

Abstract
The genome of measles virus is encapsidated by multiple copies of the nucleoprotein (N), forming helical nucleocapsids of molecular mass approaching 150 Megadalton. The intrinsically disordered C-terminal domain of N (NTAIL) is essential for transcription and replication of the virus via interaction with the phosphoprotein P of the viral polymerase complex. The molecular recognition element (MoRE) of NTAIL that binds P is situated 90 amino acids from the folded RNA-binding domain (NCORE) of N, raising questions about the functional role of this disordered chain. Here we report the first in situ structural characterization of NTAIL in the context of the entire N-RNA capsid. Using nuclear magnetic resonance spectroscopy, small angle scattering, and electron microscopy, we demonstrate that NTAIL is highly flexible in intact nucleocapsids and that the MoRE is in transient interaction with NCORE. We present a model in which the first 50 disordered amino acids of NTAIL are conformationally restricted as the chain escapes to the outside of the nucleocapsid via the interstitial space between successive NCORE helical turns. The model provides a structural framework for understanding the role of NTAIL in the initiation of viral transcription and replication, placing the flexible MoRE close to the viral RNA and, thus, positioning the polymerase complex in its functional environment.
Keywords: NMR, SAXS, ensemble description, dynamics, unfolded protein
Measles virus (MeV) is a member of the Paramyxoviridae family of the Mononegavirales order of negative sense, single stranded RNA viruses. The viral genome is encapsidated by multiple copies of the nucleoprotein (N) forming a helical nucleocapsid. Transcription and replication of the viral RNA are initiated by an interaction between N and the polymerase complex, composed of the phosphoprotein (P) and the RNA-dependent RNA polymerase (1). N consists of two domains: NCORE (residues 1–400), responsible for the interaction with the viral RNA and for maintaining the nucleocapsid structure, and a long intrinsically disordered domain, NTAIL (residues 401–525) serving as the anchor point for the polymerase complex (2, 3). The molecular recognition element (MoRE) (residues 485–502) of the disordered NTAIL interacts with the C-terminal three-helix bundle domain, XD, of P (residues 459–507) (4) and thereby recruits the polymerase complex onto the nucleocapsid template (5, 6).
The realization that intrinsically disordered proteins (IDPs) are functional despite a lack of structure (7–9) has revealed entirely new paradigms that appear to redefine our understanding of the role of conformational flexibility in molecular interactions (10–12). Until now most IDPs have been studied in isolation, or in the presence of a single interaction partner, although it is evident that a real physiological environment could influence the nature and relevance of apparent intrinsic disorder. In this context resolving the question of whether the protein is actually disordered in situ is of paramount importance. In this case the mechanistic role of the extensive disorder present in NTAIL is particularly intriguing, because the MoRE is located at a distance of 90 apparently unfolded amino acids away from the folded NCORE domain that binds the RNA (13). In order to resolve the mechanism by which the remote interaction between NTAIL and the polymerase complex initiates transcription and replication, it is necessary to develop an atomic resolution understanding of molecular disorder in the context of the intact nucleocapsid. Here we use Nuclear Magnetic Resonance (NMR) spectroscopy, small angle scattering (SAS), and electron microscopy (EM) to describe the conformational behavior and mechanistic role of NTAIL in situ.
Results
NTAIL Populates a Dynamic Equilibrium Comprising Preencoded Helical Conformers at the Phosphoprotein Recognition Site.
In this study we have developed an atomic resolution ensemble description of isolated NTAIL from MeV using recently developed tools designed to provide quantitative descriptions of conformational equilibria in IDPs on the basis of experimental NMR data (14–16). Chemical shifts (17, 18) and residual dipolar couplings (RDCs) (19, 20), measured in a weakly ordering alignment medium were combined to directly probe the level and nature of residual structure in NTAIL, revealing that while the majority of NTAIL behaves like an intrinsically disordered chain, the MoRE exists in a rapidly interconverting conformational equilibrium between an unfolded form and conformers containing one of four discrete α-helical elements situated around the interaction site (Fig. 1, Fig. S1, Tables S1 and S2). All of these α-helices are found to be stabilized by N-capping interactions mediated by side chains of four different aspartic acids or serines that precede the observed helices (21, 22). N-capping stabilization of helices or turns represents an important mechanism by which the primary sequence encodes prerecognition states in disordered proteins, and has been observed in the proteins Tau (23), Sendai virus NTAIL (19), the N-terminal transactivation domain of p53 (24), and the ribosomal protein L9 (25).
