

Summary
Salmonella pathogenicity islands 1 and 2 (SPI-1 and SPI-2) play key roles in the pathogenesis of Salmonella enterica. Previously, we showed that when Salmonella grows in LB medium HilD, encoded in SPI-1, first induces the expression of hilA, located in SPI-1, and subsequently of the ssrAB operon, located in SPI-2. These genes code for HilA and the SsrA/B two-component system, the positive regulators of the SPI-1 and SPI-2 regulons, respectively. In this study, we demonstrate that CsrA, a global regulatory RNA-binding protein, post-transcriptionally regulates hilD expression by directly binding near the Shine-Dalgarno and translation initiation codon sequences of the hilD mRNA, preventing its translation and leading to its accelerated turnover. Negative regulation is counteracted by the global SirA/BarA two-component system, which directly activates the expression of CsrB and CsrC, two non-coding regulatory RNAs that sequester CsrA, thereby preventing it from binding to its target mRNAs. Our results illustrate the integration of global and specific regulators into a multi-factorial regulatory cascade controlling the expression of virulence genes acquired by horizontal transfer events.
Keywords: SirA/BarA, Csr, HilD, two-component system, Salmonella, SPI, virulence gene regulation
Introduction
Horizontal transfer of DNA fragments, encoding for a variety of virulence factors, has been of paramount importance in the evolution of pathogenic bacteria by providing the ability to survive in and exploit and survive in different host niches and thus to cause different diseases (Ochman et al., 2000, Schmidt and Hensel, 2004). To take advantage of the genetic information contained in the acquired genes, bacteria had to adapt regulatory mechanisms to control their potentially deleterious expression under non-permissive conditions, and to induce their appropriate spatiotemporal expression inside the hosts to establish an infection.
Salmonella are Gram-negative bacteria comprising around 2500 serotypes, many of which are important human and animal pathogens. Two major clinical manifestations characterize Salmonella infections: enteritis (salmonellosis), a self-limiting local intestinal inflammatory response, and enteric fever (typhoid), a systemic infection characterized by the dissemination of the bacteria throughout the reticuloendothelial system via infected macrophages to the lymph nodes, liver and spleen (Ohl and Miller, 2001; Haraga et al., 2008). S. enterica serotype Typhimurium (S. Typhimurium) can produce enteritis in humans, calves or chickens, whereas in mice it causes a systemic infection similar to human typhoid fever produced by serotype Typhi (Ohl and Miller, 2001; Haraga et al., 2008); thus it is frequently used as a model to study Salmonella pathogenesis. Most of the Salmonella virulence genes are located within distinct genomic regions denominated Salmonella Pathogenicity Islands (SPIs), which have been acquired by independent horizontal transfer events (Groisman and Ochman, 1997; Marcus et al., 2000). SPI-1 and SPI-2, composed by 39 and 44 genes, respectively, play a key role in Salmonella pathogenesis (Hansen-Wester and Hensel, 2001). Each pathogenicity island encodes a type three secretion system (T3SS), different effector proteins, which are translocated into the host cells by their cognate T3SS, chaperones and transcriptional regulators that control the expression of the genes within each island (Hensel, 2000; Marcus et al., 2000; Hansen-Wester and Hensel, 2001). SPI-1 genes are necessary for the invasion of epithelial host cells, thus for the intestinal colonization leading to enteritis; whereas SPI-2 genes are required for bacterial replication/survival inside macrophages, and thus for the systemic disease (Hensel et al., 1998, Santos et al., 2003; Haraga et al., 2008). However, different studies have revealed that SPI-2 genes also play a role in the development of the intestinal inflammatory disease and, accordingly, they are expressed in the intestinal lumen (Bispham et al., 2001; Brown et al., 2005; Coburn et al., 2005; Coombes et al., 2005; Hapfelmeier et al., 2005; Jones et al., 2007).
In vitro, SPI-1 genes are expressed when Salmonella grows in the nutrient-rich medium Luria-Bertani (LB) (Lundberg et al., 1999; Miao and Miller, 2000), and are regulated by environmental conditions such as osmolarity, oxygen concentration and pH (Altier, 2005; Jones, 2005; Ellermeier and Slauch, 2007). Under these growth conditions, two AraC-like transcriptional regulators, HilD and HilC, encoded within SPI-1, activate hilA expression (Schechter et al., 1999; Schechter and Lee, 2001, Ellermeier et al., 2005), the central positive regulator of SPI-1, by counteracting the repression exerted by the global repressor H-NS on the hilA promoter (Schechter et al., 2003; Olekhnovich and Kadner, 2006). HilA then directly activates expression of genes encoding T3SS components (Eichelberg and Galan, 1999; Lostroh et al., 2000, Lostroh and Lee, 2001), as well as the expression of another AraC-like transcriptional regulator, InvF (Lostroh et al., 2000), which in turn activates the expression of genes encoding effector proteins (Darwin and Miller, 1999; Eichelberg and Galan, 1999).
The SPI-2 genes are expressed during late stationary phase when Salmonella grows in LB medium (Bustamante et al., 2008) or in acidic minimal media containing low concentrations of phosphate, calcium and magnesium (Deiwick et al., 1999; Miao and Miller, 2000). The SPI-2-encoded SsrA/SsrB two-component regulatory system (Cirillo et al., 1998, Deiwick et al., 1999; Worley et al., 2000), controls the expression of the SPI-2 regulon mainly by counteracting the repression exerted by H-NS on genes located within and outside the island (Walthers et al., 2007; Walthers et al., 2010).
In addition to these regulators, other regulatory proteins encoded outside of these islands have been shown to be involved in controlling the expression of the SPI-1 and SPI-2 regulons (reviewed by Altier, 2005; Jones, 2005; Ellermeier and Slauch, 2007; Fass and Groisman, 2009).
