

Abstract
We present a novel assay for rapid detection and identification of bacterial urinary tract infections using isotachophoresis (ITP) and molecular beacons. We applied on-chip ITP to extract and focus 16S rRNA directly from bacterial lysate, and used molecular beacons to achieve detection of bacteria specific sequences. We demonstrated detection of E. coli in bacteria cultures as well as in patient urine samples in the clinically relevant range 1E6-1E8 cfu/mL. For bacterial cultures we further demonstrate quantification in this range. The assay requires minimal sample preparation (a single centrifugation and dilution), and can be completed, from beginning of lysing to detection, in under 15 min. We believe that the principles presented here can be used for design other rapid diagnostics or detection methods for pathogenic diseases.
Keywords: isotachophoresis, molecular beacons, urinary tract infection, 16S rRNA, bacteria, pathogen, urine analysis
INTRODUCTION
Infectious diseases caused by bacterial pathogens remain one of the most common causes of mortality worldwide.1 Urinary tract infection (UTI) is the second most common infection in the United States affecting all patient demographics,2 with approximately 8 million visits to outpatient clinics and emergency departments, and 100,000 hospitalizations each year.3 Overall, medical expenditures for UTI in the United States are estimated to be $3.4 billion.4 Similar to most other bacterial infections, diagnosis of UTI requires a centralized clinical microbiology laboratory and trained professionals to perform bacterial culture and phenotyping, which typically takes 1–3 days. A rapid, inexpensive, definitive test capable of detecting pathogens in urine would be enormously beneficial in ensuring timely treatment, in eliminating empirical treatment, and in reducing costs and burden on the health care system.
Several nucleic acid amplification techniques such as polymerase chain reaction (PCR) and real-time PCR genotyping tests have been developed for bacterial identification.5 Such tests have recently been implemented on microchips but require elaborate off-chip preparation including the extraction and purification of nucleic acids.6 Other approaches include microarray-based tests requiring pre-amplification of the target, and immunoassays typically require sequential processes such as multiple washes, incubations and the implementation of specialized chemistries for signal amplification/transduction.7, 8
PCR-based techniques are yet to replace standard bacterial culture due to their complexity, cost and need for specially trained personnel. PCR-free assays, in which the genetic content of the sample could be directly analyzed, could offer a simple yet specific diagnostic tool, while alleviating or eliminating many of the constraints associated with genetic amplification. We here present a novel assay for UTI detection in which we use isotachophoresis (ITP) to extract, focus, and hybridize bacterial-specific 16S ribosomal RNA (rRNA) with sequence-specific molecular beacons, directly from urine pellet lysate. ITP is an electrophoretic technique in which only ions with mobilities bracketed by those of a leading electrolyte (LE) and trailing electrolyte (TE) are focused to achieve both sensitivity and selectivity.9–11 Jung et al.12 used ITP on microliter samples to separate and concentrate sample ions (as low as 100 attomolar) by up to million-fold.13 ITP has earlier been applied to urine samples, primarily for measurement of small molecules.14–21 The latter studies have been typically performed on long separation capillaries using electrochemical detection, electric potentials of 10 kV or higher, and separation times on the order of tens of minutes to hours. More recently, on-chip ITP has been applied to extraction and purification of biological samples: Schoch et al.22 demonstrated the use of ITP for extraction of short RNA from bacterial lysate using a sieving matrix, Persat et al. performed extraction of DNA from whole blood using ITP,23 and Matsumura et al. used ITP for extraction of DNA from cultured bacteria lysate.24
In this work, we adapted a chemical lysing technique compatible with ITP, and for the first time applied ITP for focusing and detection of 16S rRNA in cell cultures and patient urine samples using molecular beacons. We reported brief, preliminary results in a conference proceedings,25 and here present a detailed study of our assay. Bacteria cells contain order 10,000 ribosomes (this value varies with the growth stage of the bacteria), each consisting of several ribosomal subunits. These subunits, typically characterized by the Svedberg unit (indicating their sedimentation rate under centrifugation), consist of an RNA sequence bound to multiple proteins. 16S rRNA is a 1542 nucleotide long well-characterized bacterial-specific biosignature. –It is commonly targeted in molecular assays, due to its high abundance (5.5% by weight) in bacterial cells.26 Molecular beacons (MBs) are hairpin-shaped oligonucleotides consisting of a probe section and a self-complementary stem which brings together fluorophore and quencher molecules.27 In the absence of target, the stem sequences hybridize and the quencher is brought closer to the fluorophore, inhibiting its fluorescence. In the presence of the target, the beacon preferentially binds to the target, separating quencher from fluorophore to yield a significant fluorescent signal. MBs have mostly been applied in conjunction with real-time PCR for the quantitative detection of bacteria, viruses, single nucleotide polymorphisms and for real-time intracellular monitoring.28 Recently, Persat and Santiago combined ITP with MBs for detection of miRNA from pre-purified total RNA.29 Since a large number of urine samples that are sent for bacterial analysis are returned with a negative result, the ability to quickly rule out an infection is of high value. We therefore focus on demonstration of our assay using a universal bacterial probe, targets a highly conserved region of bacterial 16S rRNA.
To the best of our knowledge, the current study is the first demonstration of on-chip ITP for rapid pathogen detection. This assay requires minimal sample preparation (a single centrifugation and dilution), and performs extraction, focusing, and detection of 16S rRNA in a single step, and without the use of a sieving matrix. Currently, the entire assay, from beginning of lysing to detection, can be completed in under 15 min, and is sensitive within a clinically relevant range of bacteria concentration (1E6-1E8 cfu/mL). We believe that by varying the molecular beacons probe sequence, the principles presented here could be used for other rapid diagnostics, including other pathogenic diseases.
PRINCIPLE OF THE ASSAY
Figure 1a schematically presents the principles of the assay. ITP uses a discontinuous buffer system consisting of LE and TE, which are typically chosen to have respectively higher and lower electrophoretic mobility than the analytes of interest. Both sample and molecular beacons are initially mixed with the TE. When an electric field is applied, all species with mobility higher than that of the TE electromigrate into the channel. Other species (including ones with lower mobility, neutral or positively charged) remain in or near the sample reservoir. Focusing occurs within an electric field gradient at interface between the LE and TE, as sample ions cannot overspeed the LE zone but overspeed TE ions.
Figure 1.
