

Synopsis
It is long thought that PTPs (protein tyrosine phosphatases) normally function as tumour suppressors. Recent high-throughput mutational analysis identified loss-of-function mutations in six PTPs in human colon cancers, providing critical cancer genetics evidence that PTPs can act as tumour suppressor genes. PTPRT (protein tyrosine phosphatase receptor-T), a member of the family of type IIB receptor-like PTPs, is the most frequently mutated PTP among them. Consistent with the notion that PTPRT is a tumour suppressor, PTPRT knockout mice are hypersensitive to AOM (azoxymethane)-induced colon cancer. This review focuses on the physiologic and pathologic functions of PTPRT as well as the cellular pathways regulated by this phosphatase.
Keywords: cell adhesion, colon cancer, protein tyrosine phosphatase receptor-T (PTPRT), receptor protein tyrosine phosphatases type IIB, tumour suppressor
INTRODUCTION
Although protein tyrosine kinases have been long known to serve as oncogenes, such as JAK (Janus kinase), BCR-Abl and Src [1,2], the role that their cellular counterpart – PTPs (protein tyrosine phosphatases) – could play in cancer progression had not been extensively explored. The PTP superfamily, which regulates a multitude of signalling pathways, is divided into either ‘receptor-like’ [RPTP (receptor protein tyrosine phsophatase)] or ‘non-transmembrane’ [3]. Recently, a high-throughput mutational analysis of all PTPs identified six PTPs to be mutated in colon cancer [4]. Among them, PTPRT (protein tyrosine phosphatase receptor-T), also known as PTPρ, is the most frequently mutated tyrosine phosphatase. In this review, we focus on the physiologic and pathologic functions of PTPRT as well as the cellular pathways regulated by this phosphatase.
DOMAIN STRUCTURE OF PTPRT
PTPRT belongs to the type IIB RPTP subfamily that consists of PTPRK, PTPRM, PTPRT and PTPRU (protein tyrosine phosphatase receptor-K, -M, -T and -U; PTPRU is frequently referred to as PCP-2). All four type IIB RPTPs share common domain architecture: an extracellular domain, a transmembrane domain, a juxtamembrane region and two phosphatase domains (Figure 1). The extracellular domains of type II RPTPs have high-sequence identities [3]: all consist of an MAM (meprin/A5/PTP μ) domain, an Ig domain and four FNIII (fibronectin type III) repeats (Figure 1). The MAM domain is suggested to play a role in protein dimerization. The Ig domain is a disulfide structure that is found in many cell surface proteins and has been shown to mediate homophilic and heterophilic interactions between cell adhesion molecules. The FNIII motif was originally identified in the extracellular matrix protein fibronectin and later found to be present in many cell adhesion molecules [3]. Intracellular domains include the juxtamembrane domain, which has similarity to cadherin domains [3,5], and two phosphatase domains, D1 and D2. The juxtamembrane domain was specifically identified as being responsible for many of the sequence differences between the four type IIb family members, providing specificity in signal transduction [5] and potentially needed for catalytic activity in some family members [6,7]. The phosphatase domain D1 is actively responsible for the phosphatase activity of type IIb members, while the ‘pseudophosphatase’ domain D2, although catalytically inactive, is thought to be important for regulation [6].
Figure 1. Structure and mutation distribution of PTPRT.