Previous reports have shown that the global transcriptional regulator SirA positively controls the expression of the SPI-1 genes (Johnston et al., 1996; Ahmer et al., 1999; Teplitski et al., 2003). SirA is a member of the FixJ family of response regulators, forming a two-component regulatory system with the sensor kinase protein BarA (Altier et al., 2000b, Teplitski et al., 2003). SirA/BarA orthologs are present in many bacteria, including E. coli (UvrY/BarA), Pseudomonas species (GacA/GacS), Vibrio cholerae (VarA/VarS), Erwinia carotovora (ExpA/ExpS) and Legionella pneumoniae (LetA/LetS), where they control the expression of genes associated with virulence, secondary metabolism, motility, exoenzyme production, quorum sensing or biofilm formation (Goodier and Ahmer, 2001, Lapouge et al., 2008). In E. coli, UvrY/BarA activates expression of two non-translated small RNAs, CsrB and CsrC, each of which contains several motifs recognized by the RNA binding protein CsrA (Suzuki et al., 2002; Weilbacher et al., 2003; Babitzke and Romeo, 2007). CsrA is a homodimeric protein that binds to sequences overlapping the Shine-Dalgarno (SD) sequence in target mRNAs, thus blocking ribosome binding and translation, and promoting their degradation (Baker et al., 2002; Lucchetti-Miganeh et al., 2008, Timmermans and Van Melderen, 2010). CsrB and CsrC antagonize CsrA function by sequestering this protein, thus counteracting its translational repression activity (Weilbacher et al., 2003; Babitzke and Romeo, 2007; Lucchetti-Miganeh et al., 2008; Timmermans and Van Melderen, 2010).
Here we demonstrate that the SirA/BarA two-component system and the Csr post-transcriptional regulatory system regulate the expression of both the SPI-1 and SPI-2 genes in Salmonella growing in LB medium. SirA activates the expression of CsrB and CsrC, which counteract the post-transcriptional repression exerted by CsrA, whose binding sites overlap the SD sequence and translation initiation codon of hilD mRNA. HilD then activates expression of hilA and ssrAB, the genes encoding the central regulators of SPI-1 and SPI-2, respectively. This study integrates a regulatory cascade constituted by global and horizontally acquired regulatory elements that controls the expression of the SPI-1 and SPI-2 regulons.
Results
SirA is involved in the expression of the SPI-2 regulon
Recently, we demonstrated that the SPI-2 genes are expressed when S. Typhimurium grows in the nutritionally rich medium LB (Bustamante et al., 2008). Under this growth condition the SPI-1-encoded regulator HilD activates the sequential expression of the SPI-1 and SPI-2 regulons at the early and late stationary phases of growth, respectively. In this study, we investigated the possibility that, in addition to HilD, other reported SPI-1 regulators could be involved in controlling the expression of SPI-2 genes. Previous studies have shown that the global SirA/BarA two-component system positively regulates the expression of SPI-1 (Johnston et al., 1996, Ahmer et al., 1999; Altier et al., 2000b; Teplitski et al., 2003) and SPI-4 genes (Gerlach et al., 2007).
To investigate if SirA also regulates the SPI-2 genes, we analyzed the expression of a transcriptional fusion of the ssaG (SPI-2) promoter to the cat (chloramphenicol acetyl transferase) reporter gene, in wild type (WT) S. Typhimurium strain SL1344 and its ΔsirA derivative, as well as in the ΔhilC, ΔhilA, ΔhilD, ΔompR and ΔssrB mutants. Each strain was grown in LB or in N-minimal medium (N-MM), two different in vitro growth conditions shown to sustain the expression of SPI-2 genes (Deiwick et al., 1999; Miao and Miller, 2000; Bustamante et al., 2008). Expression of ssaG-cat showed a similar reduction in the ΔsirA and ΔhilD mutants grown in LB with respect to the WT strain (Fig. 1A), but was not affected when they were grown in N-MM (Fig. 1B). These results are consistent with our previous observations indicating that HilD is required for the expression of the SPI-2 genes when Salmonella grows in LB but not in N-MM (Bustamante et al., 2008). Also in accordance with our previous results (Bustamante et al., 2008), expression of ssaG was reduced in both LB and N-MM in the ΔompR and ΔssrB mutants, which lack the SPI-2 regulators OmpR and SsrB. In contrast, expression was not affected in the ΔhilA and ΔhilC mutants that lack the SPI-1 regulators HilA and HilC (Figs. 1A and B). As a control, we determined the expression of a hilA-cat transcriptional fusion in the same strains grown in LB. In agreement with previous studies showing that both SirA and HilD positively regulate hilA (SPI-1) expression (Johnston et al., 1996; Ahmer et al., 1999; Schechter et al., 1999; Schechter and Lee, 2001; Schechter et al., 2003; Teplitski et al., 2003; Ellermeier et al., 2005; Olekhnovich and Kadner, 2006), expression of the hilA-cat fusion was reduced in the ΔsirA and ΔhilD mutants, but was not affected in the ΔompR, ΔssrB and hilC mutants (Fig. 1C). Furthermore, expression of the hilA-cat fusion increased in the ΔhilA mutant (Fig. 1C), consistent with a previous study showing that HilA negatively regulates its own expression (De Keersmaecker et al., 2005). Taken together, these results indicated that SirA, in addition to HilD, OmpR and SsrB, is required for the expression of SPI-2 genes when Salmonella grows in LB.
Fig. 1.
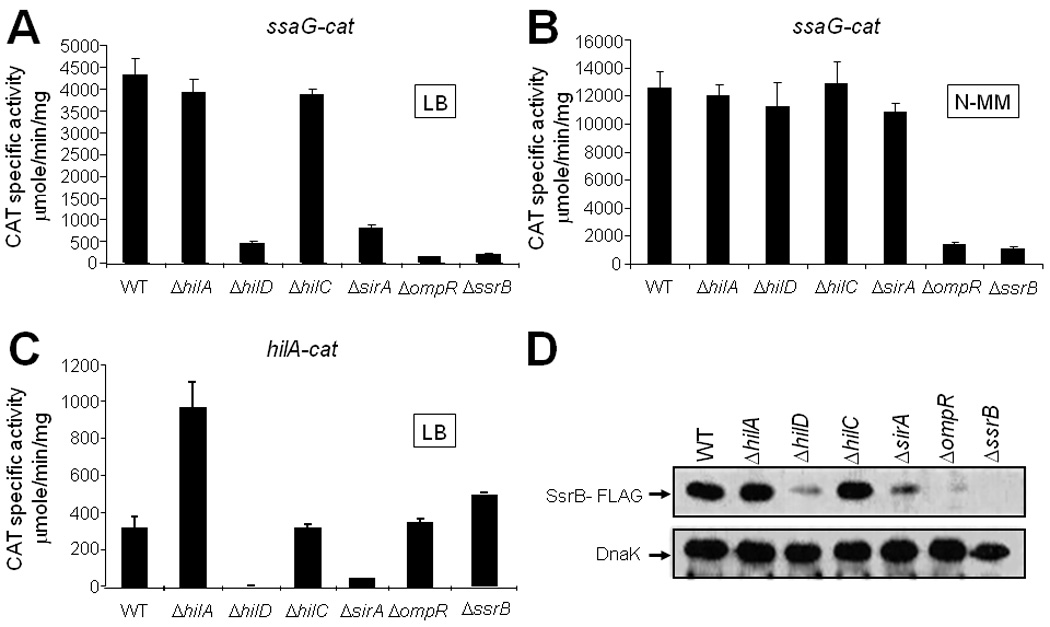
SirA is involved in the expression of the SPI-2 genes. Expression of the ssaG-cat transcriptional fusion contained in pssaG-cat1 was analyzed in WT and isogenic ΔhilA::kan, ΔhilD::kan, ΔhilC::kan, ΔsirA::kan, ΔompR::kan and ΔssrB::kan strains, grown in LB (A) or in N-MM (B) for 10 and 16 h, respectively, at 37°C. (C) Expression of the hilA-cat transcriptional fusion contained in philA-cat1 was determined in WT and isogenic ΔhilA::kan, ΔhilD::kan, ΔhilC::kan, ΔsirA::kan, ΔompR::kan and ΔssrB::kan strains, grown in LB for 5 h at 37°C. CAT specific activity was determined as described in Experimental Procedures. Data are the average of three independent experiments done in duplicate. Bars represent the standard deviations. (D) Western blot analysis of SsrB expression in WT and isogenic ΔhilA, ΔhilD, ΔhilC, ΔsirA, and ΔompR strains carrying a chromosomal FLAG-tagged ssrB gene, using monoclonal anti-FLAG antibodies. Samples were taken from cultures grown for 10 h in LB at 37°C. As a loading control, the expression of DnaK was also determined using monoclonal anti-DnaK antibodies.