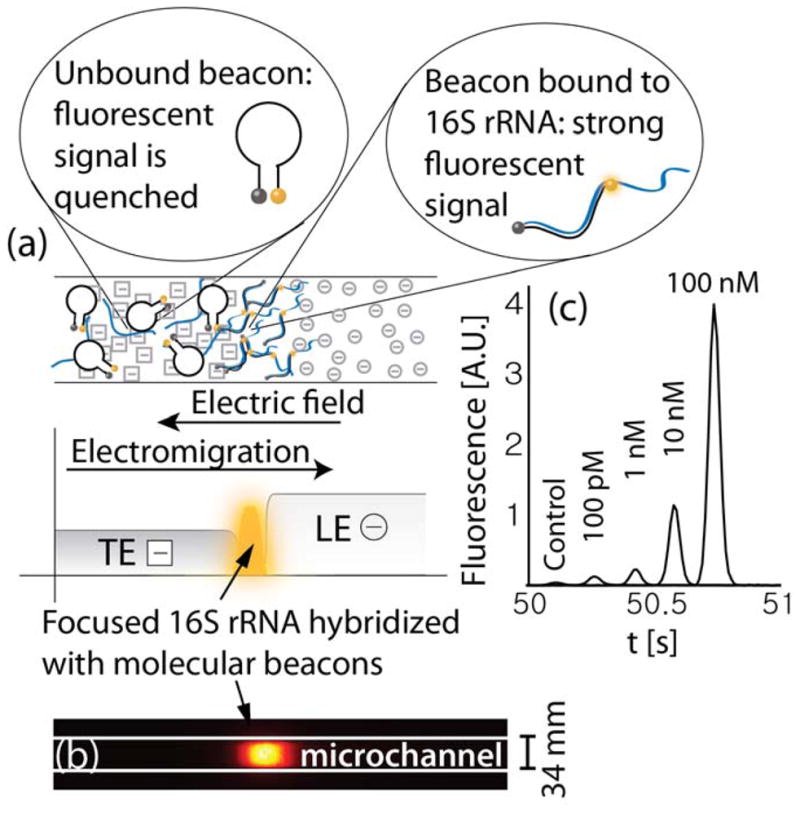
(a) Schematic showing simultaneous isotachophoretic extraction, focusing, hybridization (with molecular beacons), and detection of 16S ribosomal RNA bound to a molecular beacon. Hybridization of the molecular beacon to 16S rRNA causes a spatial separation of its fluorophore and quencher pair resulting in a strong and sequence-specific increase in fluorescent signal. (b) Raw experimental image showing fluorescence intensity of molecular beacons hybridized to synthetic oligonucleotides using ITP. (c) Detection of oligonucleotides having the same sequence as the target segment of 16S rRNA. Each curve presents the fluorescence intensity in time, as recorded by a point detector at a fixed location in the channel (curves are shifted in time for convenient visualization). 100 pM of molecular beacons and varying concentrations of targets were mixed in the trailing electrolyte reservoir. The total migration (and hybridization) time from the on-chip reservoir to the detector was less than a minute.
We designed ITP buffers to focus 16S rRNA, molecular beacons, and their (possible) complex at the interface. Their hybridization produces a sequence-specific fluorescence signal which we use to both identify and quantify bacteria. In positive control experiments, we modeled 16S rRNA using synthetic oligonucleotides with a complementary sequence to the molecular beacon probe. The probe used in this work targets a 27 nucleotide sequence common to all bacteria, and has been validated in previous work with a large cohort of clinical samples using electrochemical detection.30 A raw intensity image of molecular beacons hybridized to the synthetic oligonucleotides is presented in Figure 1b. Figure 1c presents example quantitative detection of the oligonucleotides. As the target concentration increased, a higher fraction of the beacons were hybridized and fluorescence signal (the area under the peak) increased. For the highest target concentration presented (100 nM), the fluorescence signal was approximately 100-fold higher than the control case (with no target oligonucleotides). The lowest concentration of synthetic targets we detected was 100 pM, corresponding to a fluorescent signal approximately 3-fold higher than the control case. This limit of detection for on-chip hybridization of molecular beacons is consistent with the results of Persat et al.29 for hybridization of molecular beacons with miRNA..
THEORY
In this section we present theory useful in quantitative analysis of the beacons signal. First, we define enhancement ratio, a normalized figure of merit for quantifying the increase in signal due to beacon-target hybridization. We use this definition to explore the sensitivity and limit of detection of the assay and highlight the key parameters useful in optimizing the assay.
Following a notation similar to that of Bonnet et al.,31 the fluorescence signal of a mixture of beacons and target, F, can be expressed as,
| (1) |
where cBT, cB, closed, and cB, open are the concentration of the hybridized beacons, closed stem beacons, and open stem (random coil) beacons respectively. is the total concentration of beacons, and α, β, γ are the conversion factors for fluorescent intensity associated with each respective state.
It is convenient to measure the signal with respect to the signal of a control case, F0, which contains the same concentration of beacons , but no targets
| (2) |
Here and are the concentrations of the two beacon states in the absence of any target. We define the ratio of signal to control signal as the enhancement ratio, given by
| (3) |
We use this enhancement ratio (which was also used by Persat et al.29 and Bercovici et al.25) as an internally-normalized figure of merit which is less sensitive than the absolute fluorescence values to experimental conditions such as illumination intensity, degree of photobleaching, and exposure time. While the hybridization reaction likely does not reach full equilibrium within the time scales of our experiments, it is instructive to perform equilibrium analyses to explore the limits of detection of the assay.
We now assume chemical equilibrium of the beacon and target reaction to explore maximum signal values and some trends also relevant to unsteady problems. Assuming equilibrium, the concentrations of all species can be related to the equilibrium and mass conservation equations as follows:
| (4) |
where is the total concentration of the target. Applying relations (4), and denoting β* = β + γ K23, we have
| (5) |
Bonnet et al.26 explored the analytical solution for the case of abundant targets . That mode of detection is useful, since all beacons are saturated with targets, maximizing signal-to-background ratio. However, for the same reason the enhancement ratio is not very sensitive to the target concentration . Furthermore, for a given , increasing , increases the rate of hybridization. To allow rapid quantification and sensitivity to target, we here explore the regime in which , and assume . The latter regime holds for most beacons and concentrations higher than 1 fM, as calculated based on the Gibbs free energy measured by Tsourkas et al.32 In this regime, and, from (4iii), . Assuming K23 ≪ 1 (which holds for typical molecular beacons stems33, 31), the equilibrium enhancement ratio is thus
| (6) |
For the beacon quencher pair used in this work, we estimate α/β* is approximately 80 (based on measurements at high target concentrations). The dynamic range of the assay is thus between ε = 1 (no target) and ε = 80 (for ). As we shall see below, this result, although assuming equilibrium, agrees with our experimental observations which showed a detectable range over two orders of magnitude of bacteria concentration (1E6 to 1E8 cfu/ml).