Fig. 1.

Ensemble description of the MoRE of NTAIL. (A) NTAIL preferentially adopts a dynamic equilibrium between a completely unfolded state and different partially helical conformations each represented by a single cartoon structure for clarity. All helices are stabilized by N-capping interactions through aspartic acids or serines (blue residues). The location of the helices within the MoRE is shown in the primary sequence. (B) Comparison of experimental (blue) and back-calculated (red) DN-HN RDCs from the model of NTAIL shown in (A). (C) Comparison of experimental (blue) and back-calculated (red) Cα secondary chemical shifts from the model of NTAIL shown in (A).
A crystal structure of the chimeric complex between a short construct of NTAIL and XD shows that NTAIL docks as a helix between residues Q486 and A502 (26). This helix is similar to the longest of the four helical elements present in isolated NTAIL. Changes in chemical shifts and RDCs (Fig. 2) confirm that upon binding to XD, the MoRE of NTAIL folds into a helix. However the decreasing values of secondary structure propensity (SSP) (17) and the RDCs towards the ends of the helix indicate some residual degree of dynamics in the complex. In addition, exchange line broadening persists for residues surrounding the two smallest helices (H1 and H2) present in the conformational equilibrium, even for a large excess of XD compared to NTAIL. There is therefore evidence that both conformational selection from the equilibrium free-form ensemble, and coupled folding and binding, drive the interaction between NTAIL and XD, testifying to the complexity of this highly dynamic interaction.
Fig. 2.

The MoRE of NTAIL folds upon binding to the XD domain of P protein. (A) SSP (17) of NTAIL obtained from experimental Cα and Cβ chemical shifts in free (red) and P (XD) bound (blue) form. (B) N-HN RDCs in free (red) and bound (blue) form of NTAIL.
NTAIL Remains Flexible in Intact Nuclecapsids and Binds Transiently to the Capsid Surface.
Although the MoRE folds upon binding, the remainder of the 90 amino acid long N-terminal chain between the interaction site and NCORE remains flexible (Fig. 2), again raising the intriguing question of the functional role of this long strand. To extend the investigation of NTAIL to a physiologically relevant environment, we have therefore used solution state NMR to characterize the conformational behavior and flexibility of 15N, 13C labeled nucleocapsids. From EM (Fig. 3) we estimate the molecular mass distribution of the objects in the NMR sample to fall in a range between 2 to 50 Megadalton that would normally preclude detection of solution state NMR signals of a folded globular protein (27). The heteronuclear single quantum coherence (HSQC) spectrum of the intact capsids however reveals that NTAIL remains flexible when attached to the nucleocapsid. Comparisons of 1H-15N (Fig. 3), and 13C-13C (Fig. S2) correlation spectra of the isolated NTAIL domain and intact nucleocapsids show that the NMR resonances superimpose, demonstrating that the local conformational behavior of residues 450–525 of NTAIL is retained in situ. However, signals for the first 50 amino acids (residues 401–450) are absent, while large variations of peak intensities indicate differential flexibility along the remainder of the chain, with the MoRE having particularly low intensities (Fig. 4A).
Fig. 3.

Electron microscopy and NMR studies of Measles virus nucleocapsids. (A) Electron micrograph (negative staining) of the 13C, 15N labeled nucleocapsid sample used for solution NMR studies. (B) Electron micrograph of trypsin-digested 13C, 15N labeled nucleocapsids. The solution NMR spectrum of this sample was empty. (C) Superposition of the 1H-15N HSQC spectrum of isolated NTAIL (blue) and intact nucleocapsids (red).
Fig. 4.

Dynamics of NTAIL in intact capsids. (A) Intensity profile of the 1H-15N HSQC spectrum of intact nucleocapsids. The intensity profile was calculated as the ratio of the intensities (I) in the capsid spectrum and the intensities in the spectrum of the free NTAIL domain (I0). (B) Comparison of 15N R2 relaxation rates measured on a 1 GHz spectrometer in the free form of NTAIL (blue) and in intact nucleocapsids (red). (C) N-H angular order parameter S2 averaged over an ensemble of 5,000 conformers of NTAIL that were calculated as shown in Fig. 5 and described in the Methods section.
