

Abstract
Objective
High expression of galectin 3 at sites of joint destruction in rheumatoid arthritis (RA) suggests that galectin 3 plays a role in RA pathogenesis. Previous studies have demonstrated the effects of galectins on immune cells, such as lymphocytes and macrophages. This study was undertaken to investigate the hypothesis that galectin 3 induces proinflammatory effects in RA by modulating the pattern of cytokine and chemokine production in synovial fibroblasts.
Methods
Matched samples of RA synovial and skin fibroblasts were pretreated with galectin 3 or tumor necrosis factor α (TNFα), and the levels of a panel of cytokines, chemokines, and matrix metalloproteinases (MMPs) were determined using enzyme-linked immunosorbent assays and multiplex assays. Specific inhibitors were used to dissect signaling pathways, which were confirmed by Western blotting and NF-κB activation assay.
Results
Galectin 3 induced secretion of interleukin-6 (IL-6), granulocyte–macrophage colony-stimulating factor, CXCL8, and MMP-3 in both synovial and skin fibroblasts. By contrast, galectin 3–induced secretion of TNFα, CCL2, CCL3, and CCL5 was significantly greater in synovial fibroblasts than in skin fibroblasts. TNFα blockade ruled out autocrine TNFα-stimulated induction of chemokines. The MAPKs p38, JNK, and ERK were necessary for IL-6 production, but phosphatidylinositol 3-kinase (PI 3-kinase) was required for selective CCL5 induction. NF-κB activation was required for production of both IL-6 and CCL5.
Conclusion
Our findings indicate that galectin 3 promotes proinflammatory cytokine secretion by tissue fibroblasts. However, galectin 3 induces the production of mononuclear cell–recruiting chemokines uniquely from synovial fibroblasts, but not matched skin fibroblasts, via a PI 3-kinase signaling pathway. These data provide further evidence of the role of synovial fibroblasts in regulating the pattern and persistence of the inflammatory infiltrate in RA and suggest a new and important functional consequence of the observed high expression of galectin 3 in the rheumatoid synovium.
Rheumatoid arthritis (RA) is a persistent systemic inflammatory disease characterized by inflammation involving multiple cell types, with the progressive destruction of involved joints (1). An essential component of the switch to persistence that underlies joint destruction is the production of chemokines, which recruit mononuclear cells, such as lymphocytes and monocytes, to the inflamed joint (2). Galectins, an evolutionarily conserved family of animal lectins, have diverse roles in cellular homeostasis and have been shown to modulate inflammatory responses, functioning as either proinflammatory or antiinflammatory regulators, in part through their ability to cluster and modulate signaling through glycan receptors associated with multiple ligands (3). This ability to influence immune responses has been demonstrated in animal models of a number of diseases, including arthritis (4).
Galectin 3, a chimera-type member of the galectin family, has a C-terminal carbohydrate recognition domain responsible for carbohydrate binding but exhibits an N-terminal domain that is responsible for interactions between subunits facilitating its oligomerization (5,6). The biologic functions attributed to this lectin are likely to depend on both ligand crosslinking and oligomerization (6,7). Galectin 3 has been associated with a proinflammatory role in models of fibrotic disease affecting the lung and liver (8,9) and has been shown to promote monocyte chemotaxis and macrophage activation (10-12) in addition to neutrophil activation, degranulation, and superoxide production (13-15), suggesting a critical role in the development of innate immune responses. Furthermore, a key role of galectin 3 has also been shown in the survival and progression of tumor metastases by modulating different processes, including homotypic and heterotypic cell adhesion, migration, angiogenesis, and tumor-immune escape (16).
In the context of synovitis, Ohshima and colleagues (17) have demonstrated increased levels of galectin 3 and its binding protein in RA synovial tissue compared with osteoarthritis (OA) synovial tissue. Furthermore, galectin 3 levels are increased in RA in both synovial fluid and peripheral blood compartments, where levels correlate with C-reactive protein (17). Interestingly, up-regulated expression of galectin 3 correlates with abnormal cell apoptosis in synovial tissue from patients with juvenile RA (18).
In contrast, galectin 1, a prototype member of the galectin family composed of 1 conserved carbohydrate recognition domain that can dimerize, has a predominantly antiinflammatory role, suppressing experimental models of inflammatory diseases, such as hepatitis, experimental autoimmune encephalomyelitis, uveitis, colitis, and arthritis (19-22). Furthermore, this glycan-binding protein appears to play an important role in the mechanisms involved in Treg cell–mediated suppression of immune responses (23), inhibition of T cell receptor–mediated signal transduction (24), and differential regulation of T helper cell viability (25). Intriguingly, synovial fibroblasts engineered to overexpress galectin 1 ameliorated collagen-induced arthritis and induced a bias toward a Th2-mediated cytokine profile in vivo (22).
Synovial fibroblasts have an established role as sentinel cells for immune cell activation in the joint (2), and in RA, these cells are responsible for secreting significant quantities of inflammatory cytokines (26). RA synovial fibroblasts actively contribute to destruction of cartilage and bone via secretion of matrix metalloproteinases (MMPs) and cathepsins, and via expression of RANKL, resulting in promotion of monocyte-to-osteoclast differentiation (27). The expanded population of synovial fibroblasts in RA is also a prolific source of chemokines responsible for the recruitment and retention of cells within the joint (2). It is clear that synovial fibroblasts are an important source of galectin 3 within the joint, as shown by messenger RNA (mRNA) and proteomic analyses (17,28). Following stimulation by the products of cartilage degradation, synovial fibroblasts also produce galectin 3 (29). However, there are likely to be many other sources of galectin 3 within the joint, including macrophages, which synthesize this glycan-binding protein in significant amounts (30).
Although considerable information is available on the cellular sources of galectins in the synovium, the downstream effects of galectin 3 on different cell types in rheumatoid synovium remain largely unexplored. We therefore examined the effects of exogenous galectin 3 on RA synovial fibroblasts, comparing them with genetically matched control skin fibroblasts. We show that inflammatory cytokines, such as interleukin-6 (IL-6), and neutrophil-attracting chemokines, such as IL-8, are produced equally by galectin 3– and tumor necrosis factor α (TNFα)–stimulated synovial and skin fibroblasts. However, in response to galectin 3, synovial fibroblasts, but not skin fibroblasts, secrete mononuclear cell–attracting chemokines such as CCL2, CCL3, and CCL5. The molecular basis for this selectivity is due to the differential activation of MAPK and phosphatidylinositol 3-kinase (PI 3-kinase) signaling pathways in response to galectin 3. The increased expression of galectin 3 found in fibroblast-rich areas of the synovium may therefore have significant functional consequences in terms of recruitment of monocyte and lymphocyte infiltrates.
