, USA. [Google Scholar]
- 46.Andreetti GD, Cavalca L, Sgarabotto P. Crystal and molecular structure of the amino acid spinacine dihydrate [4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridine-6-carboxylic acid dihydrate] Gazz. Chim. Ital. 1971;101:625–634. [Google Scholar]
- 47.McMullan RK, Epstein J, Ruble JR, Craven BM. The crystal structure of imidazole at 103 K by neutron diffraction. Acta Crystallogr. 1979;B35:688–691. [Google Scholar]
- 48.Madden JL, McGandy EL, Seeman NC. The crystal structure of the monoclinic form of l-Histidine. Acta Crystallogr. 1972;B28:2377–2382. [Google Scholar]
- 49.Cremer D, Pople JA. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975;97:1354–1358. [Google Scholar]
- 50.Nardelli M. Ring asymmetry parameters from out-of plane atomic displacements. Acta Crystallogr. 1983;C39:1141–1142. [Google Scholar]
- 51.Gibson SE, Guillo N, Tozer MJ. Towards control of χ-space: Conformationally constrained analogues of Phe, Tyr, Trp and His. Tetrahedron. 1999;55:585–615. [Google Scholar]
- 52.Pages RA, Burger A. 1-Amino-2-(4-imidazolyl)cyclopropanecarboxylic acid. J. Med. Chem. 1966;9:766–768. doi: 10.1021/jm00323a030. [DOI] [PubMed] [Google Scholar]
- 53.Awad WI, Fateen AK, Zayed MA. 2-Phenyl-4-arylidene-5-oxazolones. Tetrahedron. 1964;20:891–896. [Google Scholar]
- 54.Mustafa A, Asker W, Harhash AH, Fleifel AM. Reactivity of unsaturated centers in heterocycles and chalcones towards diazoalkanes. Tetrahedron. 1965;21:2215–2229. [Google Scholar]
- 55.Easton CJ, Hutton CA, Roslet PD, Tiekink ERT. A stereocontrolled synthesis of β-hydroxyphenylalanine and β-hydroxytyrosine derivatives. Tetrahedron. 1994;50:7327–7340. [Google Scholar]
- 56.Easton CJ, Hutton CA, Tan EW, Tiekink ERT. Synthesis of homochiral hydroxyl-α-amino acid derivatives. Tetrahedron Lett. 1990;31:7059–7062. [Google Scholar]
- 57.Hutton CA. Substituent effects in the stereoconvergent synthesis of β-hydroxyphenylalanine derivatives. Tetrahedron Lett. 1997;38:5899–5902. [Google Scholar]
- 58.Croft AK, Easton CJ, Kociuba K, Radom L. Strategic use of amino acid N-substituents to limit α-carbon-centered radical formation and consequent loss of stereochemical integrity. Tetrahedron: Asymmetry. 2003;14:2919–2126. [Google Scholar]
- 59.Crich D, Banerjee A. Expedient synthesis of threo-β-hydroxy-α-amino acid derivatives: Phenylalanine, Tyrosine, Histidine and Tryptophan. J. Org. Chem. 2006;71:7106–7109. doi: 10.1021/jo061159i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu J, Yadan JC. Synthesis of l-(+)-Ergothioneine. J. Org. Chem. 1995;60:6296–6301. [Google Scholar]
- 61.Battersby AR, Nicoletti M, Staunton J, Vleggaar R. Studies of enzyme-mediated reactions. Part 13. Stereochemical course of the formation of histamine by decarboxylation of (2S)-histidine with enzymes from Clostridium welchii and Lactobacillus 30a. J Chem Soc Perkin I. 1980:43–51. doi: 10.1039/p19800000043. [DOI] [PubMed] [Google Scholar]
- 62.Cativiela C, Diaz de Villegas MD, Garcia JI, Mayoral JA, Melendez E. Stereoselective synthesis of (Z)-2-(acylamino)-3-hetaryl-2-propenoic acids. An. Quim. 1985;81:56–61. [Google Scholar]
- 63.Parker AR, Moore JA, Schwab JM, Jo Davisson V. Escherichia Coli imidazoleglycerol phosphate dehydratase: Spectroscopic characterization of the enzymic product and the steric course of the reaction. J. Am. Chem. Soc. 1995;117:10605–10613. [Google Scholar]