To further probe the conformational dynamics of NTAIL, we have measured 15N R2 spin relaxation rates in isolated NTAIL and intact nucleocapsids (Fig. 4B). Isolated NTAIL shows uniform R2 relaxation rates throughout the sequence, except in the MoRE where the presence of residual helical structure results in elevated rates. 15N R2 values of NTAIL in the capsid exhibit a very different profile. In the center of the MoRE (around residue 495) R2 values are similar to the rates in the isolated NTAIL domain, indicating that the MoRE is in slow exchange, while the larger relaxation rates observed at the edges of the MoRE indicate that the same exchange rate appears faster (smaller chemical shift differences) for these sites. These results suggest that the MoRE of NTAIL slowly exchanges on and off the surface of the nucleocapsids. Analysis of the intensity of the peaks shows that more than 95% of the MoRE population is bound. The R2 values increase dramatically around residue 460, which, combined with the absence of signals of the first 50 residues of NTAIL, indicates that the first stretch of 50 amino acids of the unfolded domain is conformationally restricted. We note that the C terminus of the protein also interacts, either directly with the capsid, or folds back onto the MoRE as it interacts with the capsid.
NTAIL Exfiltrates from Inside to Outside of the Capsid Helix Through the Interstitial Space Between Successive NCORE Helical Turns.
MeV nucleocapsids have previously been visualized by EM, exhibiting a characteristic herring-bone appearance (5, 28–31). Nothing is known about the location and conformational state of NTAIL in intact nucleocapsids because NTAIL does not appear to contribute coherently to the reconstructed density from EM, however it is apparent that both the structure and dynamics of the nucleocapsids are significantly modulated by NTAIL. Whereas full-length capsids adopt flexible structures, the capsids become significantly more compact and rigid upon cleavage of the disordered tail (Fig. 3 A and B) (5, 32, 33). EM also reveals that the diameter of the capsid decreases from 200 to 190 Å and that the pitch decreases from 57.2 to 48.7 Å upon removal of NTAIL (34).
The atomic resolution structure of NCORE of MeV is unknown, however, the structure of the N-RNA complex of Respiratory Syncytial Virus (RSV), another member of the Paramyxoviridae family, was recently solved using X-ray crystallography (35), and docked into the EM density map of MeV N-RNA on the basis of secondary structural homology (34). Notably, this coarse docking places the C terminus of NCORE, and therefore the N terminus of NTAIL, at the interior of the helix capsid, raising intriguing questions about the position of NTAIL within the capsid. Due to steric hindrance, the 13 copies of NTAIL per turn of the capsid helix cannot reside in the interior of the capsid and remain flexible enough to give rise to NMR signals. We have therefore investigated whether the disordered NTAIL can escape from the interior of the MeV nucleocapsid helix, as reconstructed using EM, by building explicit models that obey random coil statistics for the conformational sampling of the primary sequence, while avoiding the NCORE domains in the capsid. This model (Fig. 5) demonstrates that NTAIL can indeed exfiltrate from the interior of the capsid via the interstitial space between the NCORE moieties. Importantly, reorientational sampling of the chain calculated over the entire ensemble (Fig. 4C), demonstrates that maximal angular freedom is only achieved after approximately 50 amino acids, providing a reasonable explanation for the lack of solution NMR signals up to residue 450. In this case the first 50 amino acids of NTAIL retain conformational disorder, which would also explain why they could not be resolved in the EM reconstruction of the capsids (34).
Fig. 5.

Proposed model of the location of NTAIL in intact nucleocapsids. The three-dimensional coordinates of the RSV N-RNA subunit docked into the EM density map of MeV N-RNA were used (34). The conformational sampling algorithm flexible-meccano was used to build chains from the C terminus of the folded domain of NCORE (successive NCORE monomers are coloured green and yellow). Amino-acid specific conformational sampling allows the chain to escape from the interstitial space of the capsid helix. (A) Representation of the conformational sampling of NTAIL from a single N protein in the capsid. Different copies of NTAIL (red) are shown to indicate the available volume sampling of the chain. The first 50 amino acids of NTAIL are shown. (B) Representation of the conformational sampling of NTAIL from a single N protein in the capsid, shown along the axis of the nucleocapsid. (C, D) Representation of the 13 NTAIL conformers from a single turn of the nucleocapsid. In the interests of clarity, (B–D) deliberately show more conformers outside the capsid, and fewer bound to the surface, than are probable at any one time (see text). The position of the RNA is shown in blue.