MATERIALS AND METHODS
Media, antibodies, and cytokines
All tissue culture reagents were purchased from Sigma (St. Louis, MO) unless stated otherwise. Fibroblasts were cultured in RPMI 1640 supplemented with 1 mM sodium pyruvate, 1% nonessential amino acid solution, 10 mM glutamine, 100 μg/ml of streptomycin, 50 units/ml of penicillin, and 10% fetal calf serum. The following signaling pathway inhibitors were used: BAY 11-7085 (5 μM; Alexis, Nottingham, UK), LY294002 (10 μM; Calbiochem, Abingdon, UK), bisindolylmaleimide I (2 μM; Calbiochem), Gö6976 (10 nM; Calbiochem), SB202190 (10 μM; Calbiochem), R406 (10 μM; supplied by one of us [DLS; Cellzome, Cambridge, UK]), and PD98059 (10 μM; Cell Signaling Technology, Beverly, MA). Anti-TNFα blocking antibodies (R&D Systems, Minneapolis, MN) were also used. Recombinant proteins used were TNFα (R&D Systems) and galectin 3 (PeproTech, London, UK).
Patients and fibroblast lines
Tissue samples were collected from 8 patients who fulfilled the American College of Rheumatology (formerly, the American Rheumatism Association) 1987 revised criteria for RA (31). All patients exhibited erosions on radiographs of the hands and feet; 7 of 8 patients were rheumatoid factor positive. Tissue samples were also obtained from 5 patients with radiographically confirmed OA. Samples of synovium and overlying skin were obtained from the knee joint of each patient at the time of joint replacement. This study was reviewed and approved by the South Birmingham Local Ethics Committee (LREC 5735); all patients gave written informed consent.
Tissue samples were minced into ~1-mm3 sections under sterile conditions, washed, and then resuspended in 10 ml of dissociation buffer (5 mM EDTA [Sigma] in phosphate buffered saline [PBS]) and incubated for 2 hours at 4°C with vigorous shaking. The resulting cell–tissue mixture was washed 3 times in fresh medium and then cultured until adherent fibroblast colonies became confluent. Nonadherent cells and tissue fragments were discarded once adherent cells had appeared. Fibroblasts were expanded by trypsin digestion and then reseeded into tissue culture flasks of twice the surface area. Fibroblast phenotype was confirmed by morphologic appearance and by immunofluorescence microscopy. All cells expressed fibronectin and prolyl 4-hydroxylase, while <0.5% of the cells stained positive for CD68, von Willebrand factor, CD31, or cytokeratin. All experiments were performed with at least 3 sets of matched fibroblast lines. All experiments used fibroblasts between passages 2 and 6.
Fibroblast stimulation assays
Trypsinized fibroblasts were seeded into flat-bottomed 96-well plates at a density of 6 × 103 cells/well and cultured for 48 hours. Culture medium was then refreshed, and after pretreatment with medium, blocking antibodies, signaling inhibitors, or DMSO carrier control, the fibroblasts were exposed to different concentrations of galectin 3 (0–10 μg/ml) or to TNFα (10 ng/ml) for 24 hours. Tissue culture supernatants were then collected and stored at −80°C until analyzed. For inhibitor studies, the numbers and morphology of the remaining adherent cells were determined by Diff-Quick staining and microscopy, with 3 cell counts per replicate, in order to rule out a direct toxic effect of inhibitors on cell viability. Limulus amebocyte lysate assay of recombinant galectin 3 indicated endotoxin levels that were consistent with control medium, many fold below the manufacturer’s specification of <1 endotoxin unit/μg (data not shown).
Enzyme-linked immunosorbent assays (ELISAs)
Tissue culture supernatants were analyzed by ELISA for IL-6 and CCL2 (monocyte chemotactic protein 1 [MCP-1]) using OptEIA kits (PharMingen, Oxford, UK) as well as for CCL5 (RANTES) using a DuoSet kit according to the recommendations of the manufacturer (R&D Systems). The lower limits of detection for the ELISA kits used were as follows: for IL-6, 15 pg/ml; for CCL2, 2 pg/ml; and for CCL5, 16 pg/ml.
Multiplex cytokine assays
Multiple cytokines were analyzed simultaneously in supernatant samples using 15-plex cytokine and chemokine kits and 3-plex MMP kits (BioSource, Fleurus, Belgium) to measure levels of basic fibroblast growth factor, granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor, vascular endothelial growth factor (VEGF), interferon-γ (IFNγ), IFNα, IL-1β, TNFα, CCL5, CCL3, CCL4, CXCL1, CXCL8, CXCL9, CXCL10, MMP-3, MMP-9, and MMP-13. The assay was performed using a Luminex 100 System, according to the recommendations of the manufacturer (Upstate Biotechnology, Lake Placid, NY).
Western blot analysis
Fibroblasts were washed with PBS and lysed with 100 μl of sodium dodecyl sulfate (SDS) loading buffer. Lysates were heated for 10 minutes at 100°C and stored at −20°C until further use. Proteins were separated on 12% SDS–polyacrylamide gel electrophoresis gels at 40V and then transferred to a 0.45-μm polyvinylidene difluoride membrane (Flowgen, Nottingham, UK) at 450 mA using a wet blotting system (Bio-Rad, Richmond, CA). The membranes were blocked with 5% bovine serum albumin (BSA) in Tris buffered saline (TBS) with 0.1% Tween for 1 hour at room temperature. The following specific primary antibodies were diluted in 0.1% Tween–TBS with 1% BSA: Akt/protein kinase B (Transduction Laboratories, Lexington, KY); phospho-Akt, p44/42 (ERK-1/2), and phospho-p44/42 (Cell Signaling Technology); p38, phospho-p38, and JNK1 (Santa Cruz Biotechnology, Santa Cruz, CA); and phospho-JNK (Promega, Madison, WI). The membranes were incubated with appropriate antibody overnight at 4°C with shaking and then washed 3 times for 15 minutes and incubated for 1 hour with species-specific horseradish peroxidase–conjugated secondary antibody (Amersham Biosciences, Roosendaal, The Netherlands) diluted to 1:5,000. After washing 3 times, the membranes were developed with an enhanced chemiluminescence system (Amersham Pharmacia Biotech, Little Chalfont, UK) and exposed on Kodak X-Omat LS film. The blots were scanned using a Syngene gel documentation system (Syngene, Cambridge, UK).