In this work, we use a point detector to record the fluorescence signal at the ITP interface, as it electromigrates through the detection point. We therefore find it useful to relate this temporal signal to the dynamics of the assay. Denoting the signal distribution in the channel (i.e. in space) as f (x), and the integration window of the detector as w(t), the signal in time is given by the convolution of the two,34
| (7) |
where xd denotes the location of the detector, and VITP is the velocity of the ITP plug (assumed here for simplicity as constant).
Since peak signal values are sensitive to noise and sampling rate, it is convenient to quantify the intensity of the signal by the total fluorescence, i.e. area under the signal curve, . The enhancement ratio ε can then be computed as ε = A/A0, where A0 corresponds to area under the signal curve for a negative control. By change of variable, η = VITPτ, and noting that the term f (xd− η) is independent of t, we can express
| (8) |
For any value of η, and any finite integration window, the term in brackets is constant, and the area under the curve is given by
| (9) |
The integral over the signal f is also constant, and therefore the total fluorescence integral A is inversely proportional to the migration velocity VITP. In a spatial image of the ITP interface taken after a fixed time from beginning of the experiment (e.g. using a CCD), the effect of the interface velocity on total fluorescence is minimal, since a fixed pre-determined exposure time is used. In contrast, a point detector (e.g. a PMT) continuously records the light intensity. Total fluorescence therefore depends directly on the velocity of the ITP zone; i.e. the duration of time in which the detector is exposed to the fluorescent peak. For a quantitative ITP assay with a point detector, it is therefore very important that the migration velocity of plug over the detection point be repeatable across experiments. In practice, this requirement translates to repeatable suppression of electroosmotic flow (EOF) and sufficient dilution of the sample to avoid sample-specific variations in buffer conductivities (which in turn alter the electric field and hence ITP velocity). In the current experiments, we used real-time current monitoring as an indicator of ITP velocity, and this is further discusseds in the results section.
EXPERIMENTAL SECTION
Experimental setup
We mounted a microfluidic chip on the stage of IX70 inverted epifluorescent microscope (Olympus, Hauppauge, NY). Constant voltage was applied using a sourcemeter (2410, Keithley Instruments, Cleveland, OH). We used a 642 nm variable-power laser diode (Stradus-642, Vortran Laser Technologies, CA) as the excitation light source. The light from the laser was coupled to the illumination port of the microscope using a multimode optical fiber (M31L05) with a fiber coupler (FiberPort PAF-X-7-A) on the laser end, and a beam collimator and expander (F230FC-A) on the microscope end, all from Thorlabs (Newton, NJ). The laser beam passed through the excitation filter of a Cy5 filter-cube (Cy5-4040A, Semrock, Rochester, NY), and was focused onto the chip using a water immersion objective (LUMPlanFL 60×, NA = 0.9, Olympus, Hauppauge, NY). Light was collected by the same objective passed through the emission filter of the filter cube. We constructed a point-confocal setup by placing a 400 μm pinhole at the focal plane of the microscope’s side-port to reject out of plane light. Light was then focused onto a photomultiplier tube (PMT) module (H6780-20, Hamamatsu Photonics, Japan) using a 1 in biconvex lens with a focal length of 50 mm (LB1471-A, Thorlabs Newton, NJ). The assembly consisting of the PMT, lens, and pinhole was mounted on three micro stages, to provide three degrees of freedom in aligning the pinhole with the laser spot, Fig. 2. The PMT signal was digitized using a data acquisition unit (C8908, Hamamatsu Photonics, Japan) and communicated via RS232 to a PC. The PMT was powered using 5V DC from a stable power source (E3631A, Agilent, Santa Clara, CA) and operated at a sampling rate of 100 Hz. We used in-house MATLAB codes (R2007b, Mathworks, Natick, MA) to simultaneously control and record the data from both the PMT and the sourcemeter.
Figure 2.
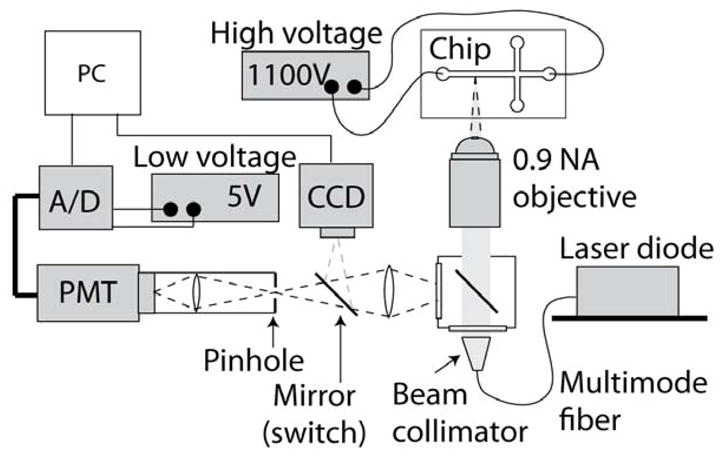
Schematic of the experimental set-up with pointwise confocal optics. We used a 0.9 numerical aperture water-immersion objective to collect the light emitted by the molecular beacons within the microfluidic chip. A 400 μm pinhole was placed at the image plane, allowing collection of light from within the 12 μm deep channel, while rejecting out-of-focus light. The light was refocused onto a PMT for detection. Excitation is performed using a variable-power laser diode coupled into the illumination port of the microscope using a multimode optical fiber. The beam is expanded and collimated before being focused onto the channel using the same objective used for light collection. A CCD camera is used for alignment of the laser and microchannel prior to each experiment.
Cell cultures and clinical samples
With approval from Stanford University Institutional Review Board, bacterial isolates and clinical urine samples were obtained from informed, qualified study participants at risk for UTI. We prepared pellets from both E. coli cultures and human urine by centrifuging 1 mL of sample at 10,000 g for 2 min, and then discarding the supernatant. The pellets were kept frozen at −80 °C.