Small Angle Scattering Confirms Transient Binding of NTAIL MoRE to Capsid Surface.
Small angle X-ray and neutron scattering (SAS) provides important information concerning the dimensions of NTAIL in intact nucleocapsids. Despite significant polydispersity in terms of length, the cross-sectional radii of gyration, RC, of the capsids can be accurately determined from these data. SAS analysis of the scattering length density distribution around the nucleocapsid symmetry axis gives RC values of (78.0 ± 0.6) Å and (69.5 ± 2.4) Å for the intact and cleaved forms respectively (Fig. 6, Fig. S3). The expected value of RC calculated from the atomic coordinates of RSV N-RNA docked into the reconstructed electron density of the cleaved MeV capsid gives very good agreement with experiment (68.0 Å), while the calculated model of the capsid with the full-length chain gives a value of 83.8 Å when the MoRE is entirely free, and 78.4 Å when the center of the MoRE is positioned less than 8 Å from any of the folded domains of the capsid. The NMR-based model of a transient interaction between the MoRE and the capsid is therefore strongly supported by the SAS data. These results also provide a steric explanation for the observed decrease in pitch between intact and cleaved capsids (34), as parts of the disordered NTAIL reside in the interstitial space between the NCORE lobes.
Fig. 6.

Small angle X-ray scattering of nucleocapsids. (A) Data from intact full-length nucleocapsids (red) and NTAIL-cleaved nucleocapsids (blue). (B) Linear fits of 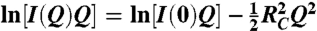 used to extract values of RC from SAXS and SANS data (Fig. S3) from the cleaved (blue) and noncleaved (red) helical capsids.
used to extract values of RC from SAXS and SANS data (Fig. S3) from the cleaved (blue) and noncleaved (red) helical capsids.
Discussion
Measurements of NMR, SAS, and EM on nucleocapsids therefore provide the basis for the development of an in situ ensemble model describing the conformational behavior of NTAIL in intact nucleocapsids. On the basis of this model we are able to provide a structural framework for understanding the dual role of the 125 amino acid intrinsically disordered NTAIL domain. The first 50 disordered amino acids form an articulated spacer that allows the MoRE to escape from the interior of the capsid via the confined interstitial space between successive turns of the helix. The remainder of the chain, on the other hand, is more mobile, and retains the conformational sampling that exists in the isolated form of the protein. This sampling includes the conformational equilibrium of rapidly interconverting helical elements in the MoRE that is predefined by the primary sequence. At the same time the MoRE exchanges on and off the surface of the nucleocapsids, with the majority of conformers in contact with the capsid. The NMR and SAS data indicate that at any given time approximately one of the 13 copies of the nucleoprotein per helical turn is completely free in solution, while the remainder are bound to the capsid surface. While we currently have no information about the position of the binding site, or whether this binding is specific, such a mode of action would provide an efficient mechanism by which NTAIL could “catch” the viral polymerase complex when in free solution, and colocalize the complex on the nucleocapsid surface, thereby initiating transcription and replication of the viral RNA. Interestingly the RNA is sequestered on the outer surface of the RSV and MeV capsids (34, 35), which would place the RNA in the immediate vicinity of NTAIL as it emerges from the interstitial space, providing a mechanistic rationalization of the entire disordered domain of the nucleocapsid. Further structural and dynamic information will be necessary in order to determine the subsequent sequence of events that follow this initial recognition step, and ultimately lead to transcription and replication.
Methods
Cloning, expression, and purification of the isotopically labeled isolated MeV NTAIL domain and the C-terminal domain of P (XD) were described previously (13). Cloning, expression, and purification procedures for MeV nucleoproteins are described in SI Text (34). Random cellular RNA forms the basis of the reconstituted nucleocapsids which are therefore of variable length.
NMR Experiments.