To determine if SirA controls ssaG expression, and thus of other SPI-2 genes, by regulating the ssrAB operon, we analyzed the expression of SsrB in WT and ΔsirA strains, as well as in the ΔhilA, ΔhilD, ΔhilC, and ΔompR mutants, which were tested as controls. To follow the expression of SsrB, the ssrB chromosomal gene was tagged with the sequence encoding a 3XFLAG epitope, rendering strains expressing a C-terminal 3XFLAG-tagged SsrB protein (SsrB-FLAG). Total protein extracts were obtained from culture samples of these strains grown in LB and analyzed by western blotting. In agreement with the results shown in Fig. 1A, expression of SsrB-FLAG was drastically reduced in the ΔsirA, ΔhilD and ΔompR mutants, but not in the ΔhilA and ΔhilC mutants (Fig. 1D). In contrast, when these strains were grown in N-MM, SsrB-FLAG expression was not affected in the ΔsirA and ΔhilD mutants, but was still highly reduced in the ΔompR mutant (data not shown). Together, these results indicated that expression of the SsrA/B system and, as a consequence, the SPI-2 genes requires the HilD, OmpR and SirA proteins when S. Typhimurium is grown in LB.
SirA controls the expression of SPI-1 and SPI-2 genes by regulating HilD expression
HilD and OmpR positively regulate ssrAB expression by directly binding to its regulatory region (Lee et al., 2000; Feng et al., 2003, 2004; Bustamante et al., 2008). Since the expression of OmpR was not affected in the absence of SirA or HilD (data not shown), we hypothesized that SirA could regulate the expression of the ssrAB operon directly, acting along with HilD and OmpR, or indirectly by controlling the expression of HilD in a cascade fashion. To distinguish between these possibilities, we first analyzed the expression of SsrB-FLAG by western blot in the ΔhilD and ΔsirA mutants containing plasmids pBAD-HilD1 or pBAD-SirA1, expressing HilD and SirA, respectively. Expression of HilD from plasmid pBAD-HilD1 restored the expression of SsrB-FLAG in both the ΔhilD and ΔsirA mutants to a level that was similar to the WT strain (Fig. 2A). In contrast, expression of SirA from plasmid pBAD-SirA1 only restored the expression of SsrB-FLAG in the ΔsirA mutant (Fig. 2A). Similar results were obtained when the expression of a C-terminal 3XFLAG-tagged HilA protein (HilA-FLAG) or of the hilA-cat fusion was determined in the same genetic backgrounds containing plasmids expressing HilD or SirA (Figs. 2B and S1).
Fig. 2.
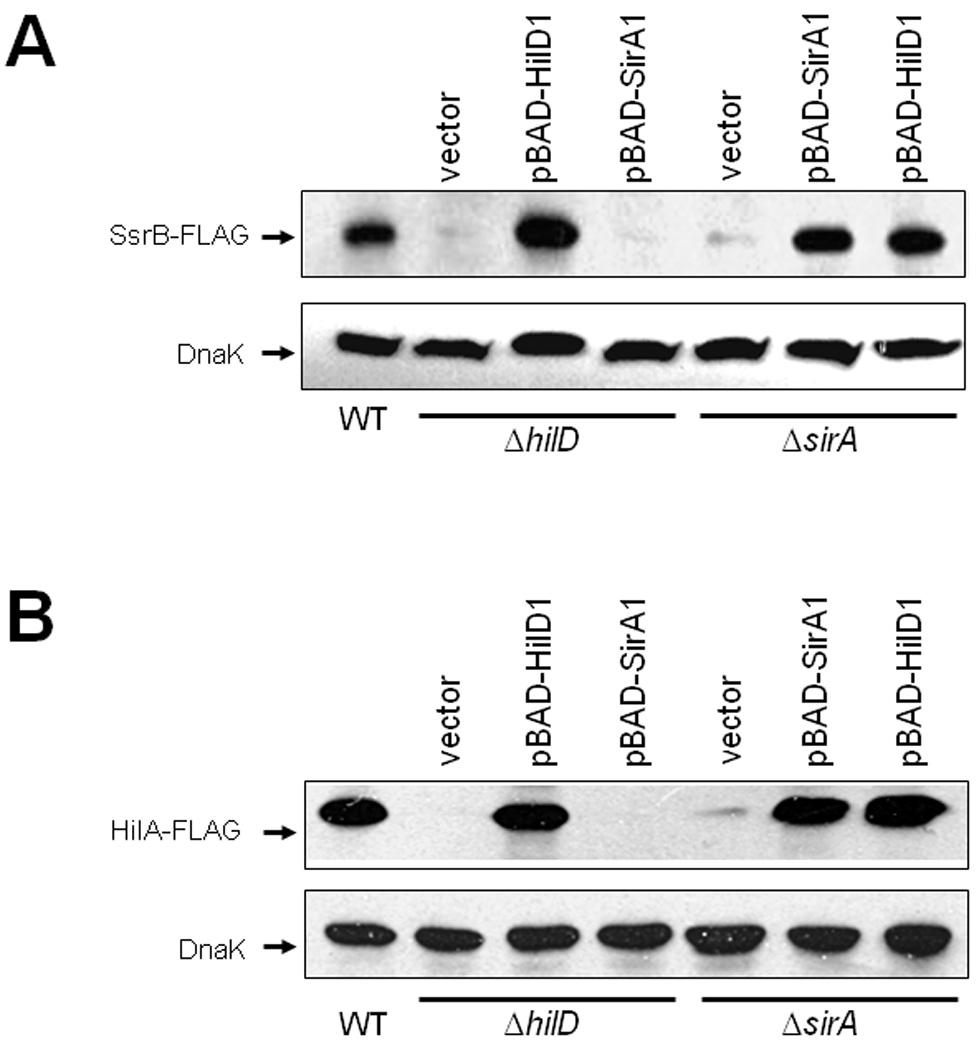
SirA activates the expression of SsrB and HilA through HilD. Expression of SsrB (A) and HilA (B) in WT, ΔhilD and ΔsirA strains carrying a chromosomal FLAG-tagged ssrB or hilA gene, and containing vector pBADMycHisC, or plasmids pBAD-HilD1 or pBAD-SirA1, was analyzed by Western blotting using monoclonal anti-FLAG antibodies. Whole-cell lysates were prepared from samples of bacterial cultures grown in LB for 10 or 5 h, for detection of SsrB-FLAG and HilA-FLAG, respectively, at 37°C. As a control, the expression of DnaK was also determined using monoclonal anti-DnaK antibodies. Expression of HilD and SirA from plasmids pBAD-HilD1 and pBAD-SirA1, respectively, was induced by adding 0.1% L-arabinose to the medium at the beginning of the bacterial cultures.