Buffers, lysing reagents, and probes
ITP
For all experiments, the leading electrolyte (LE) was composed of 250 mM HCl and 500 mM bistris, 5 mM MgCl2, and 1% 1.3 MDa poly(vinylpyrrolidone) (PVP). The trailing electrolyte (TE) was composed of 50 mM tricine and 100 mM bistris. We used a high ionic strength LE to maximize the focusing rate of species.35 Mg2+ ions were used as a second counter ion (in addition to bistris) to promote rapid hybridization of the beacons and target rRNA at the ITP interface.36 PVP was used in the LE for suppression of electroosmotic flow (EOF). The TE concentration was empirically determined to provide sufficient buffering and repeatability over a range of samples (additional details in the results section), while also promoting focusing rate.35 Tricine, bistris, and MgCl2 were obtained from Sigma-Aldrich (St. Louis, MO). PVP was obtained from ACROS Organics (Thermo Fisher Scientific, NJ).
Lysis
Lysis was composed of two steps using two sets of reagents. Lysis reagent I was composed of 10 mM Tricine, 10 mM BisTris, 2 mM EDTA (GIBCO Invitrogen, Carlsbad, CA), 0.1 % Triton-X, and 5 mg/mL lysozyme (both from Sigma-Aldrich, St. Louis, MO). Lysis reagent II was composed purely of 400 mM NaOH (Sigma-Aldrich, St. Louis, MO). This technique is based on our earlier optimized technique for bacterial lysis25 and has here been adapted for compatibility with ITP and molecular beacon hybridization.
Beacons
The beacons solution contained 50 mM tricine, 100 mM bistris, 5 mM MgCl2, and 1 nM of molecular beacons (IDT, Coralville, Iowa). We chose a 27-mer probe sequence which was shown by Liao et al.37 to detect a wide range of urinary pathogens. We added 6 base-pairs to either side of the probe to form the molecular beacon stem. The 5′ terminus was labeled with Cy5, and the 3′ terminus was labeled with Black Hole Quencher 2 (BHQ2): 5′-/Cy5/CCG AGC [CAT CGT TTA CGG CGT GGA CTA CCA GGG] GCT CGG/BHQ2/-3′ (probe sequence in brackets). For the control experiment, we designed a molecular beacon with an inverted probe sequence, 5′-/5Cy5/CGC TCG [GGG ACC ATC AGG TGC GGC ATT TGC TAC] CGA GCG/3BHQ_2/-3′. In this beacon we have also swapped cytosine for guanine at the tip of the stems, to preserve the same level of quenching by the nucleotide closest to the 5′ dye (cytosine is a weaker quencher than guanine38). The two beacons are thus very similar in their thermodynamic properties, but the inverted probe cannot hybridize to the 16S rRNA target sequence.
All solutions were prepared in UltraPure DNase/RNase free deionized (DI) water (GIBCO Invitrogen, Carlsbad, CA). Buffer stock solutions were prepared in 80 mL glass bottles (VWR, Radnor, PA) and kept at room temperature. 50 mg/mL of lysozyme were prepared from powder and kept at 4°C for no more than a week. All solutions were freshly prepared at the beginning of each set of experiments. Lysis reagent I was kept on ice when not in use.
Assay description and microchip implementation
A schematic of the steps involved in the assay is presented in Figure 3a. For experiments involving cell cultures or patient urine, we resuspended the pellet in 80 μL of DI water, and actuated the pipette several times to homogenize the solution. We added 10μL of lysis reagent I, and incubated for 5 min at room temperature. We then added 10 μL of lysis reagent II and actuated the pipette until the solution became clear and transparent. For experiments using bacterial cultures, we diluted the sample down from an initial concentration of 1E8 cfu/mL. We separated 10 μL of this lysate and mixed it with 90 μL of beacons solution. Since we initially resuspended the cells in 100 μL and then diluted 10 fold, the final target concentration is equal to its initial concentration in the 1 ml urine sample. We incubated the sample off-chip for 5 min at 60°C prior to introducing to chip (see discussion of Figure 3b).
Figure 3.
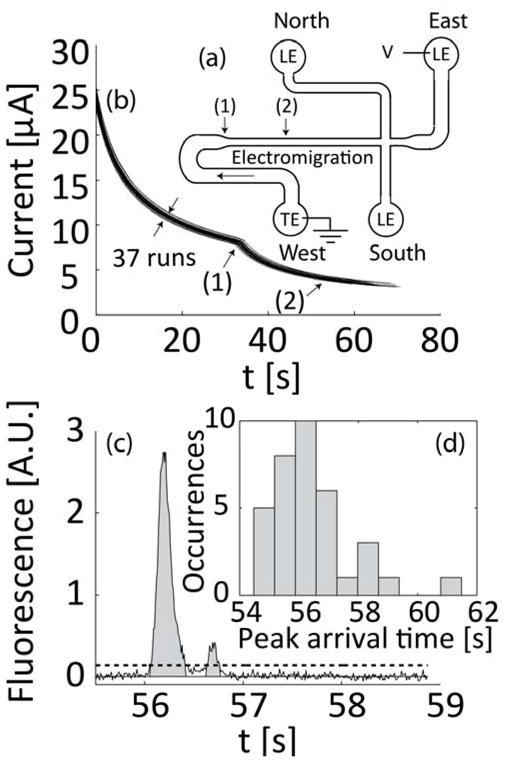
Microfluidic chip and experiments to establish analyte quantitation. (a) Schematic of the microfluidic chip showing the direction of electromigration, the area where channel width decreases (1), and location of detector (2). (b) Current was measured in real-time for all experiments. Current monitoring is important for interpretation of fluorescence signal integrals and allows detection of unwanted effects such as clogging of the channel or significant variations in initial conditions. Sample lysing (and dilution) were optimized for repeatability. Shown are overlayed time traces of 37 runs for sample concentrations of 1E6 to 1E8 cfu/ml and negative control samples. (c) Typical fluorescent signal with bacterial sample. We integrate the signal to estimate the total amount of focused sample. We determine signal baseline using the GIFTS method40 and then integrate the data in regions where values are 5 standard deviations above the baseline noise level. Integration values are measured from – the mean background noise level, as indicated by gray area). (d) Histogram showing the distribution of peak arrival time for the 37 runs presented in (b). The standard deviation of arrival times (at (1)) is less than 5% of the mean.