All NMR experiments were carried out at 25 °C. For the measurement of RDCs 13C, 15N labeled NTAIL was aligned in a liquid crystal composed of poly-ethylene glycol (PEG) and 1-hexanol (36) giving rise to a residual deuterium splitting of 21 Hz. 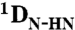 ,
, 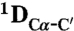 and
and 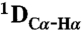 RDCs were obtained using 3D BEST-type HNCO and HN(CO)CA experiments modified to allow for coupling evolution in the 13C dimension (37). Spectra were acquired with a sweep width of 7.5 kHz and 512 complex points in the 1H dimension and a sweep width of 1.32 kHz and 36 complex points in the 15N dimension. For the 13C dimension, the spectra were acquired with a sweep width of 1.2 kHz and 60 complex points (HNCO-type spectra) and 3 kHz and 60 complex points [HN(CO)CA-type spectrum]. Estimates of experimental errors on the RDCs were obtained through repeated measurements: 1.0 Hz (
RDCs were obtained using 3D BEST-type HNCO and HN(CO)CA experiments modified to allow for coupling evolution in the 13C dimension (37). Spectra were acquired with a sweep width of 7.5 kHz and 512 complex points in the 1H dimension and a sweep width of 1.32 kHz and 36 complex points in the 15N dimension. For the 13C dimension, the spectra were acquired with a sweep width of 1.2 kHz and 60 complex points (HNCO-type spectra) and 3 kHz and 60 complex points [HN(CO)CA-type spectrum]. Estimates of experimental errors on the RDCs were obtained through repeated measurements: 1.0 Hz (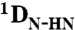 ), 2.0 Hz (
), 2.0 Hz (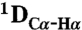 ) and 0.5 Hz (
) and 0.5 Hz (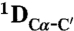 ). Spectra were processed in NMRPipe (38) and analyzed using Sparky (39) and CCPN (40).
). Spectra were processed in NMRPipe (38) and analyzed using Sparky (39) and CCPN (40).
The complex between NTAIL and XD was obtained by preparing a sample containing 0.14 mM 15N, 13C NTAIL and 1.4 mM unlabeled XD. The complex was aligned in a liquid crystal composed of PEG and 1-hexanol giving rise to a residual deuterium splitting of 26 Hz. 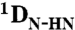 were obtained for NTAIL in the complex using a 2D IPAP SOFAST-HMQC (41) experiment containing 1,024 complex points in the 1H dimension and 150 complex points in the 15N dimension. All RDCs (free and bound form of NTAIL) were measured at a 1H resonance frequency of 600 MHz. 15N R2 relaxation rates of NTAIL in its free form and in the context of intact nucleocapsids were measured at a 1H frequency of 1,000 MHz. Standard pulse sequences were used and the spectra were recorded with a sweep width of 14 kHz and 1,024 complex points in the 1H dimension and a sweep width of 3 kHz and 100 complex points in the 15N dimension (42).
were obtained for NTAIL in the complex using a 2D IPAP SOFAST-HMQC (41) experiment containing 1,024 complex points in the 1H dimension and 150 complex points in the 15N dimension. All RDCs (free and bound form of NTAIL) were measured at a 1H resonance frequency of 600 MHz. 15N R2 relaxation rates of NTAIL in its free form and in the context of intact nucleocapsids were measured at a 1H frequency of 1,000 MHz. Standard pulse sequences were used and the spectra were recorded with a sweep width of 14 kHz and 1,024 complex points in the 1H dimension and a sweep width of 3 kHz and 100 complex points in the 15N dimension (42).
Asteroids Description of the Molecular Recognition Element of NTAIL from NMR Data.
Experimental RDCs and Cα chemical shifts were used in a combined approach to obtain an ensemble description of the MoRE of NTAIL using the minimal ensemble approach (19). A representative ensemble description of the NTAIL MoRE (defined between residues 485–502) was obtained by generating ensembles of NTAIL each consisting of 10,000 conformers using flexible-meccano (14) with varying helix lengths and positions within the MoRE. One hundred and twenty different ensembles were created to cover the entire MoRE with helices with a minimum length of four residues and a maximum length of 18 residues. Furthermore, an ensemble without helices comprising 50,000 conformers was generated. The alignment tensor of each conformer in the ensembles was calculated using PALES (43, 44) and ensemble-averaged RDCs were obtained for each of the 121 ensembles. Ensemble-averaged chemical shifts were calculated using SPARTA (45) using 1,000 conformers, except for the completely unfolded ensemble where 5,000 conformers were used.