NF-κB activation assay
To assess the activation of transcription factor NF-κB (p65 subunit), nuclear extracts from galectin 3 or TNFα-stimulated fibroblasts were prepared using a Nuclear Extract kit according to the recommendations of the manufacturer (Active Motif, Rixensart, Belgium). The protein content of each sample was measured by bicinchoninic acid assay (Pierce, Rockford, IL), and equal amounts of protein were obtained from each sample to perform the TransAM p65 assay (Active Motif). The TransAM kit uses 96-well microtiter plates coated with an oligonucleotide containing the NF-κB consensus-binding sequence. The active form of the p65 subunit in whole cell lysates was detected using antibodies specific for an epitope that is accessible only when the subunit is activated and bound to its target DNA. Detection of the activated DNA-bound form of p65 was performed according to the recommendations of the manufacturer, and the results were calculated as the fold increase compared with unstimulated cells.
Statistical analysis
Nonparametric distribution was assumed for all assays. For the difference between paired (stimulation, inhibitor, and blockade) experiments, significance was assessed using the Wilcoxon signed rank test with 2-tailed P values. Results are shown as the mean ± SD or the mean ± SEM where appropriate.
RESULTS
Production of proinflammatory cytokines and MMPs in both synovial fibroblasts and skin fibroblasts after stimulation with galectin 3
We first investigated the effects of stimulation with exogenous recombinant galectin 3 on synovial fibroblasts by measuring the production of a panel of cytokines, growth factors, and MMPs. By using genetically matched skin fibroblasts (as control fibroblasts), we eliminated the confounders that are frequently present in studies of fibroblast lines obtained from different subjects of widely varying age, disease subtype, and prior therapy. Galectin 3 stimulated significant increases in levels of IL-6, GM-CSF, and MMP-3 production by both synovial and skin fibroblasts (Figures 1A–C) in a manner similar to that seen with TNFα stimulation (32). Unexpectedly, a small, but consistent, quantity of TNFα was secreted by synovial fibroblasts, but not skin fibroblasts, stimulated with galectin 3 (Figure 1D).
Figure 1.
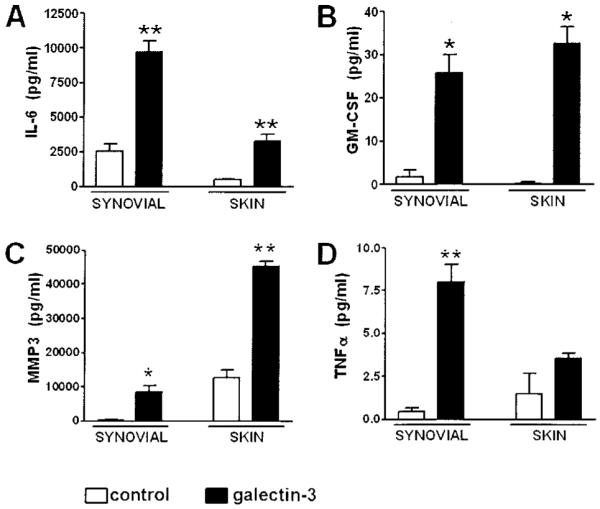
Production of proinflammatory cytokines and matrix metalloproteinases in synovial and skin fibroblasts after stimulation with galectin 3. Matched samples of synovial and skin fibroblasts from patients with rheumatoid arthritis were seeded and treated with medium (control) or recombinant galectin 3 (10 μg/ml). A, Production of interleukin-6 (IL-6) in synovial and skin fibroblasts after 24 hours of stimulation, as analyzed by enzyme-linked immunosorbent assay. B–D, Production of granulocyte–macrophage colony-stimulating factor (GM-CSF) (B), matrix metalloproteinase 3 (MMP-3) (C), and tumor necrosis factor α (TNFα) (D) in synovial and skin fibroblasts after 24 hours of stimulation, as analyzed by multiplex bead assay. Bars show the mean and SD of duplicate assays combined from at least 3 patients. * = P < 0.05; ** = P < 0.01, versus control medium.
Selective production of mononuclear cell–attracting chemokines in synovial, but not skin, fibroblasts after stimulation with galectin 3
To determine whether galectin 3 affected the production of chemokines associated with the arthritogenic process, we analyzed chemokine production in synovial fibroblasts compared with matched skin fibroblasts. Stimulation with galectin 3 greatly increased levels of secretion of CXCL8 (IL-8), a prototypic neutrophil chemoattractant, from both synovial and skin fibroblasts (Figure 2A). In complete contrast, increased levels of chemokines responsible for the recruitment of mononuclear cell populations (e.g., CCL2, CCL3, and CCL5) were produced by galectin 3–stimulated synovial fibroblasts only (and not by skin fibroblasts) (Figures 2C, E, and G). In particular, significant levels of CCL5 were seen only in RA synovial fibroblasts stimulated with 10 μg/ml of galectin 3.
Figure 2.
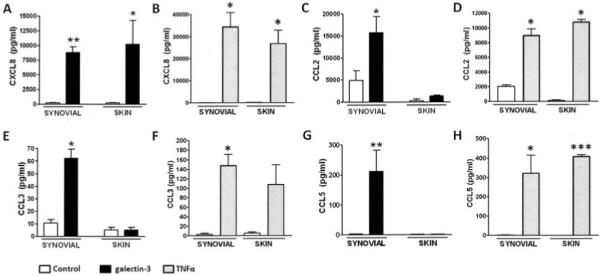
Selective production of chemokines capable of attracting a mononuclear cell population in synovial fibroblasts, but not skin fibroblasts, after stimulation with galectin 3. Matched samples of synovial and skin fibroblasts from patients with rheumatoid arthritis were seeded and treated with medium (control), recombinant galectin 3 (10 μg/ml), or recombinant tumor necrosis factor α (TNFα; 10 ng/ml). Galectin 3 induced the production of chemokines in synovial fibroblasts only, while TNFα induced the production of chemokines in all fibroblasts regardless of tissue of origin. A–D, Production of CXCL8 (A and B) and CCL2 (C and D) in synovial and skin fibroblasts after 24 hours of stimulation with galectin 3 (A and C) or TNFα (B and D), as analyzed by multiplex bead assay. E–H, Production of CCL3 (E and F) and CCL5 (G and H) in synovial and skin fibroblasts after 24 hours of stimulation with galectin 3 (E and G) or TNFα (F and H), as analyzed by enzyme-linked immunosorbent assay. Bars show the mean and SD of duplicate assays combined from at least 5 patients. * = P < 0.05; ** = P < 0.01; *** = P < 0.001, versus control medium.