For chip loading, we pipetted 2 μL of the sample/beacons mixture into the South reservoir of the chip containing 20 μL of TE. For the positive control experiments in which a synthetic target was used (Figure 1c), we skipped the lysing and incubation steps, and directly introduced 2 μL of 1 nM beacons, and 2 μL of varying concentration of synthetic target into the TE (South) reservoir. In all cases, we used commercially available microfluidic chips made of borosilicate (NS-95) from Caliper Life Sciences (Mountain View, CA). The channel is isotropically etched to a depth of 12 μm and consists of a 54 μm wide section which constricts into a 34 μm wide section. The total length of the channel is 34.6 mm, with the initial (wide) section 11.5 mm in length. The chip layout is depicted schematically in Figure 4a.
Figure 4.
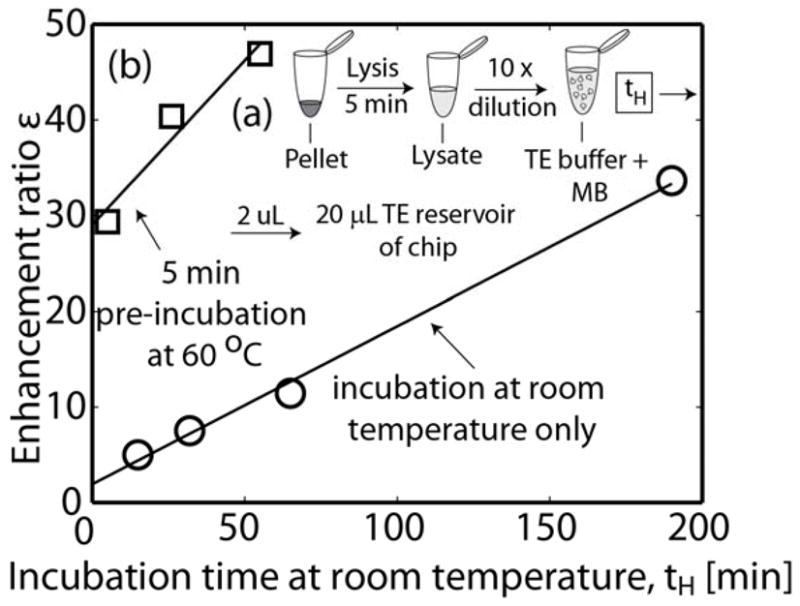
Sample preparation schematic and assay enhancement ratio as a function of initial off-chip incubation time, tH. (a) Schematic of sample lysis and dilution. A pellet (from either bacterial culture or infected patient urine) is lysed at room temperature for 5 min. The lysate is diluted 10× with buffer containing molecular beacons and MgCl2. The mixture of sample and beacons is then incubated at either room temperature or 60°C. 2 μL are introduced into the microfluidic chip reservoir. (b) Enhancement ratio as a function of off-chip hybridization time, using a bacterial culture sample. Shown are pre-ITP incubations at either room temperature or for 5 min at 60°C followed by incubation at room temperature. There is a tradeoff between total assay time and sensitivity. Based on these data, we chose off-chip, pre-ITP incubation at 60°C for 5 min. The assay can be completed in under 15 min from beginning of lysing to signal detection.
At the beginning of each set of runs, we cleaned the channel by flowing 200 mM NaOH for 5 min, and then rinsed the channel with DI for 2 min. For each experiment, we filled the North, East and South reservoirs with 20 μL of LE and applied vacuum to the West reservoir (connected to the longest channel) until all channels were filled. We then rinsed the West reservoir with DI water, and filled it with 20 μL of TE and 2 μL of the sample/beacons mixture. We use a semi-infinite sample injection (where sample is mixed with the TE) as it allows increasing the total amount of focuses sample by continuously focusing new sample at the interface.35 This is in contrast to finite injection, where the amount of sample is limited to a preset injection volume. We then applied 1.1 kV between the East and West reservoirs and detected at a distance of 19 mm from the West reservoir. Note we do not use or need the “cross” channel intersection of the chip, and the experiment can be realized with a single, straight channel.
RESULTS AND DISCUSSION
Repeatability of ITP velocity and temporal fluorescence signals
The analysis presented in the theory section suggests that repeatability of ITP velocity is important for quantitative measurements using a point detector. To monitor ITP velocity for each experiment, we used a sourcemeter to apply constant voltage while measuring current. Initially, the channel was filled entirely with LE. As the ITP interface electromigrated, the lower conductivity TE replaced the LE. This resulted in an increase of the overall resistance of the channel and a gradual decrease in current. Figure 3b presents 37 overlaid curves of current versus time for experiments performed in the same day, and using both bacterial sample concentrations ranging from 1E6 to 1E8 cfu/ml and negative control samples. The expected, abrupt change in slope of the curve near 35 s (labeled (1)) corresponds to the time when the ITP interface moves into the narrow region of the channel (c.f. Figure 3a). Figure 3d presents the distribution of the peak’s arrival time at the detector for these 37 experiments. The standard deviation of arrival time at the detector is less than 3% of the mean.
Application of constant current is often preferred over constant voltage for electrophoretic assays, as it allows maintaining constant electric fields, and its results are simpler to interpret and analyze. However, application of constant current has a significant disadvantage: For a given maximum power supply voltage, the maximum allowable current must be set according to maximum resistance in channel. This maximum resistance is only achieved at the end of the run. Since the ITP velocity is directly proportional to the current, this results in significantly longer assay times. For example, in figure 3b we show that, under constant voltage, current decreases from approximately 25 μA at t = 0, to 2.5 μA at t = 60 s. Performing the same assay under constant current conditions (and using the same power source) conditions would require to set the current at 2.5 μA throughout, resulting in approximately 7 fold increase in assay time. The results in figure 3 indicate assay time can be minimized using constant voltage, without compromising repeatability.
Using equation (9), we can propagate this uncertainty to the enhancement ratio, for which the standard deviation is also 3%. Figure 3c presents a typical raw signal recorded by the PMT. We observed in several experiments dual peaks such as the one presented in the figure (with one peak have a significantly lower intensity and lower area then the other). We hypothesize that some 16S rRNA degradation takes place, resulting in RNA fragments. Although theoretically all species should overlap under peak mode ITP, a previous study showed that carbonic acid (naturally present in the buffers) can result in separation of species at the interface.39 To account for this, and since peak intensities are highly sensitive to sampling rate and dispersion, we use ‘total fluorescence’ as the integral of the signal (area under the curve). As discussed in the theory section, we then describe the signal enhancement factor as ε = A/A0, where A is the area under the signal peak, and Ao is the value for the control case. To integrate the signal peak, we first determined the baseline using the GIFTS method.40 The signal magnitude is the value above this baseline. We then integrate the signal only in regions where the signal is 5 standard deviations above the baseline (integration area indicated in gray).