The number of helices, N, necessary to describe the experimental data, and the position and length of the helices, were determined by incrementing N. For each step, the genetic algorithm ASTEROIDS (16) was used to select N helical ensembles and their associated populations such that the predicted population weighted RDCs (Fig. 1B, Fig. S1) and chemical shifts (Fig. 1C) were in agreement with the experimental values using:
| [1] |
Ok and OU are the simulated ensemble-averaged observables for the kth helical and unfolded ensemble, respectively, and pk is the population associated with the kth ensemble. A χ2 function is calculated over all residues of the MoRE. Model selection is achieved by optimization of the population and a scaling factor for the RDCs corresponding to the degree of alignment (Table S2). Experimental Cα chemical shift uncertainty used in the combined target function was estimated as 0.3 ppm. The ASTEROIDS selection used 2,000 successive generations and was repeated 10 times for each run to ensure a well defined solution for each value of N (16). Standard F-statistics were used to test the significance of one model over the other (Table S1).
Modelling of NTAIL in the Context of the Capsid.
NTAIL was built onto the atomic resolution model of NCORE derived from docking of the RSV NCORE structure into the EM density of MeV N-RNA capsids. Disordered NTAIL conformers were built using the flexible-meccano algorithm that sequentially constructs peptide chains by randomly sampling amino acid specific dihedral angle distributions (14). Steric clashes are avoided with the folded domains of all copies of NCORE in the capsid. Angular order parameters relative to the capsid frame were calculated over 5,000 conformers in a single ensemble of independent copies of NTAIL from the same N protein as described (46).
Small Angle X-Ray Scattering.
SAXS experiments were carried out on intact and trypsin-digested nucleocapsids at concentrations of 0.35 mM (intact capsids) and 0.25 mM (digested capsids). All sample volumes were adjusted to 50 μL, and were measured on the high brilliance beamline ID02 at the European Synchrotron Radiation Facility (ESRF) Grenoble, France, using a quartz capillary with 2 mm optical path-length. Scattering data were recorded at a sample-detector distance of 1.5 m at a photon wavelength λ = 0.996 Å (E = 12.46 keV). Both samples and the buffer were exposed for five times 0.1 s. No radiation damage was observed in any case. The corrected one-dimensional intensities I(Q) (Q = (4π/λ) sin θ, where 2θ is the scattering angle) from the buffers were subtracted from the respective sample intensities using the SAXS Utilities program (47).
Small Angle Neutron Scattering.
Small Angle Neutron Scattering (SANS) experiments were carried out on intact and trypsin-digested nucleocapsids at concentrations of 0.35 mM (intact capsids) and 0.05 mM (digested capsids). All sample volumes were adjusted to 200 μL and were measured on the instrument D22 at the Institute Laue-Langevin (ILL) (Grenoble, France) in Hellma® quartz cuvettes 100QS with 1 mm optical path length. Scattering data were recorded at instrumental configurations (collimator/detector) 2 m/2 m, 8 m/8 m, and 17.6 m/17.6 m at a neutron wavelength λ = 6 Å. At each configuration, the samples, the buffers, the empty beam, an empty quartz cuvette, as well as a boron sample (electronic background) were measured. Exposure times varied from 30 min to 3 h according to sample and collimator/detector setup. Transmissions were measured during 2 min for each sample. Raw data were reduced using a standard ILL software package (48), normalized to an absolute scale after the various detector corrections and azimuthally averaged to obtain the one-dimensional scattering curve.
Calculation of Cross-Sectional Radius of Gyration from Scattering Data.
Assuming capsid structures (hollow cylinders with an overall length much larger than the diameter), scattering curves were analyzed in terms of rod-like shaped particles. Cross-sectional radii of gyration, RC, were extracted from linear fits of SAXS and SANS data according to (49):
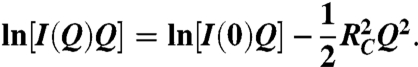 |
[2] |
I(0) is the cross-sectional part of the scattering. The range of validity of the approximation was reasonably fulfilled in both cases (intact form: 0.92 ≤ RCQ ≤ 1.43; cleaved form: 0.79 ≤ RCQ ≤ 1.22).
The experimentally determined cross-sectional radii of gyration (Eq. 2) were compared to the ones calculated from the atomic resolution structure of RSV docked into the EM density map of MeV N-RNA using the radial coordinates ri of the N atoms in a unit sectorial element around the cylindrical axis of symmetry:
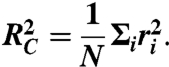 |
[3] |
Supplementary Material
Acknowledgments.