These results show that HilD is able to activate the expression of ssrAB and hilA in the absence of SirA, whereas SirA requires the presence of HilD, suggesting that SirA controls HilD expression, which in turn directly regulates hilA and ssrAB. In agreement with this notion, the presence of pT3-HilD1, but not of pK3-SirA1, activated expression of hilA-cat, hilC-cat and rtsA-cat transcriptional fusions in the absence of any other Salmonella-specific regulatory protein, as it was determined in an E. coli Δ uvrY mutant lacking UvrY, the E. coli ortholog of SirA (Fig. S2). In contrast, expression of a csrB-cat transcriptional fusion was activated in the presence of pK3-SirA1, but not of pT3-HilD1 (Fig. S2). These results are in agreement with previous studies showing that HilD directly regulates hilA, hilC and rtsA (Schechter et al., 1999; Schechter and Lee, 2001; Ellermeier et al., 2005) and that SirA directly controls the expression of csrB (Teplitski et al., 2003, Teplitski et al., 2006).
The results described above suggest that the SirA/BarA system controls HilD expression. To further determine the extent to which the SirA/BarA system controls hilD expression, we analyzed the activity of a hilD-cat transcriptional fusion and measured hilD mRNA levels by q-RT-PCR in WT, ΔsirA and ΔbarA strains. As HilD has also been reported to positively regulate its own expression (Olekhnovich and Kadner, 2002; Ellermeier et al., 2005), the expression of hilD was also tested in the hilD mutant. Expression of ΔhilD was similarly reduced in the ΔsirA and ΔbarA mutants and to a greater extent in the ΔhilD mutant (Figs. 3A and B). These results, together with those from previous studies (Lucas and Lee, 2001; Ellermeier et al., 2005; Mizusaki et al., 2008), demonstrate that the SirA/BarA system positively regulates the expression of hilD.
Fig. 3.
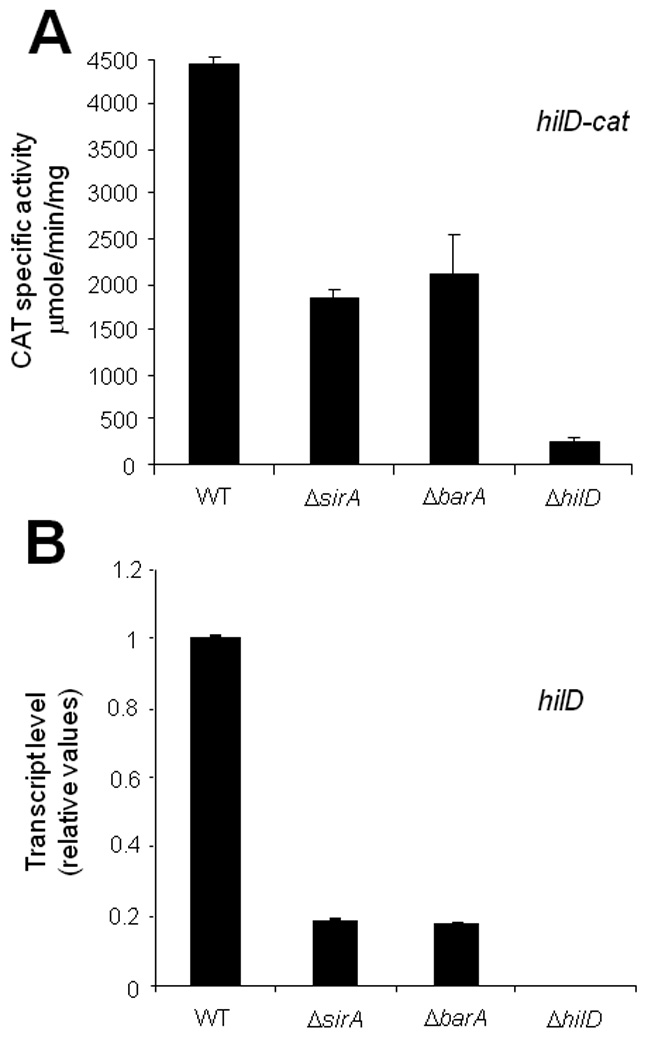
SirA and its cognate sensor kinase protein BarA positively regulate the expression of hilD. Expression of hilD was determined in WT and isogenic ΔsirA, ΔbarA::kan and ΔhilD strains, by monitoring expression of the hilD-cat transcriptional fusion contained in philD-cat1 (A) or by quantifying the hilD mRNA by q-RT-PCR (B), from samples of bacterial cultures grown for 5 h in LB at 37°C. dnaK was used as the internal control to normalize the results obtained for the expression of hilD by q-RT-PCR. Data for the CAT specific activity are the average of three independent experiments done in duplicate. The data for the q-RT-PCR assays indicate the relative expression of hilD, with respect to its expression in the WT strain, and are the average of two independent experiments done in triplicate. Bars represent the standard deviations.