- 64.Toshiyuki SJ. New imidazole derivative, method for producing the same, and method for producing histidine amide derivative using the imidazole derivative. JP Patent 2010031004. 2010 [Google Scholar]
- 65.Bloemhoff W, Kerling KET. Polypeptides, X.V. Synthesis of l- and d-homo-histidine. Rec. Trav. Chim. Pays-Bas. 1975;94:182–185. [Google Scholar]
- 66.Altman J, Wilchek M, Lipp R, Schunack W. An improved synthesis of l-homo-histidine. Synth. Commun. 1989;19:2069–2076. [Google Scholar]
- 67.Lee Y, Martasek P, Roman LJ, Masters BSS, Silverman RB. Imidazole-containing amino acids as selective inhibitors of nitric Oxide synthase. Biorg. Med. Chem. 1999;7:1941–1951. doi: 10.1016/s0968-0896(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 68.Pirrung MC, Pei TJ. Synthesis of (+)-Homo-histidine. J. Org. Chem. 2000;65:2229–2230. doi: 10.1021/jo991630q. [DOI] [PubMed] [Google Scholar]
- 69.Browne LJ, Gude C, Rodriguez H, Steele RE, Bhatnager A. Fadrozole hydrochloride: A potent, selective, nonsteroidal inhibitor of aromatase for the treatment of estrogen-dependent desease. J. Med. Chem. 1991;34:725–736. doi: 10.1021/jm00106a038. [DOI] [PubMed] [Google Scholar]
- 70.Court TT, Chester PW, Ila S. Amino-substituted heterocycles as rennin inhibitors. US Patent 5,643,879. 1997 [Google Scholar]
- 71.Mazur RH, Schlatter JM, Goldkamp AH. Structure-taste relationships of some dipeptides. J. Am. Chem. Soc. 1969;91:2684–2691. doi: 10.1021/ja01038a046. [DOI] [PubMed] [Google Scholar]
- 72.Janusz JM, Young PA, Blum RB, Riley CM. High-Potency Dipeptide Sweeteners. 2. l-Aspartylfuryl-, Thienyl-, and Imidazolylglycine Esters. J. Med. Chem. 1990;33:1676–1682. doi: 10.1021/jm00168a022. [DOI] [PubMed] [Google Scholar]
- 73.van Batenburg OD, Kerling KET, Havinga E. Studies on polypeptides. Part XIX. Synthesis and enzymic activity of an RNase S’ analog in which the 4-imidazolylglycyl residue takes the position and the role of histidine-12. FEBS Lett. 1976;68:228–230. doi: 10.1016/0014-5793(76)80442-5. [DOI] [PubMed] [Google Scholar]
- 74.van Batenburg OD, Kerling KET. Improved synthesis of tert-butyloxycarbonyl-l-histidine. Int. J. Pept. Prot. Res. 1976;8:1–2. doi: 10.1111/j.1399-3011.1976.tb02473.x. [DOI] [PubMed] [Google Scholar]
- 75.Furukawa M, Arimoto M, Nakamura S, Ejima A, Higashi O, Tagawa H. Semisynthetic β-lactam antibiotics. I. Synthesis and and antibacterial activity of 7β-[2-aryl-2-(aminoacetamido)acetamido]cephalosporins. J. Antibiot. 1986;9:1225–1235. doi: 10.7164/antibiotics.39.1225. [DOI] [PubMed] [Google Scholar]
- 76.Etoga JLG, Ahmed SK, Patel S, Bridges RJ, Thompson CM. Conformationally-restricted amino acid analogues bearing a distal sulfonic acid show selective inhibition of system x_c over the vesicular glutamate transporter. Bioorg. Med. Chem. Lett. 2010;20:2680–2683. doi: 10.1016/j.bmcl.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chruma JJ, Liu L, Zhou W, Breslow R. Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics. Bioorg. Med. Chem. 2005;13:5873–5883. doi: 10.1016/j.bmc.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 78.Nenajdenko VG, Zakurdaev EP, Prusov EV, Balenkova ES. Convenient synthesis of melatonin analogues: 2- and 3-substituted-N-acetylindolylalkylamines. Tetrahedron. 2004;60:11719–11724. [Google Scholar]
- 79.Stalker RA, Munsch TE, Tran JD, Nie X, Warmuth R, Beatty A, Aakeröy CB. Asymmetric synthesis of two new conformationally constrained lysine derivatives. Tetrahedron. 2002;58:4837–4849. [Google Scholar]