We thank Dr. Phil Callow (ILL) for providing local support on the instrument D22, Dr. Theyencheri Narayanan for support using the ID02 beamline at the ESRF, and Stéphanie Costanzo for preparation of the isolated NTAIL domain and the C-terminal domain of P. We would like to thank the Partnership for Structural Biology for access to biochemical and biophysical platforms, and Irina Gutsche for discussions. This work was supported by the Agence Nationale de la Recherche through ANR PCV 2007 ProteinMotion (M.B.), ANR “Physico-Chimie du Vivant” ANR-08-PCVI-0020-01 (S.L.), ANR “Jeunes Chercheuses Jeunes Chercheurs” 2010 ProteinDisorder (M.R.J), and the Finovi foundation of Lyon (M.B. and R.W.H.R). M.R.J. benefited from a long-term EMBO fellowship and support from Lundbeckfonden, Denmark. The authors acknowledge use of the 1 GHz spectrometer installed at the French high field NMR center in Lyon through the Très Grandes Infrastructures de Recherche, and acknowledge access to CERM research infrastructure (EC contracts 026145 and 261863).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103270108/-/DCSupplemental.
References
- 1.Curran J, Kolakofsky D. Replication of paramyxoviruses. Adv Virus Res. 1999;54:403–422. doi: 10.1016/s0065-3527(08)60373-5. [DOI] [PubMed] [Google Scholar]
- 2.Kingston RL, Baase WA, Gay LS. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J Virol. 2004;78:8630–8640. doi: 10.1128/JVI.78.16.8630-8640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curran J, et al. The hypervariable C-terminal tail of the Sendai paramyxovirus nucleocapsid protein is required for template function but not for RNA encapsidation. J Virol. 1993;67:4358–4364. doi: 10.1128/jvi.67.7.4358-4364.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johansson K, et al. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J Biol Chem. 2003;278:44567–44573. doi: 10.1074/jbc.M308745200. [DOI] [PubMed] [Google Scholar]
- 5.Longhi S, et al. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 6.Bourhis J, et al. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004;99:157–167. doi: 10.1016/j.virusres.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Dunker AK, et al. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 8.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 9.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 10.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 12.Bracken C, Iakoucheva LM, Romero PR, Dunker AK. Combining prediction, computation and experiment for the characterization of protein disorder. Curr Opin Struct Biol. 2004;14:570–576. doi: 10.1016/j.sbi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Gely S, et al. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J Mol Recognit. 2010;23:435–447. doi: 10.1002/jmr.1010. [DOI] [PubMed] [Google Scholar]
- 14.Bernadó P, et al. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen MR, et al. Quantitative determination of the conformational properties of partially folded and intrinsically disordered proteins using NMR dipolar couplings. Structure. 2009;17:1169–1185. doi: 10.1016/j.str.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Nodet G, et al. Quantitative description of backbone conformational sampling of unfolded proteins at amino acid resolution from NMR residual dipolar couplings. J Am Chem Soc. 2009;131:17908–17918. doi: 10.1021/ja9069024. [DOI] [PubMed] [Google Scholar]
- 17.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen MR, Salmon L, Nodet G, Blackledge M. Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J Am Chem Soc. 2010;132:1270–1272. doi: 10.1021/ja909973n. [DOI] [PubMed] [Google Scholar]
- 19.Jensen MR, et al. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: application to the molecular recognition element of Sendai virus nucleoprotein. J Am Chem Soc. 2008;130:8055–8061. doi: 10.1021/ja801332d. [DOI] [PubMed] [Google Scholar]
- 20.Jensen MR, Blackledge M. On the origin of NMR dipolar waves in transient helical elements of partially folded proteins. J Am Chem Soc. 2008;130:11266–11267. doi: 10.1021/ja8039184. [DOI] [PubMed] [Google Scholar]
- 21.Serrano L, Fersht AR. Capping and alpha-helix stability. Nature. 1989;342:296–299. doi: 10.1038/342296a0. [DOI] [PubMed] [Google Scholar]
- 22.Serrano L, Sancho J, Hirshberg M, Fersht AR. Alpha-helix stability in proteins. I. Empirical correlations concerning substitution of side-chains at the N and C-caps and the replacement of alanine by glycine or serine at solvent-exposed surfaces. J Mol Biol. 1992;227:544–559. doi: 10.1016/0022-2836(92)90906-z. [DOI] [PubMed] [Google Scholar]