Of note, a concentration of 10 μg/ml was selected because it corresponds to levels recently found to be pathologically relevant in an experimental model of Crohn’s disease (33). The effect of galectin 3 was titratable (Figure 3) and became saturating beyond the chosen concentration of 10 μg/ml. CCL5 production was very low in RA skin fibroblasts and was absent in 2 of the 4 lines tested. OA synovial fibroblasts stimulated with galectin 3 produced both IL-6 and CCL5, but in lesser quantities than did RA synovial fibroblasts (Figure 3). Similar results were also seen for CCL4; results for other cytokines, chemokines, and growth factors are shown in Table 1.
Figure 3.
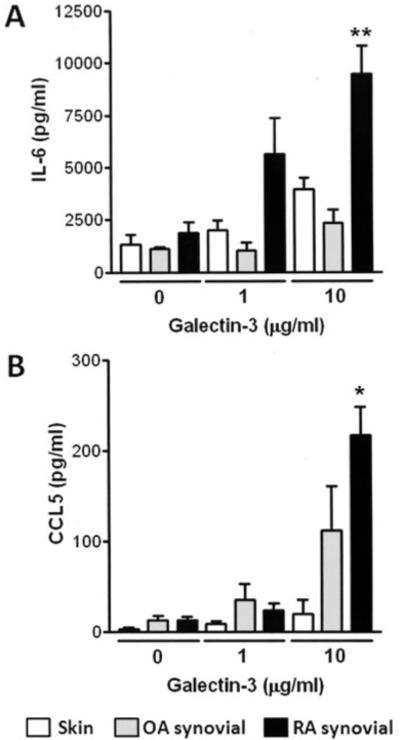
Effect of galectin 3 concentration on interleukin-6 (IL-6) and CCL5 production in rheumatoid arthritis (RA) skin fibroblasts, osteoarthritis (OA) synovial fibroblasts, and RA synovial fibroblasts. Skin, OA synovial, and RA synovial fibroblasts were seeded and treated with recombinant galectin 3 at the concentrations shown. Production of IL-6 (A) and CCL5 (B) after 24 hours of stimulation was analyzed by enzyme-linked immunosorbent assay. Bars show the mean and SD of duplicate assays combined from at least 4 patients. * = P < 0.05; ** = P < 0.01, versus OA and skin fibroblasts stimulated with 10 μg/ml of galectin 3.
Table 1.
Cytokine, chemokine, and growth factor levels in galectin 3–stimulated and unstimulated RA synovial and skin fibroblasts*
| Analyte concentration, pg/ml | ||||
|---|---|---|---|---|
|
|
||||
| RA synovial fibroblasts |
RA skin fibroblasts |
|||
| Unstimulated | Galectin 3–stimulated | Unstimulated | Galectin 3–stimulated | |
| bFGF | <10 | <10 | <10 | <10 |
| G-CSF | – | – | – | – |
| VEGF | 10.1–50 | 50.1–100 | – | <10 |
| IFNγ | – | – | – | – |
| IFNα | – | – | – | – |
| IL-1β | <10 | <10 | <10 | <10 |
| CCL4 | <10 | 10.1–50 | <10 | <10 |
| CXCL1 | <10 | 10.1–50 | – | <10 |
| CXCL9 | 10.1–50 | 10.1–50 | 10.1–50 | 10.1–50 |
| CXCL10 | – | <10 | – | – |
| MMP-9 | <10 | <10 | <10 | <10 |
| MMP-13 | – | – | – | – |
Supernatants from rheumatoid arthritis (RA) synovial fibroblasts were cultured in medium alone (unstimulated) or with 10 μg/ml of galectin 3, and were assayed by multiplex enzyme-linked immunosorbent assay (ELISA) for cytokines and chemokines, and by ELISA for IL-6 (lower detection limit 15 pg/ml). Duplicate wells for each sample were analyzed. Values are those obtained after background levels were subtracted. bFGF = basic fibroblast growth factor; G-CSF = granulocyte colony-stimulating factor; – = undetectable or <1 pg/ml; VEGF = vascular endothelial growth factor; IFNγ = interferon-γ; MMP-9 = matrix metalloproteinase 9.
Chemokine production in both synovial and skin fibroblasts after stimulation with TNFα
In order to confirm that the differential production of these chemokines in synovial fibroblasts, but not skin fibroblasts, was not due to any intrinsic differences in the fibroblasts from the 2 different sites, both synovial and skin fibroblasts were exposed to 10 ng/ml of TNFα before collection of supernatants for analysis. Results confirmed that TNFα was capable of the generic induction of chemokines in all fibroblasts regardless of tissue of origin (Figure 2). This finding suggested that the induction of mononuclear cell–attracting chemokines by synovial fibroblasts was a selective property of the biology of these fibroblasts as compared with skin fibroblasts when stimulated specifically with galectin 3.
TNFα induction by galectin 3 is not the mechanism underlying selective chemokine secretion
The production of low levels of TNFα in galectin 3–stimulated synovial fibroblasts but not in galectin 3–stimulated skin fibroblasts suggested that the differential ability of synovial fibroblasts to respond to galectin 3 might be through the production of TNFα promoting chemokine secretion via an autocrine mechanism. We therefore pretreated fibroblast cultures with a blocking antibody to TNFα before exposing synovial fibroblasts to either TNFα (10 ng/ml) or galectin 3 (10 μg/ml) (Figure 4). While IL-6, CCL2, and CCL5 secretion was completely abrogated by antibody blockade when cells were exposed to TNFα, no effect was seen in response to galectin 3 stimulation (Figure 4). These findings ruled out the possibility that autocrine production of TNFα by synovial fibroblasts might regulate the differential production of chemokines induced by galectin 3.