SirA binds to the csrB and csrC regulatory regions, but not to those of hilD, hilA, hilC or ssrAB
The results described above are consistent with the notion that SirA acts through HilD to indirectly control the expression of hilA and ssrAB. In this regard, results from a previous study suggested that SirA most likely acts along with HilD or through HilD to control hilA expression and thus SPI-1 regulation (Ellermeier et al., 2005). Moreover, it is known that SirA regulates the expression of csrB and csrC and that these non-coding RNAs positively regulate Salmonella invasion and expression of SPI-1 genes (Altier et al., 2000b; Teplitski et al., 2003; Fortune et al., 2006; Teplitski et al., 2006). However, it has also been shown that SirA binds to the regulatory regions of hilA and csrB, but that it does not bind to the regulatory regions of hilD and csrC (Teplitski et al., 2003, Teplitski et al., 2006).
To further dissect and clarify the mechanism by which SirA regulates the expression of SPI-1 and SPI-2 genes, we analyzed the interaction of this protein with the regulatory regions of genes under the control of the SirA cascade. SirA was purified as an MBP-SirA fusion protein expressed from plasmid pMAL-SirA1 (as described in Experimental Procedures section in Supporting Information). SPI-1 protein secretion was restored in the ΔsirA mutant carrying plasmid pMAL-SirA1 (data not shown), confirming the functionality of MBP-SirA. Binding of phosphorylated and nonphosphorylated purified MBP-SirA to a 32P-5’-end-labeled PCR product spanning the regulatory region of csrB was tested by electrophoretic mobility shift assays (EMSAs) to further verify the functionality of the fusion protein. MBP-SirA similarly shifted the csrB fragment whether it was phosphorylated or not, starting at a concentration of 1.5 µM (Fig. S3A), indicating that MBP-SirA phosphorylation is not required for DNA binding in vitro, which is consistent with previous results showing that phosphorylation only increases the DNA-binding affinity of SirA-His6X by approximately twofold (Teplitski et al., 2003). SirA phosphorylation in vivo seems to be critical for its function, since when its predicted phosphorylated residue, Asp54, is substituted for an alanine, or in the absence of its cognate sensor kinase BarA, csrB expression is not induced (data not shown). Therefore, it is likely that the in vitro conditions used to test SirA DNA-binding, or the fusion of SirA to the MBP or to the His6X tag, rendered a SirA protein that specifically binds to its target DNA sequences regardless of its phosphorylation state.
In contrast, a DNA fragment containing the regulatory region of sigD, which was used as a negative control, was not shifted even at a concentration of 3 µM (Fig. S3A). Specific binding was confirmed by competing MBP-SirA binding to the labeled csrB fragment with 10- to 50-fold excess of unlabeled specific (csrB) and nonspecific (sigD) fragments by EMSA. As shown in figure S3B, only the unlabeled csrB fragment was able to compete with the interaction of MBP-SirA with the labeled fragment of csrB.
We next performed EMSA s with phosphorylated MBP-SirA and labeled DNA fragments containing the regulatory regions of hilD, hilA, hilC, ssrAB and csrC. The DNA fragments used for the EMSAs were those used to construct the corresponding transcriptional fusions that showed SirA-mediated regulation (Figs. 5 and S4). The fragments spanning the hilD, hilA, hilC, ssrAB and sigD regulatory regions were not shifted by phosphorylated MBP-SirA up to a concentration of 3 µM (Figs. 4A–D). Similar results were obtained using non-phosphorylated MBP-SirA (data not shown). At higher concentrations (≥ 5 µM), phosphorylated or nonphosphorylated MBP-SirA bound to all of these fragments, including sigD, suggesting that MBP-SirA binds nonspecifically at these concentrations (data not shown). In contrast, csrC was bound by MBP-SirA, starting at a concentration of 2 µM (Fig. 4E). Specific binding to the csrC regulatory region was also confirmed by competitive EMSAs, as described above for csrB. Only a 10- to 50-fold excess of unlabeled csrC fragment, but not of the unlabeled nonspecific sigD fragment, was able to disrupt the interaction of MBP-SirA with the csrC labeled fragment (Fig. 4F), confirming the specific binding of MBP-SirA to csrC. SirA-mediated regulation of csrB and csrC in the strain used in this study (S. Typhimurium SL1344), was confirmed by analyzing the expression of csrB-cat and csrC-cat transcriptional fusions in the WT and ΔsirA strains (Fig. S4).
Fig. 5.