Effect of off-chip incubation on on-chip assay
We analyzed the effect of initial off-chip incubation times at 60°C to complement our assay’s subsequent, on-chip, ITP-aided incubation. Figure 4b presents the effect of off-chip incubation times on the enhancement ratio of the assay. Shown are measured, post-ITP enhancement ratios as a function of off-chip incubation time, tH. We tested two incubation schemes: (1)incubation at room temperature only, and (2) initial 5 min incubation at 60 °C (approximately 20°C below the melting temperature of the probe), followed by continued incubation at room temperature. Testing the sample immediately after the 5 min incubation at 60°C resulted in an enhancement ratio equivalent to more than 2 h of incubation at room temperature. Clearly, there is a tradeoff between the sensitivity of the assay, and the total time to complete the assay. For this work, we chose to limit the hybridization time to 5 min at 60°C (and thereafter kept the sample on ice to minimize further hybridization). This procedure enabled detection over a range of clinically relevant bacterial concentration while keeping the total assay time under 15 min.
Detection of E. coli from cell cultures and patient urine samples
Figure 5a presents measured enhancement ratio versus bacteria concentration for patient-derived bacterial isolates grown in culture media, as well as for infected human urine samples. For each sample, the bacteria concentration indicated in the bottom horizontal scale was determined separately by cell plating, and is given in cfu/ml units. For convenience, we present in the top horizontal scale the concentrations of target molecules in molar units. We here assume 20,000 copies of 16S rRNA per bacterial cell, but note that the number of ribosomes per cell, and consequently the number or rRNA copies, may vary between approximately 7000 and 70,000, depending on the growth stage of the bacteria.26, 41 We note this range in copy number translates to variations in the measured enhancement ratios for the case of clinical samples. As bacteria concentration increases, a larger fraction of the molecular beacons is hybridized and the fluorescent signal and enhancement ratio increase. The enhancement ratios obtained for the clinical urine samples were overall in good agreement with the values obtained for bacteria cultures, except for the sample having a concentration of 1E8 cfu/mL. The pellet for this sample was larger, indicative of a large number of white blood cells, and consistent with the setting of a significant infection. After the standard lysis step of 5 min, there remained visible cellular aggregates suggesting that lysis was incomplete for this particularly turbid clinical sample. For consistency with other runs, we did not lengthen the lysis time and tested the sample using the same time line as described in experimental setup setction. This issue of white blood cells content and its effect on signal merits further study.
Figure 5.
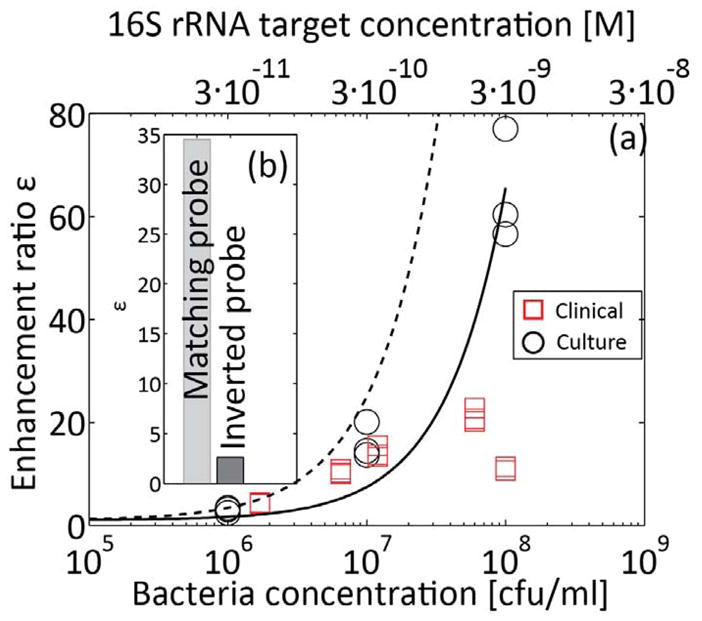
Quantitative detection of E. coli 16S rRNA sequence from bacterial cultures, and bacterial detection in patient urine samples.. The solid line is presented to aid visualization, and corresponds to a best linear fit on bacteria cultures results. The dashed line corresponds to the equilibrium model (equation (6)) for α/β* 80. 16S rRNA was extracted and focused with molecular beacons using ITP, and detected on chip. (a) Measured enhancement ratio for cultured bacteria samples (circles) and urine samples (squares) at clinically relevant bacterial concentrations. The top horizontal axis shows estimate molar concentration of target in the initial urine sample. The bacteria concentrations presented on the x-axis were evaluated separately using cell plating. The lysing procedure described in Figure 4 was used for all samples, and followed by incubation with molecular beacons for 5 min at 60°C. The limit of detection is approximately 1E6 cfu/mL, with an enhancement ratio of ~3 (b) We checked the specificity of the probe by performing the assay with a molecular beacon having an inverted probe sequence (from 3′ to 5′). While some non-specific hybridization was observed, the enhancement ratio using the inverted probe is significantly lower.
Given the current choice of molecular beacon concentrations, the total assay time was approximately 15 min, and was sensitive in the clinically relevant range of (1E6-1E8 cfu/mL). The enhancement ratio for 1E6 cfu/ml was approximately 3. For a concentration of 1E5 cfu/ml (data not presented) the signal intensity was indistinguishable from the negative control case, with an enhancement ratio of approximately 1. This result is consistent with the analysis we presented in the theory section, predicting sensitivity across two orders of magnitude of sample concentration. While it may possible to improve sensitivity by reducing the level of dilution, we found this adversely affects assay repeatability. We hypothesize that this is due to the effect of higher concentrations of acids in the lysate42 affecting the conditions in the reservoir. We believe some of these acids overspeed the TE and create additional ITP zones between the LE and TE.9
Shown together with the data in Figure 5a are a linear regression best fit to the data values, as well as the predicted enhancement ratio dependence predicted by Eq. (6). We attribute the difference between the data and the prediction to the equilibrium assumption inherent in the equation. We believe the reactions kinetics are not fully at equilibrium. For example, a bacteria concentration of 1E7 cfu/ml corresponds to 1 pM of target in the reservoir. From measurements of the interface width, we estimate ITP increases the concentration at the interface by 10,000, to a value of order 10 nM at the interface. The half-time for DNA beacons hybridization at this concentration is at best 20 min (at room temperature, corresponding to a rate constant of 1E5 M−1s−1 at high ionic strength36), which is longer than the sum of off-chip and on-chip hybridization time. Furthermore, the model assumes all target sequences are available for hybridization, which may not hold due to incomplete lysis, and secondary structure of the rRNA. We hope to explore the kinetics of this assay and develop models which couple reaction kinetics and ITP dynamics in future study.