Figure 4.
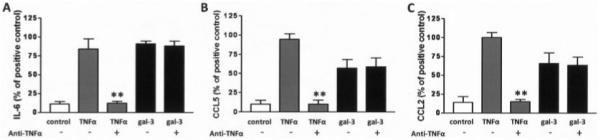
Tumor necrosis factor α (TNFα) induction by galectin 3 (gal-3) is not the mechanism underlying selective chemokine secretion by synovial fibroblasts. Synovial fibroblasts from patients with rheumatoid arthritis (RA) were seeded and treated with medium (control), recombinant TNFα (10 ng/ml), or recombinant galectin 3 (10 μg/ml). Duplicate wells of cytokine-treated fibroblasts were pretreated with an anti-TNFα blocking antibody. A, Production of interleukin-6 (IL-6), a representative proinflammatory cytokine, after 24 hours of stimulation, as analyzed by enzyme-linked immunosorbent assay (ELISA). B and C, Production of CCL2 (B) and CCL5 (C), representative chemokines, after 24 hours of stimulation, as analyzed by ELISA. Bars show the mean and SD of duplicate assays combined from at least 4 patients. ** = P < 0.01.
Regulation of synovial fibroblast production of IL-6 and CCL5 via distinct signaling pathways
In order to explore the molecular basis of the distinct pattern of chemokine secretion observed in synovial fibroblasts stimulated with galectin 3, we used signaling inhibitors that have been shown to block candidate pathways in the fibroblast inflammatory program. For this purpose, IL-6 and CCL5 were measured as a prototype cytokine and mononuclear chemokine, respectively. Notably, we observed involvement of the MAPKs p38, JNK, and ERK-1 in IL-6 secretion, as shown by pharmacologic intervention with SB202190, R406, and PD98059, respectively (34,35) (Figure 5A). In addition, the use of BAY 11-7085, an irreversible inhibitor of TNFα-induced IκBα phosphorylation (36), indicated that NF-κB was an important downstream mediator of IL-6 production by synovial fibroblasts (Figure 5A). Bisindolylmaleimide I is a broad-range inhibitor of the protein kinase C (PKC) family of signaling molecules, while Gö6976 selectively inhibits classic PKC isoenzymes (α and β), allowing dissection of the contribution of different PKC family members to signaling cascades (34). However, no effect of these inhibitors was seen.
Figure 5.
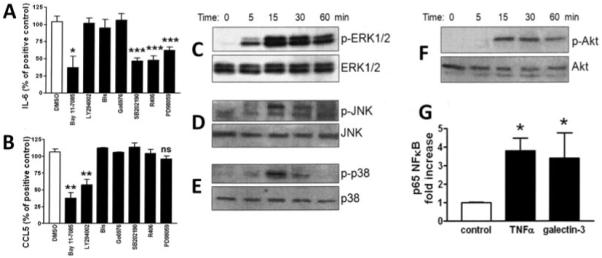
Regulation of synovial fibroblast production of IL-6 and CCL5 via distinct signaling pathways. Synovial fibroblasts from patients with RA were seeded and treated with either DMSO carrier control or the specified pathway inhibitors for 1 hour. All wells were then treated with recombinant galectin 3 (10 μg/ml). A and B, Production of IL-6 (A) and CCL5 (B) after 24 hours of stimulation, as analyzed by ELISA. Bars show the mean and SD of duplicate assays combined from at least 3 patients. C–F, Western blots of cell lysates of synovial fibroblasts obtained from patients with RA and stimulated with galectin 3 for the indicated time periods. Cell lysates were labeled using primary antibodies to ERK (C), JNK (D), p38 (E), and Akt (F) and their active phosphorylated forms. Results are representative of 4 lines or are from 4 lines combined. G, Binding of the activated p65 NF-κB subunit to an NF-κB consensus sequence in nuclear extracts from RA synovial fibroblasts stimulated with medium (control), TNFα (10 ng/ml), or galectin 3 (10 μg/ml), as analyzed using a TransAm NF-κB ELISA kit. Bars show the mean and SD. * = P < 0.05; ** = P < 0.01; *** = P < 0.001, versus DMSO or control medium. Bis = bisindolylmaleimide I; NS = not significant (see Figure 4 for other definitions).
Notably, LY294002, an inhibitor of PI 3-kinase that acts upstream of Akt (37), and BAY 11-7085 significantly decreased levels of secretion of the chemokine CCL5, suggesting participation of PI 3-kinase and NF-κB in CCL5 production by fibroblasts. Thus, while NF-κB appears to be a key common pathway in the secretion of both inflammatory chemokines and cytokines, upstream pathways controlling the selective secretion of chemokines (PI 3-kinase) and cytokines (MAPK) do not appear to overlap significantly. Since signaling pathway inhibitors have the potential to affect cell viability via off-target effects, we carefully monitored the effect of inhibitors on fibroblast number and morphology. No effect was seen on these parameters at the concentrations of inhibitors used in these assays (data not shown).
Activation of selective signaling pathways in galectin 3–stimulated synovial fibroblasts
In order to dissect the molecular mechanisms involved in galectin 3 modulation of cytokine and chemokine secretion, we analyzed the relevant signaling pathways that were shown to be important in IL-6 and CCL5 secretion. Western blotting was used to assess phosphorylation and activation of ERK, JNK, and p38 MAPKs, with activation of ERK MAPK evident within 5 minutes of galectin 3 stimulation, and definite phospho-JNK and phospho-p38 evident by 15 minutes (Figures 5C–E). Akt phosphorylation, consequent upon PI 3-kinase activation, was also evident by 15 minutes (Figure 5F). In order to independently confirm NF-κB activation, nuclear extracts from galectin 3–stimulated synovial fibroblasts were tested for binding of the activated p65 NF-κB subunit to an NF-κB consensus sequence using an NF-κB ELISA kit. Figure 5G shows that similar fold changes in binding of active p65 subunit were seen with both TNFα and galectin 3 treatment.