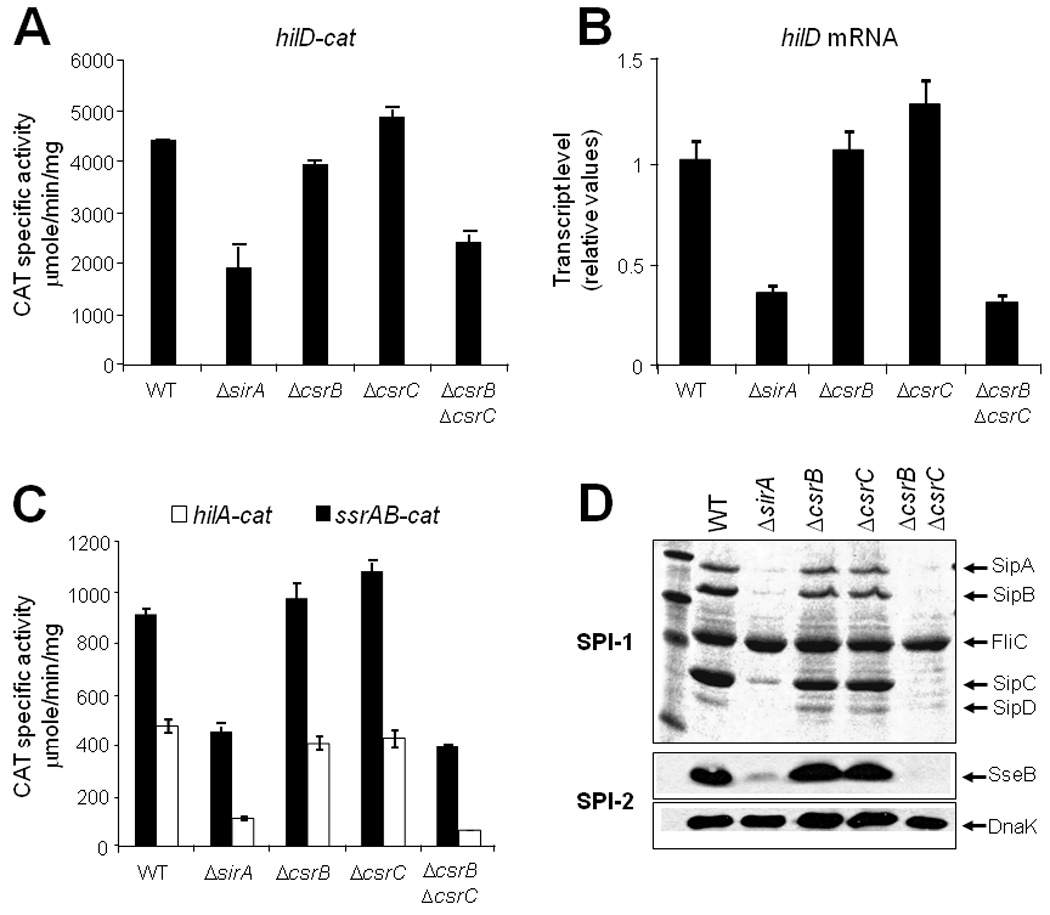
SirA and CsrB/C are similarly required for the expression of hilD and therefore of the SPI-1 and SPI-2 genes. Expression of the transcriptional fusions hilD-cat (A), hilA-cat and ssrAB-cat (C) contained in philD-cat1, philA-cat1 and pssrAB-cat1, respectively, was determined in WT S. Typhimurium and its isogenic ΔsirA, ΔcsrB::kan, ΔcsrC::kan and ΔcsrB ΔcsrC::kan mutant strains. CAT specic activity was determined from samples collected from bacterial cultures grown for 5 h in LB at 37°C (for hilD and hilA fusions) or 10 h (for ssrAB fusion), as described in Experimental procedures. The data are the average of three independent experiments done in duplicate. Bars represent the standard deviations. The hilD mRNA was quantified by q-RT-PCR (B) using total RNA samples from the same strains mentioned above and as described in figure 3. Supernatants and whole cell extracts from culture samples of the same strains grown for 9 h at 37°C, were used to analyze the secretion of the SPI-1-encoded proteins SipA, SipB, SipC and SipD (D, upper) and the expression of the SPI-2 effector protein SseB (D, lower), respectively. FliC is a flagellar protein whose secretion is not SPI-1 dependent. As a loading control, DnaK expression levels were also determined using the same whole cell extracts and a monoclonal anti-DnaK antibody.
Fig. 4.

SirA specifically binds to csrC but not to hilD, hilA, hilC or ssrAB. EMSAs were performed to analyze SirA binding to hilD (A), hilA (B), hilC (C), ssrAB (D) and csrC (E). 32P-5’-end-labeled DNA fragments containing the regulatory region of the respective gene were incubated with increasing concentrations of phosphorylated MBP-SirA (0, 0.5, 1.0, 1.5, 2.0 and 3.0µM). These DNA fragments correspond to those contained in the hilD-cat, hilA-cat, hilC-cat, ssrAB-cat and csrC-cat transcriptional fusions, which were regulated by SirA. As a negative control, a fragment containing the regulatory region of sigD, was included in each DNA-binding reaction. SirA binding to csrC was further analyzed by competitive EMSA (F). The 32P-5’-end-labeled DNA fragment containing the regulatory region of csrC was mixed with 1, 2 or 3 µM of phosphorylated MBP-SirA in the absence or presence of 10- to -50-fold excess of unlabeled specific (csrB) or nonspecific (sigD) competitors. The DNA-protein complexes were resolved in a non-denaturing 6% polyacrylamide gel.
The lack of binding of MBP-SirA to the hilD, hilA and ssrAB regulatory regions is in agreement with the genetic evidence indicating that the SirA/BarA system regulates the expression of hilD, and thus of ssrAB and hilA, indirectly. In contrast, specific binding of MBP-SirA to both the csrB and csrC regulatory regions supports the notion that the SirA-mediated expression of hilD is regulated by the Csr system.
SirA controls the expression of HilD through the CsrB and CsrC RNA molecules
In other bacteria, SirA/BarA orthologs control gene expression in a cascade fashion by directly activating the expression of csrB and csrC, encoding the small untranslated RNA molecules CsrB and CsrC, respectively (Babitzke and Romeo, 2007; Lapouge et al., 2008; Lucchetti-Miganeh et al., 2008).
Therefore, to determine if SirA regulates expression of hilD and the SPI-1 and SPI-2 genes through CsrB and CsrC, single ΔcsrB and ΔcsrC mutants, as well as a ΔcsrB ΔcsrC double mutant, were constructed as described in Experimental Procedures. Expression of the hilD-cat (SPI-1), hilA-cat (SPI-1), ssrAB-cat (SPI-2) and ssaG-cat (SPI-2) transcriptional fusions was determined in these mutants and compared with their expression in WT and ΔsirA strains; additionally, the level of the hilD mRNA was measured by q-RT-PCR. Expression of hilD was not appreciably affected in the single ΔcsrB or ΔcsrC mutants; however, expression in the ΔcsrB ΔcsrC double mutant strain was reduced to the level observed in the ΔsirA mutant (Figs. 5A and B). As expected, expression of the hilA, ssrAB and ssaG fusions was also reduced in the ΔcsrB ΔcsrC double mutant and in the ΔsirA mutant, but not in the single ΔcsrB and ΔcsrC mutants (Fig. 5C and data not shown). Furthermore, the secretion of the SPI-1-encoded proteins SipA, SipB, SipC and SipD (Fig. 5D, upper), as well as the expression of the SPI-2-encoded effector protein SseB (Fig. 5D, lower), were considerably reduced in the ΔcsrB ΔcsrC double mutant and, as expected, in the ΔsirA mutant, but not in the single ΔcsrB and ΔcsrC mutants. In agreement with these results, a previous study reported that the expression of hilA and sipC (a SPI-1 gene), decreased in a ΔcsrB ΔcsrC double mutant, while it was not affected in ΔcsrB and ΔcsrC single mutants (Fortune et al., 2006). It has been demonstrated that the loss of CsrB causes a compensatory increase in CsrC levels and vice versa (Weilbacher et al., 2003; Fortune et al., 2006), which probably explains why the ΔcsrB ΔcsrC double mutant, but not the single mutants, affected expression of the SPI-1 and SPI-2 genes.