While detection of a nucleic acid sequence does not necessarily indicate viable bacteria, (if, for example, the genetic content is preserved after cell death), previous studies on electrochemical detection of 16S rRNA37, 30 showed that all cases of 16S rRNA detection were confirmed by the ability to culture the bacteria by plating, which is indicative of viable bacteria. Conversely, all the samples that were sensor negative were also culture negative. Thus we conclude that the majority of 16S rRNA that we are able to detect is derived from viable bacteria. We hypothesize that RNA of dead cells may quickly degrade in urine (e.g. via RNAse), thus significantly reducing or eliminating the signal associated with their 16S rRNA content.
We also performed several control runs to ensure that the fluorescence signal was the result of probe-target hybridization. Before each set of experiments we performed ITP using the TE and LE buffers alone, and prepared new buffers from stock solution and/or replaced the microfluidic chip if contamination was observed. Between experiments using sample, we routinely performed a control run (with beacons, but without sample) to establish the baseline signal. Further, we performed experiments with cell lysate and no beacons and found it contributes negligibly to enhancement ratio. Lastly, we tested a patient-derived bacterial isolate using an inverted molecular beacon (figure 5b). Some increase in fluorescence was observed, likely due to non-specific binding with other regions of the rRNA. The enhancement ratio was approximately 15 fold lower than with the correct beacon.
CONCLUSIONS AND FUTURE WORK
We demonstrated and characterized a new assay for rapid detection of UTI using ITP and molecular beacons. We use on-chip ITP to selectively focus 16S rRNA and molecular beacons directly from bacterial lysate. We perform detection of the focused hybridized complex using a point detector.
We presented detection of E. coli in bacteria cultures as well as in patient urine samples in the clinically relevant range 1E6-1E8 cfu/mL. For bacterial cultures we further presented quantification in this range. Since central clinical microbiology laboratories, including our institution, do not provide quantitative measure of concentration above 1E5 cfu/ml, we routinely perform quantitative plating and have found that vast majority of our UTI patients have bacterial concentrations of 1E6 cfu/ml or greater. Our assay therefore covers an important range of the UTI samples that are seen clinically. However, further improvements to sensitivity are required in order to encompass the entire clinically relevant range of roughly 1E5-1E8 cfu/mL.43, 44 Lower molecular beacons concentrations may result in improved enhancement ratios at low bacteria concentrations (see equation (6)), but would require significantly longer hybridization times, and perhaps loss of quantitation at higher concentrations (since nearly all beacons would be hybridized above a certain target concentration). We hypothesize the most promising method of achieving high sensitivity while maintaining a short assay time is to improve hybridization rate. This may be possible by, for example, further optimization of probe sequence, stem sequence, and chemistry of fluorophore/quencher pair (i.e., improving α/β* in equation (6)). Alternatively, this could be achieved by saturating the sample with a high concentration of beacons in the reservoir (to increase hybridization rate), and using highly specific ITP to selectively focus only the hybridized product (excluding free beacons, to avoid a high negative control signal).
In the current assay our initial samples were pellets obtained from urine sample by centrifugation. In order to achieve an automated analysis system any centrifugation steps ideally should be eliminated. Improvement of assay sensitivity may also enable detection of bacteria directly from urine with little or no off-chip sample preparation (all other steps in the assay are dilutions and mixing, functions which can presumably be implemented on chip45, 46). We demonstrated the assay using a universal prove targeting a highly conserved region of bacterial 16S rRNA. This type of test could be highly beneficial in quickly ruling out bacterial infections. We believe that by changing the molecular beacons probe sequence, the principles presented here can be directly used for detection of bacteria-specific sequences, as well as for the design of a variety of other rapid diagnostics or detection methods for pathogenic diseases.
Acknowledgments
We gratefully acknowledge the support NSF grant ECCS 0901292 and of of NIH/NIAID grant U01 AI 082457 to J.C.L. J.G.S. acknowledges funding from DARPA sponsored Micro/Nano Fluidics Fundamentals Focus (MF3) Center under contract number N66001-10-1-4003, and to DARPA grant N660001-09-C-2082. G.V.K was supported by a postdoctoral fellowship from the Natural Science and Engineering Research Council, Canada.