DISCUSSION
The recruitment and retention of a cellular infiltrate rich in hematopoietic cells, such as monocytes, lymphocytes, and granulocytes, is critical to the persistence of inflammation in the RA synovium. Synovial fibroblasts play a central role in the survival of a variety of hematopoietic cell subpopulations (32,37-39). The role of cytokine-stimulated fibroblasts in elaborating chemokines is well documented, including recent data demonstrating selective pathway activation by IL-18 in the induction of CXCL12 (stromal cell–derived factor 1), CCL2, and VEGF (40).
Synovial fibroblasts are subject to a proinflammatory cytokine network with direct cell contact with other infiltrating cells, such as T lymphocytes (41,42), leading to high levels of expression of many inflammatory chemokines. Monocytes and T cells appear to be recruited by a range of CXC and CC chemokines found at high levels in the synovium. For example, CCL2 (MCP-1) produced by synovial fibroblasts is considered to be a prototypical chemokine for the recruitment of monocytes (43,44). CCL3 (macrophage inflammatory protein 1α [MIP-1α]), CCL4 (MIP-1β), and CCL5 (RANTES) are chemotactic for monocytes and lymphocytes, are expressed at high levels in inflamed rheumatoid synovium and are also produced by synovial fibroblasts (45,46). Fibroblasts are therefore an important cell type in the switch to persistent inflammation, since they respond to many hematopoietic cells and their derived cytokines by secreting a range of proinflammatory cytokines and chemokines that recruit and maintain the inflammatory infiltrate (2).
Our findings extend the range of factors to which synovial fibroblasts respond to include galectin 3. Little is known about the effects of galectins on fibroblast biology in the synovium. Galectin 9 has recently been shown to exert a proapoptotic effect on synovial fibroblasts from RA synovium and has been shown to be present at high levels in RA synovial fluid. Furthermore, previous studies have demonstrated that recombinant galectin 9 ameliorates disease in mice with collagen-induced arthritis through modulation of the balance of Th17 and Treg cells (47,48). It is therefore clear that galectins may play both proinflammatory and antiinflammatory roles in the RA synovium.
A paracrine role for galectin 3 has been shown previously, demonstrating a proproliferative effect on fibroblasts via cell surface carbohydrate receptors (49). Lippert et al (33) identified galectin 3 as a protein present in conditioned media from colonic epithelial cells that is responsible for lamina propria fibroblast IL-8 secretion and NF-κB activation. Notably, the galectin 3 activity detected in their assays was equivalent to >10 μg/ml of recombinant protein, indicating that the concentration chosen for our assays was biologically relevant.
Janelle-Montcalm and colleagues (50) have demonstrated a direct pathogenetic role of galectin 3 in models of arthritis by injecting exogenous galectin 3 into the knee joints of mice. Galectin 3 induced knee swelling and resulted in changes in the histologic scores of cartilage and subchondral bone. In vitro studies of chondrocytes demonstrated induction of the proteoglycan-degrading enzymes ADAMTS-5 and MMP-3, while in osteoblasts, production of osteocalcin was inhibited with involvement of the PI 3-kinase and ERK pathways (50). No analysis of synovial fibroblasts was performed, but these findings clearly demonstrate an important role for galectin 3 in the modulation of inflammatory pathways in stromal cells of the joint, leading to production of, among other factors, MMPs, a finding we confirmed in the present study (Figure 1C).
Data concerning the signaling pathways used by galectin 3 to achieve its various biologic effects are beginning to emerge. Signaling of galectin 3 via PI 3-kinase has been demonstrated in macrophages, where an autocrine feedback loop is implicated in alternative macrophage activation (12). Galectin 3 also plays important roles in tumor cell survival and metastasis. Bladder carcinoma cells are protected against apoptosis via PI 3-kinase/Akt pathway up-regulation, which is abolished in galectin 3–deficient cells (51). MAPK activation has also been shown to be a component of galectin 3 signaling; exogenous galectin 3 regulates neutrophil apoptosis via p38 phosphorylation (14), while ERK MAPK is one of a group of genes that are up-regulated in the galectin 3–mediated progression of lung carcinoma (52). Galectin 3 may also play a role in the induction of JNK protein via increased transcription or mRNA stabilization, as demonstrated by JNK deficiency in mast cells from galectin 3–null mutant mice (53). The PI 3-kinase and MAPK pathways thus appear to be the major mediators of galectin 3 signaling. The findings of the present study show that this extends to effects on cytokine and chemokine secretion.
Our results are consistent with those of previous studies of the regulation of chemokine secretion by PI 3-kinase and MAPK pathways. PI 3-kinase is frequently implicated in the production of mononuclear cell–attracting chemokines such as CCL2 and CCL5 in stromal cells. PI 3-kinase and JNK are implicated in IL-18 regulation of CCL2 production by synovial fibroblasts, whereas JNK has been implicated in VEGF production (40). Although TWEAK induction of CCL5 in osteoblasts occurs via PI 3-kinase independently of NF-κB, suggesting that the NF-κB transcription pathway may be partially redundant (54), our data suggest a major role of both PI 3-kinase and NF-κB in galectin 3 induction of CCL5. Moreover, given that the percentage inhibition by the inhibitors we used was ~50% and ~70%, respectively (Figure 5A and B), and that signaling pathway inhibitors such as LY294002 have variable target specificities (34), it is possible that additional pathways may be involved.
The 3 MAPK pathways are pivotal in proinflammatory cytokine activation of synovial fibroblasts and other RA synovial cells (55), with TNFα, IL-1β, and IL-6 all capable of activating the 3 main pathways (56). Previous work using knockout mice demonstrated that JNK MAPK is crucial to the production of MMPs such as the collagenases, which corresponds to our findings (57). Similarly, ERK MAPK is implicated in the regulation of IL-6 and TNFα secretion in other cell types (56). The p38 MAPK has also been implicated in the regulation of MMPs by synovial fibroblasts (58).
A key finding of the present study is the significant difference observed in the activation of regulatory pathways for cytokine and chemokine production after stimulation with galectin 3 in synovial fibroblasts but not skin fibroblasts. This offers a convincing explanation for the differences in leukocyte subsets observed between inflamed synovial and skin lesions and suggests that galectin 3 specifically drives a highly active PI 3-kinase pathway exclusively in synovial fibroblasts. Our results indicate that RA synovial fibroblasts are more responsive to galectin 3 stimulation than are OA fibroblasts, which is consistent with a role of galectin 3 in the persistence of inflammation in the RA synovium, where, in contrast to OA tissue, high levels of galectin 3 are seen (17). The selective activation of the PI 3-kinase signaling pathway provides further evidence to support the concept that synovial fibroblasts display a unique phenotype and provides a rational target for site-specific therapy based on PI 3-kinase inhibition in synovial fibroblasts (59). In conclusion, our data represent the first evidence that galectin 3 plays a selective role in modulating cytokine and chemokine production by synovial fibroblasts, thus illuminating a novel target for intervention in RA patients.