To further investigate if SirA acts only through CsrB/C or has an additional CsrB/C-independent regulatory role on the expression of hilD or the SPI-1 and SPI-2 genes, we analyzed the expression of the hilD-cat, hilA-cat and ssaG-cat fusions in the ΔsirA mutant containing plasmids pMPM-K3 (empty vector) or pK3-CsrB1, expressing CsrB from a constitutive promoter. Expression of hilD, hilA and ssaG was restored in the ΔsirA mutant, but not in the ΔhilD mutant, in the presence of pK3-CsrB1 (Figs. 6A–C). In agreement with the notion that SirA controls the expression of hilD and, as a consequence, of the SPI-1 and SPI-2 genes through CsrB/C, a plasmid expressing SirA was not able to restore the secretion of the SPI-1-encoded proteins (Fig. 7, upper), or the expression of the SPI-2-encoded SseB protein (Fig. 7, lower), in a ΔsirA ΔcsrBΔcsrC triple mutant. In contrast, SPI-1-mediated protein secretion and the expression of SseB, were restored by a plasmid expressing HilD in this triple mutant (Fig. 7). Taken together, these results support the model in which SirA indirectly regulates hilD expression, and thus the SPI-1 and SPI-2 genes, by directly activating transcription of csrB and csrC.
Fig. 6.

The presence of CsrB restores the expression of hilD, hilA and ssaG in the absence of SirA but not in the absence of HilD. Expression of transcriptional fusions hilD-cat (A) hilA-cat (B) and ssaG-cat (C) contained in philD-cat1, philA-cat1 and pssaG-cat1, respectively, was determined in WT and isogenic ΔsirA and hilD strains containing vector pMPM-K3 or plasmid pK3-CsrB1, which expresses CsrB from a constitutive promoter. The CAT specific activity was determined from samples collected from bacterial cultures grown for 5 h (for hilD and hilA fusions) or 10 h (for the ssaG fusion) in LB at 37°C. Data are the average of three independent experiments done in duplicate. Error bars represent the standard deviations.
Fig. 7.

SirA, but not HilD, requires the presence of CsrB and CsrC to induce secretion of SPI-1-encoded proteins and expression of the SPI-2 effector protein SseB. Secretion of the SPI-1-encoded proteins (upper) and expression of SseB (lower) was analyzed in WT and sirA::Tn10d ΔcsrB ΔcsrC::kan triple mutant strains, containing vector pBADMycHisA, or plasmids pBAD-HilD1 or pBAD-SirA1, expressing HilD and SirA, respectively, from an arabinose-inducible promoter, as described in figure 5. 0.1% L-arabinose was added (+) to the medium for inducing expression of HilD and SirA.
CsrA represses expression of HilD by binding to the hilD leader transcript
In E. coli, CsrB and CsrC regulate gene expression by binding to and sequestering the RNA binding protein CsrA, thus counteracting the negative posttranscriptional control exerted by CsrA on its target mRNAs (Babitzke and Romeo, 2007; Lucchetti-Miganeh et al., 2008; Timmermans and Van Melderen, 2010). To evaluate the regulatory role of CsrA on the expression of the SPI-1 and SPI-2 genes, we first analyzed the expression of these genes when CsrA is overexpressed. As CsrB and CsrC antagonize CsrA activity, the phenotypes associated with CsrA overexpression was predicted to mimic the phenotypes observed in the ΔcsrB ΔcsrC double mutant strain. The transcriptional activity of the hilD-cat, hilA-cat and ssrAB-cat, as well as of the sirA-cat, csrB-cat and csrC-cat transcriptional fusions was determined in a WT strain containing plasmids pMPM-K3 (empty vector) or pK3-CsrA1. Additionally, the level of the hilD mRNA was measured by q-RT-PCR and the expression of HilA-FLAG and SsrB-FLAG proteins was determined by Western blot, in these same genetic backgrounds. Overproduction of CsrA drastically reduced the expression of hilD, hilA and ssrAB (Figs. 8A, B and E); while the expression of sirA, csrB and csrC was not affected (Figs. 8C and D). Furthermore, the presence of pK3-CsrA1 drastically reduced the expression of HilA-FLAG and SsrB-FLAG, as well as the secretion of the SPI-1-encoded proteins in WT S. Typhimurium (Fig. 8F and data not shown).
Fig. 8.

CsrA negatively regulates expression of hilD, hilA, ssrAB. Expression of transcriptional fusions hilD-cat (A), hilA-cat and ssrAB-cat (B), sirA-cat (C), and csrB-cat and csrC-cat (D) contained in plasmids philD-cat1, philA-cat1, pssrAB-cat1, psirA-cat1, pcsrB-cat1 and pcsrC-cat1, respectively, was determined in WT and ΔcsrA strains carrying vector pMPM-K3 or plasmid pK3-CsrA1, expressing CsrA from a constitutive promoter. CAT specific activity was determined from samples of bacterial cultures grown in LB for 5 h (for hilD, hilA, sirA, csrB and csrC fusions) or 10 h (for the ssrAB fusion) at 37°C. The data are the average of three independent experiments done in duplicate. Error bars represent the standard deviations. The level of the hilD mRNA (E) was quantified by q-RT-PCR in WT S. Typhimurium and its ΔcsrA mutant carrying plasmids pMPM-K3 (vector) or plasmid pK3-CsrA1, as described in figure 3. Expression of HilA and SsrB in WT S. Typhimurium carrying chromosomal FLAG-tagged ssrB or hilA genes and containing plasmids pMPM-T3 (vector) or plasmid pT3-CsrA expressing CsrA from a constitutive promoter, was analyzed by Western blot (F), as described in figure 2.
To further confirm the role of CsrA, we analyzed the expression of SPI-1 and SPI-2 genes in a csrA deletion mutant (Table 1) carrying the hilD-cat, hilA-cat, ssrAB-cat, sirA-cat, csrB-cat and csrC-cat reporter fusions. The level of the hilD mRNA was also measured by q-RT-PCR in this mutant. Consistent with the observed negative regulatory role of csrA overexpression, expression of hilD, hilA and ssrAB significantly increased in the ΔcsrA mutant (Figs. 8A, B and E), whereas the expression of the sirA, csrB and csrC was not affected (Figs. 8C and D).
Table 1.