References
- 1.Morens DM, Folkers GK, Fauci AS. Nature. 2004;430:242–249. doi: 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vital and Health Statistics. 1999:13. [Google Scholar]
- 3.Ronald A. Disease-a-Month. 2003;49:71–82. doi: 10.1067/mda.2003.8. [DOI] [PubMed] [Google Scholar]
- 4.Freedman AL. The Journal of Urology. 2005;173:949–954. doi: 10.1097/01.ju.0000152092.03931.9a. [DOI] [PubMed] [Google Scholar]
- 5.Millar BC, Xu JR, Moore JE. Current Issues in Molecular Biology. 2007;9:21–39. [PubMed] [Google Scholar]
- 6.Mariella R. Biomedical Microdevices. 2008;10:777–784. doi: 10.1007/s10544-008-9190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Call DR. Critical reviews in microbiology. 2005;31:91–99. doi: 10.1080/10408410590921736. [DOI] [PubMed] [Google Scholar]
- 8.Wong CW, Heng CL, Yee LW, Soh SW, Kartasasmita CB, Simoes EA, Hibberd ML, Sung WK, Miller LD. Genome Biol. 2007;8:R93. doi: 10.1186/gb-2007-8-5-r93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everaerts FM, Beckers JL, Verheggen TP. Isotachophoresis: theory, instrumentation, and applications. 3. Elsevier Scientific Publishing Company; Amsterdam, The Netherlands: 1976. [Google Scholar]
- 10.Kendall J, Jette ER, West W. J Am Chem Soc. 1926;48:3114–3117. [Google Scholar]
- 11.Longsworth LG. J Am Chem Soc. 1930;52:1897–1910. [Google Scholar]
- 12.Jung BG, Zhu YG, Santiago JG. Anal Chem. 2007;79:345–349. doi: 10.1021/ac060949p. [DOI] [PubMed] [Google Scholar]
- 13.Jung B, Bharadwaj R, Santiago JG. Anal Chem. 2006;78:2319–2327. doi: 10.1021/ac051659w. [DOI] [PubMed] [Google Scholar]
- 14.Pantuckova P, Urbanek M, Krivankova L. Electrophoresis. 2007;28:3777–3785. doi: 10.1002/elps.200700189. [DOI] [PubMed] [Google Scholar]
- 15.Prochazkova A, Krivankova L, Bocek P. J Chromatogr, A. 1999;838:213–221. doi: 10.1016/s0021-9673(99)00114-4. [DOI] [PubMed] [Google Scholar]
- 16.Krivankova L, Samcova E, Bocek P. Electrophoresis. 1984;5:226–230. [Google Scholar]
- 17.Sadecka J, Netriova J. J Liq Chromatogr Relat Technol. 2005;28:2887–2894. [Google Scholar]
- 18.Flottmann D, Hins J, Rettenmaier C, Schnell N, Kuci Z, Merkel G, Seitz G, Bruchelt G. Microchim Acta. 2006;154:49–53. [Google Scholar]
- 19.Bexheti D, Anderson EI, Hutt AJ, Hanna-Brown M. J Chromatogr, A. 2006;1130:137–144. doi: 10.1016/j.chroma.2006.06.110. [DOI] [PubMed] [Google Scholar]
- 20.Mikus P, Kubacak P, Valaskova I, Havranek E. Talanta. 2006;70:840–846. doi: 10.1016/j.talanta.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt K, Hagmaier V, Bruchelt G, Rutishauser G. Urological Research. 1980;8:177–180. doi: 10.1007/BF00256414. [DOI] [PubMed] [Google Scholar]
- 22.Schoch RB, Ronaghi M, Santiago JG. Lab Chip. 2009;9:2145–2152. doi: 10.1039/b903542g. [DOI] [PubMed] [Google Scholar]
- 23.Persat A, Marshall LA, Santiago JG. Anal Chem. 2009;81:9507–9511. doi: 10.1021/ac901965v. [DOI] [PubMed] [Google Scholar]
- 24.Matsumura T, Bercovici M, Santiago JG. Lab Chip. 2010 doi: 10.1039/c004120c. [DOI] [PubMed] [Google Scholar]
- 25.Bercovici M, Kaigala GV, Liao JC, Santiago JG. The 14th International Conference on Miniaturized Systems for Chemistry and Life Science; Groningen, The Netherlands. 2010. pp. 797–799. [Google Scholar]
- 26.Neidhardt FC, Umbarger HE. ASM Press. 2. Washington, D.C: [Google Scholar]
- 27.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 28.Wang K, Tang Z, Yang CJ, Kim Y, Fang X, Li W, Wu Y, Medley CD, Cao Z, Li J, Colon P, Lin H, Tan W. Angew Chem Int Ed. 2008:2–17. doi: 10.1002/anie.200800370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persat A, Santiago JG. Anal Chem. 2011 doi: 10.1021/ac103225c. in press. [DOI] [PubMed] [Google Scholar]
- 30.Liao JC, Mastali M, Li Y, Gau V, Suchard MA, Babbitt J, Gornbein J, Landaw EM, McCabe ER, Churchill BM, Haake DA. Journal of Molecular Diagnostics. 2007;9:158–168. doi: 10.2353/jmoldx.2007.060052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Proc Natl Acad Sci U S A. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsourkas A. Nucleic Acids Res. 2003;31:1319–1330. doi: 10.1093/nar/gkg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Acad Sci U S A. 1998;95:8602. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bharadwaj R, Santiago JG, Mohammadi B. Electrophoresis. 2002;23:2729–2744. doi: 10.1002/1522-2683(200208)23:16<2729::AID-ELPS2729>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 35.Khurana TK, Santiago JG. Anal Chem. 2008;80:6300–6307. doi: 10.1021/ac800792g. [DOI] [PubMed] [Google Scholar]
- 36.Kuhn H, Demidov VV, Coull JM, Fiandaca MJ, Gildea BD, Frank-Kamenetskii MD. J Am Chem Soc. 2002;124:1097–1103. doi: 10.1021/ja0041324. [DOI] [PubMed] [Google Scholar]
- 37.Liao JC, Mastali M, Gau V, Suchard MA, Moller AK, Bruckner DA, Babbitt JT, Li Y, Gornbein J, Landaw EM, McCabe ERB, Churchill BM, Haake DA. Journal of Clinical Microbiology. 2006;44:561–570. doi: 10.1128/JCM.44.2.561-570.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marras SAE, Kramer FR, Tyagi S. Nucleic Acids Res. 2002;30:8. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khurana TK, Santiago JG. Lab Chip. 2009;9:1377–1384. doi: 10.1039/b815460k. [DOI] [PubMed] [Google Scholar]
- 40.Feng X, Zhu Z, Cong P. Natural Computation. ICNC’09. Fifth International Conference; 2009; 2009. pp. 496–499. [Google Scholar]
- 41.Bremer H, Dennis PP, et al. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. American Society for Microbiology; Washington, DC: 1987. pp. 1527–1542. [Google Scholar]
- 42.Carstensen EL, Cox HA, Mercer WB, Natale LA. Biophys J. 1965;5:289–300. doi: 10.1016/s0006-3495(65)86717-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunin CM, VanArsdale White L, Hua Hua T. Annals of Internal Medicine. 1993;119:454–460. doi: 10.7326/0003-4819-119-6-199309150-00002. [DOI] [PubMed] [Google Scholar]
- 44.Wilson ML, Gaido L. Clinical Infectious Diseases. 2004;38:1150–1158. doi: 10.1086/383029. [DOI] [PubMed] [Google Scholar]
- 45.Jacobson SC, McKnight TE, Ramsey JM. Anal Chem. 1999;71:4455–4459. [Google Scholar]
- 46.Walker GM, Monteiro-Riviere N, Rouse J, O’Neill AT. Lab Chip. 2007;7:226. doi: 10.1039/b608990a. [DOI] [PubMed] [Google Scholar]