ACKNOWLEDGMENTS
We gratefully acknowledge John Harrison and Jessica Taylor (Cellzome) for help with the synthesis of R406 and Ulrich Kruse (Cellzome) for technical expertise in cell signaling.
Supported by research grants from the Medical Research Council and a European Union Marie Curie Actions fellowship. The Birmingham University Rheumatology Research Group is a member of the European Union AutoCure Consortium.
Dr. Raza has received research grants for the study of rheumatoid arthritis from Wyeth, UCB Celltech, and Cellzome UK. Dr. Simmons owns stock or stock options in Cellzome UK.
REFERENCES
- 1.Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36:372–8. doi: 10.1016/s1357-2725(03)00259-0. [DOI] [PubMed] [Google Scholar]
- 2.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 3.Van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 4.Rabinovich GA, Liu FT, Hirashima M, Anderson A. An emerging role for galectins in tuning the immune response: lessons from experimental models of inflammatory disease, autoimmunity and cancer. Scand J Immunol. 2007;66:143–58. doi: 10.1111/j.1365-3083.2007.01986.x. [DOI] [PubMed] [Google Scholar]
- 5.Hsu DK, Zuberi RI, Liu FT. Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J Biol Chem. 1992;267:14167–74. [PubMed] [Google Scholar]
- 6.Nieminen J, Kuno A, Hirabayashi J, Sato S. Visualization of galectin-3 oligomerization on the surface of neutrophils and endothelial cells using fluorescence resonance energy transfer. J Biol Chem. 2007;282:1374–83. doi: 10.1074/jbc.M604506200. [DOI] [PubMed] [Google Scholar]
- 7.Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol. 2006;176:778–89. doi: 10.4049/jimmunol.176.2.778. [DOI] [PubMed] [Google Scholar]
- 8.Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A. 2006;103:5060–5. doi: 10.1073/pnas.0511167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishi Y, Sano H, Kawashima T, Okada T, Kuroda T, Kikkawa K, et al. Role of galectin-3 in human pulmonary fibrosis. Allergol Int. 2007;56:57–65. doi: 10.2332/allergolint.O-06-449. [DOI] [PubMed] [Google Scholar]
- 10.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–64. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 11.Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM, et al. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.107.159160. E-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Mackinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180:2650–8. doi: 10.4049/jimmunol.180.4.2650. [DOI] [PubMed] [Google Scholar]
- 13.Yamaoka A, Kuwabara I, Frigeri LG, Liu FT. A human lectin, galectin-3 (epsilon bp/Mac-2), stimulates superoxide production by neutrophils. J Immunol. 1995;154:3479–87. [PubMed] [Google Scholar]
- 14.Fernandez GC, Ilarregui JM, Rubel CJ, Toscano MA, Gomez SA, Beigier BM, et al. Galectin-3 and soluble fibrinogen act in concert to modulate neutrophil activation and survival: involvement of alternative MAPK pathways. Glycobiology. 2005;15:519–27. doi: 10.1093/glycob/cwi026. [DOI] [PubMed] [Google Scholar]
- 15.Nieminen J, St-Pierre C, Sato S. Galectin-3 interacts with naive and primed neutrophils, inducing innate immune responses. J Leukoc Biol. 2005;78:1127–35. doi: 10.1189/jlb.1204702. published erratum appears in J Leukoc Biol 2008;83:797. [DOI] [PubMed] [Google Scholar]
- 16.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 17.Ohshima S, Kuchen S, Seemayer CA, Kyburz D, Hirt A, Klinzing S, et al. Galectin 3 and its binding protein in rheumatoid arthritis. Arthritis Rheum. 2003;48:2788–95. doi: 10.1002/art.11287. [DOI] [PubMed] [Google Scholar]
- 18.Harjacek M, Diaz-Cano S, De Miguel M, Wolfe H, Maldonado CA, Rabinovich GA. Expression of galectins-1 and -3 correlates with defective mononuclear cell apoptosis in patients with juvenile idiopathic arthritis. J Rheumatol. 2001;28:1914–22. [PubMed] [Google Scholar]
- 19.Bianco GA, Toscano MA, Ilarregui JM, Rabinovich GA. Impact of protein-glycan interactions in the regulation of autoimmunity and chronic inflammation. Autoimmun Rev. 2006;5:349–56. doi: 10.1016/j.autrev.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Santucci L, Fiorucci S, Cammilleri F, Servillo G, Federici B, Morelli A. Galectin-1 exerts immunomodulatory and protective effects on concanavalin A-induced hepatitis in mice. Hepatology. 2000;31:399–406. doi: 10.1002/hep.510310220. [DOI] [PubMed] [Google Scholar]
- 21.Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol. 2006;176:6323–32. doi: 10.4049/jimmunol.176.10.6323. [DOI] [PubMed] [Google Scholar]
- 22.Rabinovich GA, Daly G, Dreja H, Tailor H, Riera CM, Hirabayashi J, et al. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J Exp Med. 1999;190:385–98. doi: 10.1084/jem.190.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2007;109:2058–65. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- 24.Chung CD, Patel VP, Moran M, Lewis LA, Miceli MC. Galectin-1 induces partial TCR ζ-chain phosphorylation and antagonizes processive TCR signal transduction. J Immunol. 2000;165:3722–9. doi: 10.4049/jimmunol.165.7.3722. [DOI] [PubMed] [Google Scholar]
- 25.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–34. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 26.Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology: role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–7. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller-Ladner U, Ospelt C, Gay S, Distler O, Pap T. Cells of the synovium in rheumatoid arthritis: synovial fibroblasts [review] Arthritis Res Ther. 2007;9:223. doi: 10.1186/ar2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dasuri K, Antonovici M, Chen K, Wong K, Standing K, Ens W, et al. The synovial proteome: analysis of fibroblast-like synoviocytes. Arthritis Res Ther. 2004;6:R161–8. doi: 10.1186/ar1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neidhart M, Zaucke F, von Knoch R, Jungel A, Michel BA, Gay RE, et al. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein. Ann Rheum Dis. 2005;64:419–24. doi: 10.1136/ard.2004.023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato S, Hughes RC. Regulation of secretion and surface expression of Mac-2, a galactoside-binding protein of macrophages. J Biol Chem. 1994;269:4424–30. [PubMed] [Google Scholar]