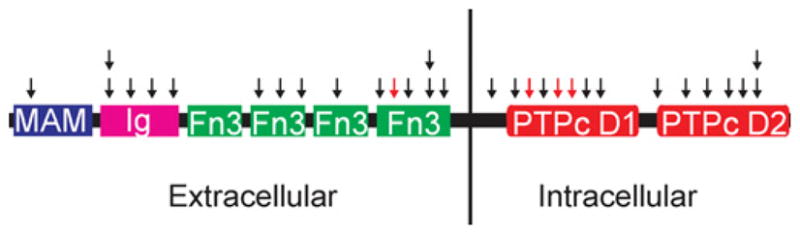
Black arrows indicate missense mutations and red arrows indicate nonsense or frame-shift mutations. Boxes represent functional domains. MAM, meprin/A5/PTP μ; Ig, immunoglobulin; Fn3, fibronectin type III; PTPc D1, first phosphatase catalytic domain; PTPc D2, second phosphatase catalytic domain
PTPRT AS A TUMOUR SUPPRESSOR
PTPRT is mutated in colon, lung, stomach and skin (melanoma) cancers. Several lines of genetic evidence suggest that PTPRT normally functions as a tumour suppressor gene: first, a significant fraction of the mutations found in tumour samples are nonsense and insertion and deletion mutations, resulting in premature truncation of PTPRT and therefore loss of its function [4]. Secondly, consistent with Knudson’s Two-Hit Hypothesis for the inactivation of tumour suppressor activity [2,8,9], approximately one-third of the tumours with PTPRT mutations harbour two different mutations in one tumour [4]. Thirdly, many of the tumour-derived missense mutations in either the D1 or the D2 phosphatase domains lead to reduced phosphatase activity of PT-PRT [4]; similarly, such mutations in the extracellular domain result in defective cell adhesion [10,11]. Fourthly, overexpression of PTPRT activity inhibited cell growth, acting as a putative tumour suppressor in cancer cell culture [4]. Most importantly, knockout mice lacking both alleles of PTPRT, grown in a strain normally resistant to AOM (azoxymethane)-induced colon cancer, became highly susceptible to AOM-induced adenomas (Figure 2) [12]. Another large-scale screen also independently identified PTPRT as a putative tumour suppressor: after mobilizing transposable elements in mouse somatic cells, the authors documented which insertion sites led to tumour formation, ultimately including PT-PRT in its list [13].
Figure 2. PTPRT-knockout mice are hypersensitive to AOM induces colon tumours.

(A) Schematic of PTPRT targeting strategy. The exon 22, which encoded the phosphatase catalytic motif of PTPRT, was deleted. (B) Southern blotting of genomic DNA from mice with indicated PTPRT genotypes. Arrowhead indicates the targeted alleles. (C) Gross morphology of two tumours (arrowheads) in a PTPRT−/− mouse (right) compared with a colon of a PTPRT+/+ (WT) mouse. (D) Representative images of haematoxylin and eosin staining of AOM-induced tumours. (E). Numbers of colon tumours developed in each of the PTPRT+/+, +/− and −/− C57BL/J6 mice (n = 20 in each group). Each circle, square or triangle represents one mouse. (C) and (E) are reproduced from [12] with permission.
ROLE Of PTPRT IN CELL ADHESION
Similar to PTPRM and PTPRK, we demonstrated that the extracellular domain of PTPRT also mediated homophilic cell–cell adhesion [10]. This homophilic interaction is very specific, as the extracellular part of PTPRT does not interact with PTPRM, PTPRK or PTPRU [10,14]. The specificity of the homophilic cell–cell adhesion seems to be determined by the MAM and Ig domains, because chimaeric PTPRT that substituted its own MAM or Ig domains for those of PTPRU would not aggregate with cells expressing wild-type PTPRT [14]. Consistent with the notion that the MAM and Ig domains are critical for PTPRT’s cell adhesion function, deletion mutants devoid of either domain abolish its ability to mediate cell aggregation [10]. Surprisingly, we found that the four FN III domains of PTPRT are also required for its cell–cell adhesion function [11]. Importantly, all the tumour-derived mutations located in the extracellular domains of PTPRT show defects in cell–cell adhesion [10,11].
The crystal structures of PTPRM revealed some clues to how PTPRT mutations affect cell–cell adhesion, as these mutated residues in PTPRT were able to be correlated to key amino acids in PTPRM [15,16]. According to these data, some of these mutations were predicted to disturb the structure as a whole, affecting the overall folding patterns, the stability of the extracellular portion of the protein or the ability for the protein to correctly localize to the membrane [16]. However, one mutated residue in the Ig domain was thought to be specifically responsible for changing the capability for protein–protein interaction, underscoring the importance of this function in normal physiology [15]. Moreover, crystal structures also inform the importance of the ectodomain in dimerization as well as ensuring correct spacing between adjacent cells at adherens junctions so that interactions with substrates can occur properly [16].