- 23.Mukrasch MD, et al. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J Am Chem Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 24.Wells M, et al. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luisi DL, Wu WJ, Raleigh DP. Conformational analysis of a set of peptides corresponding to the entire primary sequence of the N-terminal domain of the ribosomal protein L9: evidence for stable native-like secondary structure in the unfolded state. J Mol Biol. 1999;287:395–407. doi: 10.1006/jmbi.1999.2595. [DOI] [PubMed] [Google Scholar]
- 26.Kingston RL, Hamel DJ, Gay LS, Dahlquist FW, Matthews BW. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc Natl Acad Sci USA. 2004;101:8301–8306. doi: 10.1073/pnas.0402690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szymczyna B, Gan L, Johnson J, Williamson J. Solution NMR studies of the maturation intermediates of a 13 MDa viral capsid. J Am Chem Soc. 2007;129:7867–7876. doi: 10.1021/ja071118j. [DOI] [PubMed] [Google Scholar]
- 28.Finch JT, Gibbs AJ. Observations on the structure of the nucleocapsids of some paramyxoviruses. J Gen Virol. 1970;6:141–150. doi: 10.1099/0022-1317-6-1-141. [DOI] [PubMed] [Google Scholar]
- 29.Lund GA, Tyrrell DL, Bradley RD, Scraba DG. The molecular length of measles virus RNA and the structural organization of measles nucleocapsids. J Gen Virol. 1984;65:1535–1542. doi: 10.1099/0022-1317-65-9-1535. [DOI] [PubMed] [Google Scholar]
- 30.Fooks AR, et al. Measles virus nucleocapsid protein expressed in insect cells assembles into nucleocapsid-like structures. J Gen Virol. 1993;74:1439–1444. doi: 10.1099/0022-1317-74-7-1439. [DOI] [PubMed] [Google Scholar]
- 31.Bhella D, Ralph A, Murphy LB, Yeo RP. Significant differences in nucleocapsid morphology within the Paramyxoviridae. J Gen Virol. 2002;83:1831–1839. doi: 10.1099/0022-1317-83-8-1831. [DOI] [PubMed] [Google Scholar]
- 32.Schoehn G, et al. The 12 A structure of trypsin-treated measles virus N-RNA. J Mol Biol. 2004;339:301–312. doi: 10.1016/j.jmb.2004.03.073. [DOI] [PubMed] [Google Scholar]
- 33.Bhella D, Ralph A, Yeo RP. Conformational flexibility in recombinant measles virus nucleocapsids visualised by cryo-negative stain electron microscopy and real-space helical reconstruction. J Mol Biol. 2004;340:319–331. doi: 10.1016/j.jmb.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Desfosses A, Goret G, Estrozi LF, Ruigrok RWH, Gutsche I. Nucleoprotein-RNA orientation in the measles virus nucleocapsid by three-dimensional electron microscopy. J Virol. 2011;85:1391–1395. doi: 10.1128/JVI.01459-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tawar RG, et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science. 2009;326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 36.Rückert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 37.Lescop E, Schanda P, Brutscher B. A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J Magn Reson. 2007;187:163–169. doi: 10.1016/j.jmr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 38.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR . 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 39.Goddard T, Kneller D. SPARKY 3. San Francisco: University of California; [Google Scholar]
- 40.Vranken WF, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 41.Kern T, Schanda P, Brutscher B. Sensitivity-enhanced IPAP-SOFAST-HMQC for fast-pulsing 2D NMR with reduced radiofrequency load. J Magn Reson. 2008;190:333–338. doi: 10.1016/j.jmr.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 42.Farrow N, et al. Backbone dynamics of a free and a phosphopeptide-complexed SRC homology-2 domain studied by N-15 NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 43.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 44.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 45.Shen Y, Bax A. Protein backbone chemical shifts predicted from searching a database for torsion angle and sequence homology. J Biomol NMR . 2007;38:289–302. doi: 10.1007/s10858-007-9166-6. [DOI] [PubMed] [Google Scholar]
- 46.Markwick PRL, et al. Toward a unified representation of protein structural dynamics in solution. J Am Chem Soc. 2009;131:16968–16975. doi: 10.1021/ja907476w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sztucki M, Narayanan T. Development of an ultra-small-angle X-ray scattering instrument for probing the microstructure and the dynamics of soft matter. J App Crystallogr. 2007;40:S459–S462. [Google Scholar]
- 48.Gosh R, Egelhaaf S, Rennie A. A computing guide for small-angle scattering experiments. 2006. Institute Laue Langevin internal report.
- 49.Porod G. In: Small Angle X-ray Scattering. Glatter O, Kratky O, editors. New York: Academic Press; 1982. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.