- 31.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 32.Filer A, Parsonage G, Smith E, Osborne C, Thomas AM, Curnow SJ, et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54:2096–108. doi: 10.1002/art.21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lippert E, Falk W, Bataille F, Kaehne T, Naumann M, Goeke M, et al. Soluble galectin-3 is a strong, colonic epithelial-cell-derived, lamina propria fibroblast-stimulating factor. Gut. 2007;56:43–51. doi: 10.1136/gut.2005.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cha HS, Boyle DL, Inoue T, Schoot R, Tak PP, Pine P, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther. 2006;317:571–8. doi: 10.1124/jpet.105.097436. [DOI] [PubMed] [Google Scholar]
- 36.Cowburn AS, Deighton J, Walmsley SR, Chilvers ER. The survival effect of TNF-α in human neutrophils is mediated via NF-κB-dependent IL-8 release. Eur J Immunol. 2004;34:1733–43. doi: 10.1002/eji.200425091. [DOI] [PubMed] [Google Scholar]
- 37.Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–8. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan A, Filer A, Parsonage G, Kollnberger S, Gundle R, Buckley CD, et al. Mediation of the proinflammatory cytokine response in rheumatoid arthritis and spondylarthritis by interactions between fibroblast-like synoviocytes and natural killer cells. Arthritis Rheum. 2008;58:707–17. doi: 10.1002/art.23264. [DOI] [PubMed] [Google Scholar]
- 39.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107:305–15. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amin MA, Mansfield PJ, Pakozdi A, Campbell PL, Ahmed S, Martinez RJ, et al. Interleukin-18 induces angiogenic factors in rheumatoid arthritis synovial tissue fibroblasts via distinct signaling pathways. Arthritis Rheum. 2007;56:1787–97. doi: 10.1002/art.22705. [DOI] [PubMed] [Google Scholar]
- 41.McInnes IB, Leung BP, Liew FY. Cell-cell interactions in synovitis: interactions between T lymphocytes and synovial cells. Arthritis Res. 2000;2:374–8. doi: 10.1186/ar115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dayer JM, Burger D. Cytokines and direct cell contact in synovitis: relevance to therapeutic intervention. Arthritis Res. 1999;1:17–20. doi: 10.1186/ar5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90:772–9. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villiger PM, Terkeltaub R, Lotz M. Production of monocyte chemoattractant protein-1 by inflamed synovial tissue and cultured synoviocytes. J Immunol. 1992;149:722–7. [PubMed] [Google Scholar]
- 45.Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. 2001;98:39–45. doi: 10.1006/clim.2000.4957. [DOI] [PubMed] [Google Scholar]
- 46.Hosaka S, Akahoshi T, Wada C, Kondo H. Expression of the chemokine superfamily in rheumatoid arthritis. Clin Exp Immunol. 1994;97:451–7. doi: 10.1111/j.1365-2249.1994.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seki M, Sakata KM, Oomizu S, Arikawa T, Sakata A, Ueno M, et al. Beneficial effect of galectin 9 on rheumatoid arthritis by induction of apoptosis of synovial fibroblasts. Arthritis Rheum. 2007;56:3968–76. doi: 10.1002/art.23076. [DOI] [PubMed] [Google Scholar]
- 48.Seki M, Oomizu S, Sakata KM, Sakata A, Arikawa T, Watanabe K, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol. 2008;127:78–88. doi: 10.1016/j.clim.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 49.Inohara H, Akahani S, Raz A. Galectin-3 stimulates cell proliferation. Exp Cell Res. 1998;245:294–302. doi: 10.1006/excr.1998.4253. [DOI] [PubMed] [Google Scholar]
- 50.Janelle-Montcalm A, Boileau C, Poirier F, Pelletier JP, Guevremont M, Duval N, et al. Extracellular localization of galectin-3 has a deleterious role in joint tissues. Arthritis Res Ther. 2007;9:R20. doi: 10.1186/ar2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oka N, Nakahara S, Takenaka Y, Fukumori T, Hogan V, Kanayama HO, et al. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer Res. 2005;65:7546–53. doi: 10.1158/0008-5472.CAN-05-1197. [DOI] [PubMed] [Google Scholar]
- 52.Abdel-Aziz HO, Murai Y, Takasaki I, Tabuchi Y, Zheng HC, Nomoto K, et al. Targeted disruption of the galectin-3 gene results in decreased susceptibility to NNK-induced lung tumorigenesis: an oligonucleotide microarray study. J Cancer Res Clin Oncol. 2008;134:777–88. doi: 10.1007/s00432-007-0345-3. [DOI] [PubMed] [Google Scholar]
- 53.Chen HY, Sharma BB, Yu L, Zuberi R, Weng IC, Kawakami Y, et al. Role of galectin-3 in mast cell functions: galectin-3-deficient mast cells exhibit impaired mediator release and defective JNK expression. J Immunol. 2006;177:4991–7. doi: 10.4049/jimmunol.177.8.4991. [DOI] [PubMed] [Google Scholar]
- 54.Ando T, Ichikawa J, Wako M, Hatsushika K, Watanabe Y, Sakuma M, et al. TWEAK/Fn14 interaction regulates RANTES production, BMP-2-induced differentiation, and RANKL expression in mouse osteoblastic MC3T3-E1 cells. Arthritis Res Ther. 2006;8:R146. doi: 10.1186/ar2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schett G, Tohidast-Akrad M, Smolen JS, Schmid BJ, Steiner CW, Bitzan P, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal–regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–12. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 56.Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–14. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 57.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, et al. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J Immunol. 1997;158:3165–73. [PubMed] [Google Scholar]
- 59.Sanchez-Pernaute O, Ospelt C, Neidhart M, Gay S. Epigenetic clues to rheumatoid arthritis. J Autoimmun. 2008;30:12–20. doi: 10.1016/j.jaut.2007.11.006. [DOI] [PubMed] [Google Scholar]