Consistent with the role of PTPRT in cell–cell adhesion, Besco et al. [17] showed that PTPRT interacts with E-cadherin (endothelial cadherin). The authors then show that this interaction leads to E-cadherin dephosphorylation, affecting the stability of junctional complexes [17].
PTPRT IN SIGNALLING PATHWAYS
Identification of the substrates of PTPRT is an important step to understand its tumour suppressor function. Since PTPRT’s phosphatase activity is often attenuated in cancers, proteins that retain phosphorylated tyrosine residues when PTPRT is mutated in the crucial D1 and D2 domains may be involved in tumorigenesis. Studies to this end ultimately identified proteins that had been previously speculated to be proto-oncogenes, specifically paxillin and STAT3 (signal transducer and activator of transcription 3) [12,18]. In each case, a phosphopeptide profile was collected from wild-type cancer cells and cells from the same lineage that express either the intracellular fragment of PTPRT (that contain both the phosphatase domains needed for enzymatic activity) or the extracellular fragment (which lacks such activity). Those phosphopeptides that still had tyrosine phosphorylation in the absence of a functional copy of PTPRT warranted further review as possible substrates.
This finding of paxillin as a putative substrate for PTPRT complements the canonical function of the type IIB family members, which often interact with adherens junction proteins [17], as paxillin is known to be involved in cell–cell adhesion [19]. We determined that PTPRT was responsible for dephosphorylating Tyr88 [12]. The importance of Tyr88 phosphorylation was especially seen in xenograft models, where cancer cells with mutated Tyr88 residues (a ‘knock-in’ cell line with phenylalanine substituted for Tyr88 – Y88F – prevented phosphorylation from occurring) were not able to grow xenografts [12]. Similarly, the Y88F mutants in cell lines experienced reduction in anchorage-independent growth and cell migration, as well as changes in Akt (also known as protein kinase B), SHP2 (short heterodimer partner 2) and p130CAS signalling due to alterations in their respective phosphorylation levels [12]. Consistent with the premise that pY88 paxillin plays an oncogenic role, this phosphorylated residue is up-regulated in majority of human colon cancer tissues in comparison with the matched adjacent normal tissues [12].
Similarly, PTPRT was also found to reverse Tyr705 phosphorylation on STAT3, a modification associated with STAT3 activation [18]. Substrate-trapping assays reinforced this finding. Then, we showed that PTPRT’s phosphatase activity was crucial in regulating STAT3’s translocation into the nucleus and activation of target genes, many of which are involved in cell survival and tumorigenesis [especially BCL-XL and SOCS3 (suppressor of cytokine signalling 3)] [18].
ROLE OF PTPRT IN NEUROLOGICAL DEVELOPMENT AND ITS REGULATION
McAndrew et al. discovered PTPRT due in part to the role that many other RPTPs play in neural physiology [20,21]. The authors demonstrated that expression of PTPRT was largely limited to the brain and spinal cord. In situ hybridization indicated the hippocampus and dentate gyrus as specific areas of the brain, especially in post-migratory cells instead of the dividing and mobile neurons of prenatal nervous system development. Therefore, the authors then hypothesized that PTPRT might be more involved in synaptic regulation rather than nervous system growth and development. Lim et al. [22] delved into this speculation in their study about PT-PRT and its regulation. They showed that PTPRT fosters synaptic formation by interacting with a series of synaptic-related proteins (including neuroligin family members and neurexin) and binding with other PTPRT molecules on other cells; both interactions occur via PTPRT’s extracellular domains. This paper was also crucial as it explored PTPRT’s regulation, as the authors demonstrate that this activity is attenuated by expression of Fyn kinase (a Src kinase family member), decreasing synapse formation. Specifically, Fyn phosphorylates PTPRT in its catalytic domain, causing inactivating interactions between PTPRT molecules on the same cell, a loss of phosphatase activity and ultimately an inability for PTPRT to interact with its target proteins.