- 80.Lelais G, Micuch P, Lefebvre DJ, Rossi F, Seeback D. Preparation of Protected β2- and β3-Homo-cysteine, β2- and β3-Homo-histidine, and β2-Homo-serine for solid-phase Syntheses. D. Helv. Chim. Acta. 2004;87:3131–3159. [Google Scholar]
- 81.Kumar A, Ghilagaber S, Knight J, Wyatt PB. The homologation of Histidine. Tetrahedron Lett. 2002;43:6991–6994. [Google Scholar]
- 82.Roux S, Ligeti M, Buisson DA, Rousseaux B, Cintrat JC. Synthesis of orthogonally protected aza-histidine: Application to the synthesis of a GHK analogue. Amino Acids. 2010;38:279–286. doi: 10.1007/s00726-009-0248-5. [DOI] [PubMed] [Google Scholar]
- 83.Ikeda Y, Kawahara S, Taki M, Kuno A, Hasegawa T, Taira K. Synthesis of a novel histidine analog and its efficient incorporation into a protein in vivo. Protein Eng. 2003;16:699–706. doi: 10.1093/protein/gzg084. [DOI] [PubMed] [Google Scholar]
- 84.Sheenan JC, Robinson CA. The synthesis of triazole analogues of histamine and related compounds. J. Am. Chem. Soc. 1949;71:1436–1440. doi: 10.1021/ja01172a083. [DOI] [PubMed] [Google Scholar]
- 85.Boyd RE, Press JB, Rasmussen CR, Raffa RB. α2 Adrenoceptor Agonists as Potential Analgesic Agents. 1. (Imidazolylmethyl)oxazoles and thiazoles. J. Med. Chem. 1999;42:5064–5071. doi: 10.1021/jm990005a. [DOI] [PubMed] [Google Scholar]
- 86.Hruby VJ, Boteju LW. In: Molecular Biology and Biotechnology. Meyers RA, editor. VCH; New York, NY, USA: 1995. pp. 658–664. [Google Scholar]
- 87.Qian X, Köver KE, Shenderovich MD, Lou BS, Misicka A, Zalewska T, Horvath R, Davis P, Bilsky EJ, Porreca F, et al. Newly discovered stereochemical requirements in the side-chain conformation of δ-opioid agonists for recognizing opioid δ-receptors. J. Med. Chem. 1994;37:1746–1757. doi: 10.1021/jm00038a004. [DOI] [PubMed] [Google Scholar]
- 88.Nikiforovich GV, Prakash OM, Gehrig CA, Hruby VJ. Solution conformations of the peptide backbone for DPDPE and its β-MePhe4-substituted analogues. Int. J. Peptide Prot. Res. 1993;41:347–361. doi: 10.1111/j.1399-3011.1993.tb00451.x. [DOI] [PubMed] [Google Scholar]
- 89.Huang Z, He YB, Raynor K, Tallent M, Reisine T, Goodman M. Main chain and side chain chiral methylated somatostatin analogs: Syntheses and conformational analyses. J. Am. Chem. Soc. 1992;114:9390–9401. [Google Scholar]
- 90.Tóth G, Russell KC, Landis G, Kramer TH, Fang L, Knapp R, Davis P, Burks TF, Yamamura HI, Hruby VJ. Ring substituted and other conformationally constrained tyrosine analogues of [D-Pen2, D-Pen5] enkephalin with δ opioid receptor selectivity. J. Med. Chem. 1992;35:238491. doi: 10.1021/jm00091a006. [DOI] [PubMed] [Google Scholar]
- 91.Hruby VJ, Krystenansky JL, McKee R, Pelton JT. Hormonal Control of Gluconeogenesis. CRC Press; Boca Raton, FL, USA: 1986. Signal Transduction; pp. 2–20. [Google Scholar]
- 92.Sànchez MS, Alberdi LMT, Rioseras MJ, Ferriera MR, Gonzales FB. The Pictet-Spengler reaction on l-Histidine. Preparation of Conformationally restricted (+)-Pilocarpine analogs. Bull. Chem. Soc. Jpn. 1998;66:191–195. [Google Scholar]
- 93.Lakaszuk A, Demaegdt H, Feytens D, Vanderheyden P, Vauquelin G, Tourwè DJ. The replacement of His(4) in Angiotensin IV by Conformationally residues provides highly potent and selective analogues. J. Med. Chem. 2009;52:5612–5618. doi: 10.1021/jm900651p. [DOI] [PubMed] [Google Scholar]