OTHER TYPE IIB FAMILY MEMBERS AS TUMOUR SUPPRESSORS
PTPRT is not the only type IIB family member implicated as a tumour suppressor, as PTPRK, PTPRM and PTPRU are also implicated in tumorigenesis [9]. After first being associated with a known tumour suppressor region, loss of PTPRK function gradually became associated with a variety of cancers. PTPRK was first identified as a possible tumour suppressor in a study that mapped it to the long arm of the sixth chromosome, specifically at 6q22.2–q22.3, a region that had already been known to be deleted in a variety of neoplasms [23]. These data were followed by a microsatellite study of 6q22–6q23 in primary CNS (central nervous system) lymphomas, confirming that PTPRK was the responsible gene [24]. The authors in that paper further investigate the possibility of PTPRK acting as a tumour suppressor, demonstrating that mRNA transcripts were absent and protein expression levels were decreased in tumour samples versus control tissue [24]. Their results also had clinical significance in that decreased PTPRK expression correlated with decreased patient survival, furthering the case that PTPRK is a tumour suppressor [24]. Another study associated PTPRK with HL (Hodgkin Lymphoma): PTPRK (transcription of which depends on TGF-β) antagonized the growth of HL cells positive for the Epstein–Barr virus, but this specific subset of HL overcomes PTPRK’s inhibition by down-regulating TGF-β signalling [25]. Moreover, this tumour suppressor role can be extended to skin cancers, as transcriptional levels of PTPRK and PTPRU were decreased in melanoma tumour samples according to data generated from RT–PCR (reverse transcription–PCR) [26] and microarray [27].
Much work has been done to elucidate the role that PTPRM plays in suppressing GBM (glioblastoma multiforme). After identifying a decrease in PTPRM expression in GBM samples (a high-grade astrocytoma) versus low-grade astrocytomas, Burgoyne et al. [28] decided to investigate the migratory abilities of cells without functional PTPRM, since the classification of GBM as ‘high grade’ depends on the dispersal of its constituent cells. In a wound-scratch assay, cells without PTPRM expression due to shRNA (small-hairpin RNA) knockdown experienced greater migratory capability than cells that expressed PTPRM [28]. A complementary paper showed that, although overexpression of PTPRM was key in decreasing cellular migration, similar results were seen when PTPRM fragments were also knocked down [29]. Other RPTPs are known to be cleaved proteolytically, and the authors identified PTPRM cleavage products in GBM cell lines, one of which translocates to the nucleus; cleavage products were seen preferentially instead of full-length PTPRM in tumour samples [29]. Then, these fragments – still catalytically active –were shown to be important for GBM cell proliferation and movement [29]. Moreover, a subsequent study demonstrated that these fragments are useful diagnostically, as probes against the extracellular fragment were used to detect GBM tumours in vivo [30].
Although one of least studied phosphatases in the RPTP type IIB family, evidence suggests that PTPRU might also be a tumour suppressor. This RPTP is notable for its role in obstructing colon carcinoma development [31]. Particularly, PTPRU antagonizes Beta-catenin’s transcriptional activation activity, preventing transcription of target genes. As a result, when expressed in a colon carcinoma cell line with increased Beta-catenin stability, PTPRU resulted in decreased cellular movement and replication, acting as a tumour suppressor. It is worth noting that PTPRU is also mutated in colon cancer tumour samples [32].
CONCLUSIONS
Recent studies have elaborated on the role that RPTP type IIB family members play in suppressing tumour growth, especially PTPRT. Although originally discovered as a primarily neurological protein, PTPRT has been shown to play integral roles in cell adhesion and intracellular signalling, due in part to extensive studies done on its extracellular and intracellular domains respectively. When this normal physiologic role becomes abrogated, however, cellular transformation often occurs, demonstrating its importance as a tumour suppressor. Further studies need to be made on PTPRT and its type IIB family members to identify additional phosphatase targets to obtain a better understanding of progression to cancer.
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [grant number R01-CA127590].
Abbreviations used
- AOM
azoxymethane
- E-cadherin
endothelial cadherin
- FNIII
fibronectin type III
- GBM
glioblastoma multiforme
- MAM
meprin/A5/PTP μ
- PTP
protein tyrosine phosphatase
- PTPRK/M/T/U
protein tyrosine phosphatase receptor-K/-M/-T/-U respectively
- RPTP
receptor protein tyrosine phsophatase
- STAT3
signal transducer and activator of transcription 3
References
- 1.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 2.Motiwala T, Jacob ST. Role of protein tyrosine phosphatases in cancer. Prog Nucleic Acid Res Mol Biol. 2006;81:297–329. doi: 10.1016/S0079-6603(06)81008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, Van Der Heijden MS, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 5.Besco J, Popesco M, Davuluri R, Frostholm A, Rotter A. Genomic structure and alternative splicing of murine type IIB receptor protein tyrosine phosphatases (PTPkappa, mu, rho and PCP-2) BMC Genomics. 2004;5:14. doi: 10.1186/1471-2164-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brady-Kalnay SM. Protein tyrosine phosphatases. In: Beckerle M, editor. Cell Adhesion: Frontiers in Molecular Biology. Oxford University Press; Oxford: 2001. pp. 217–258. [Google Scholar]
- 7.Besco J, Frostholm A, Popesco M, Burghes A, Rotter A. Genomic organization and alternative splicing of the human and mouse RPTPrho genes. BMC Genomics. 2001;2:1. doi: 10.1186/1471-2164-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacob ST, Motiwala T. Epigenetic regulation of protein tyrosine phosphatases: potential molecular targets for cancer therapy. Cancer Gene Ther. 2005;12:665–672. doi: 10.1038/sj.cgt.7700828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, Becka S, Zhang P, Zhang X, Brady-Kalnay SM, Wang Z. Tumor-derived extracellular mutations of PTPRT/PTPP are defective in cell adhesion. Mol Cancer Res. 2008;6:1106–1113. doi: 10.1158/1541-7786.MCR-07-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang P, Becka S, Craig SEL, Lodowski DT, Brady-Kalnay S, Wang Z. Cancer-derived mutations in the fibronectin III repeats of PTPRT/PTPρ inhibit cell–cell aggregation. Cell Commun Adhes. 2009;16:146–153. doi: 10.3109/15419061003653771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Zhang X, Guda K, Lawrence E, Sun Q, Watanabe T, Iwakura Y, Asano M, Wei L, Yang Z, et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci USA. 2010;107:2592–2597. doi: 10.1073/pnas.0914884107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 14.Becka S, Zhang P, Craig SE, Lodowski DT, Wang Z, Brady-Kalnay SM. Characterization of the adhesive properties of the type IIb subfamily receptor protein tyrosine phosphatases. Cell Commun Adhes. 2010;17:34–47. doi: 10.3109/15419061.2010.487957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aricescu AR, Hon W, Siebold C, Lu W, Van Der Merwe PA, Jones EY. Molecular analysis of receptor protein tyrosine phosphatase [mu]-mediated cell adhesion. EMBO J. 2006;25:701–712. doi: 10.1038/sj.emboj.7600974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, Van Der Merwe PA, Jones EY. Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science. 2007;317:1217–1220. doi: 10.1126/science.1144646. [DOI] [PubMed] [Google Scholar]
- 17.Besco JA, Hooft van Huijsduijnen R, Frostholm A, Rotter A. Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPρ/PTPRT) Brain Res. 2006;1116:50–57. doi: 10.1016/j.brainres.2006.07.122. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, Polakiewicz RD, Kinzler KW, Vogelstein B, Velculescu VE, Wang ZJ. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci USA. 2007;104:4060–4064. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deakin NO, Turner CE. Paxillin comes of age. J Cell Sci. 2008;121:2435–2444. doi: 10.1242/jcs.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAndrew PE, Frostholm A, White RA, Rotter A, Burghes AHM. Identification and characterization of RPTPρ, a novel RPTP μ/κ-like receptor protein tyrosine phosphatase whose expression is restricted to the central nervous system. Mol Brain Res. 1998;56:9–21. doi: 10.1016/s0169-328x(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 21.Ensslen-Craig SE, Brady-Kalnay SM. Receptor protein tyrosine phosphatases regulate neural development and axon guidance. Dev Biol. 2004;275:12–22. doi: 10.1016/j.ydbio.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Lim S, Kwon S, Lee MK, Moon J, Jeong DG, Park E, Kim SJ, Park BC, Lee SC, Ryu S, et al. Synapse formation regulated by protein tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J. 2009;28:3564–3578. doi: 10.1038/emboj.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Siebert R, Matthiesen P, Yang Y, Ha H, Schlegelberger B. Cytogenetical assignment and physical mapping of the human R-PTP-κ gene (PTPRK) to the putative tumor suppressor gene region 6q22.2–q22.3. Genomics. 1998;51:309–311. doi: 10.1006/geno.1998.5323. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura M, Kishi M, Sakaki T, Hashimoto H, Nakase H, Shimada K, Ishida E, Konishi N. Novel tumor suppressor loci on 6q22–23 in primary central nervous system lymphomas. Cancer Res. 2003;63:737–741. [PubMed] [Google Scholar]
- 25.Flavell JR, Baumforth KRN, Wood VHJ, Davies GL, Wei W, Reynolds GM, Morgan S, Boyce A, Kelly GL, Young LS, Murray PG. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein–Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood. 2008;111:292–301. doi: 10.1182/blood-2006-11-059881. [DOI] [PubMed] [Google Scholar]
- 26.McArdle L, Rafferty M, Maelandsmo GM, Bergin O, Farr CJ, Dervan PA, O’Loughlin S, Herlyn M, Easty DJ. Protein tyrosine phosphatase genes downregulated in melanoma. J Invest Dermatol. 2001;117:1255–1260. doi: 10.1046/j.0022-202x.2001.01534.x. [DOI] [PubMed] [Google Scholar]
- 27.McArdle L, Rafferty MM, Satyamoorthy K, Maelandsmo GM, Dervan PA, Herlyn M, Easty DJ. Microarray analysis of phosphatase gene expression in human melanoma. Br J Dermatol. 2005;152:925–930. doi: 10.1111/j.1365-2133.2005.06454.x. [DOI] [PubMed] [Google Scholar]
- 28.Burgoyne AM, Palomo JM, Phillips-Mason PJ, Burden-Gulley SM, Major DL, Zaremba A, Robinson S, Sloan AE, Vogelbaum MA, Miller RH, Brady-Kalnay SM. PTP μ suppresses glioma cell migration and dispersal. Neuro-Oncology. 2009;11:767–778. doi: 10.1215/15228517-2009-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, Robinson S, Sloan AE, Miller RH, Brady-Kalnay SM. Proteolytic cleavage of protein tyrosine phosphatase μ regulates glioblastoma cell migration. Cancer Res. 2009;69:6960–6968. doi: 10.1158/0008-5472.CAN-09-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burden-Gulley SM, Gates TJ, Burgoyne AM, Cutter JL, Lodowski DT, Robinson S, Sloan AE, Miller RH, Basilion JP, Brady-Kalnay SM. A novel molecular diagnostic of glioblastomas: detection of an extracellular fragment of protein tyrosine phosphatase μ. Neoplasia. 2010;12:305–316. doi: 10.1593/neo.91940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, Yang W, Zhang R, Chen L, Tang L, Zhai B, Liu S, Cao H, Man X, Wu H. Protein-tyrosine phosphatase PCP-2 inhibits β-catenin signaling and increases E-cadherin-dependent cell adhesion. J Biol Chem. 2006;281:15423–15433. doi: 10.1074/jbc.M602607200. [DOI] [PubMed] [Google Scholar]
- 32.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]