Bacterial strains used in this study
| Strain | Genotype or description | Reference or source |
|---|---|---|
| S. Typhimurium | ||
| SL1344 | xyl, hisG, rpsL, SmR | (Hoiseth and Stocker, 1981) |
| MJW112 | ΔssrB::kan | M. Worley and F. Heffron |
| VV341 | ΔhilA::kan-339 | (Bajaj et al., 1996) |
| JPTM3 | ΔompR::kan | (Bustamante et al., 2008) |
| JPTM5 | ΔhilD::kan | (Bustamante et al., 2008) |
| JPTM6 | ΔhilC::kan | (Bustamante et al., 2008) |
| JPTM23 | ΔsirA::kan | This study |
| JPTM24 | ΔhilA | This study |
| JPTM25 | ΔhilD | This study |
| JPTM26 | ΔhilC | This study |
| JPTM27 | ΔsirA | This study |
| JPTM28 | ΔompR | This study |
| JPTM29 | ΔssrB | This study |
| JPTM30 | ssrB::3XFLAG-kan | This study |
| JPTM31 | ssrB::3XFLAG-kan ΔhilA | This study |
| JPTM32 | ssrB::3XFLAG-kan ΔhilD | This study |
| JPTM33 | ssrB::3XFLAG-kan ΔhilC | This study |
| JPTM34 | ssrB::3XFLAG-kan ΔsirA | This study |
| JPTM35 | ssrB::3XFLAG-kan ΔompR | This study |
| JPTM36 | hilA::3XFLAG-kan | (Bustamante et al., 2008) |
| JPTM37 | hilA::3XFLAG-kan ΔhilD | This study |
| JPTM38 | hilA::3XFLAG-kan ΔsirA | This study |
| JPTM39 | ΔbarA::kan | This study |
| JPTM40 | ΔcsrB::kan | This study |
| JPTM41 | ΔcsrC::kan | This study |
| JPTM42 | ΔcsrB | This study |
| JPTM43 | ΔcsrB ΔcsrC::kan | This study |
| JPTM44 | ΔcsrA::kan | This study |
| CJO35 | sirA::Tn10d | (Johnston et al., 1996) |
| JPTM45 | sirA::Tn10d ΔcsrB ΔcsrC::kan | This study |
| E. coli | ||
| MC4100 | Cloning strain, K12 derivative | (Casadaban, 1976) |
| BL21/pLys | Strain for expression of | |
| recombinant proteins | Invitrogen | |
| DH10β | Laboratory strain | Invitrogen |
| JPMC46 | ΔuvrY::kan | This study |
| JPMC47 | ΔuvrY:: | This study |
In agreement with these results, it was previously shown that overproduction of CsrA reduced the expression of hilD and hilA (Altier et al., 2000a). This study also reported the generation of a csrA mutant in S. Typhimurium strain 14028s, which surprisingly showed a reduction in the expression of SPI-1 genes; however, this mutant presented a severe growth defect, which may have affected the growth-phase-dependent induction of the SPI-1 genes. Our ΔcsrA mutant showed a less severe growth defect, which was restored when CsrA was expressed in trans (Fig. S5). It remains to be seen if the different effects on growth and on the expression of SPI-1 and SPI-2 genes caused by the absence of CsrA could also be attributed to distinct features of the strains used, SL1344 (this study) and 14028s (Altier et al., 2000a), or to random secondary suppressor mutations acquired as a consequence of eliminating a pleiotropic regulatory protein.
The results described above, indicating that SirA/BarA and CsrB/C activate expression of hilA and ssrAB through HilD, also suggested that CsrA could directly repress hilD expression. To test this possibility, we performed EMSAs to determine whether CsrA binds to hilD, hilA, ssrAB or csrB transcripts. The addition of 1 µM CsrA led to reduced mobility of the hilD, ssrAB and csrB transcripts, indicating that CsrA bound to these RNAs (Fig. 9A). In contrast, CsrA did not bind to hilA RNA (Fig. 9A). We next performed experiments to determine the affinity of CsrA for hilD, ssrAB and csrB mRNAs. CsrA bound to the hilD transcript as a distinct band (Fig. 9B). A nonlinear least-squares analysis of these data yielded an apparent Kd value of 5 nM CsrA for hilD mRNA. The specificity of CsrA-hilD RNA interaction was investigated by performing competition experiments with specific (hilD) and non-specific (E. coli phoB) unlabeled RNA competitors. Unlabeled hilD was an effective competitor, whereas phoB did not compete with the CsrA-hilD RNA interaction (Fig. 9B). These results establish that CsrA binds specifically to the hilD transcript. Similar binding experiments were performed with the ssrAB and csrB transcripts. The affinity of CsrA for ssrAB mRNA (Kd = 340 nM) was much lower than for hilD. In addition, while unlabeled ssrAB RNA was able to compete with the labeled transcript for CsrA binding, it did so poorly (Fig. 9C). As expected, CsrA bound specifically to csrB mRNA with high affinity (Kd = 20 pM). Moreover, multiple shifted species were observed (Fig. 9D). While the stoichiometry of the shifted complexes was not examined, the first shifted species likely contains one CsrA dimer bound per transcript, with the slower migrating species containing multiple CsrA molecules bound to each transcript.
Fig. 9.

CsrA interaction with the hilD mRNA. (A) EMSAs of CsrA with hilA, hilD, ssrAB or csrB RNA. 5'-end-labeled RNA (250 pM) of each gene was incubated in the absence (−) or presence (+) of 1 µM CsrA. (B) Determination of the apparent equilibrium binding constant (Kd) of CsrA-hilD RNA interaction. 5'-end-labeled hilD RNA (100 pM) was incubated with the concentration of CsrA shown at the bottom of each lane. Positions of bound (B) and free (F) RNA are shown. For the competition assay, labeled hilD RNA (100 pM) was incubated with CsrA in the absence or presence of specific (hilD) or non-specific (phoB) competitor RNA. (C) Determination of the apparent Kd of CsrA-ssrAB RNA interaction. 5'-end-labeled ssrAB RNA (100 pM) was incubated with the concentration of CsrA shown at the bottom of each lane. Positions of bound (B) and free (F) RNA are shown. For the competition assay, labeled ssrAB RNA (100 pM) was incubated with CsrA in the absence or presence of specific (ssrAB) or non-specific (phoB) competitor RNA. (D) Determination of the apparent Kd of CsrA-csrB RNA interaction. 5'-end-labeled csrB RNA (10 pM) was incubated with the concentration of CsrA shown at the bottom of each lane. Positions of bound (B) and free (F) RNA are shown. For the competition assay, labeled csrB RNA (10 pM) was incubated with CsrA in the absence or presence of specific (ssrAB) or non-specific (phoB) competitor RNA. (E) CsrA-hilD RNA footprint analysis. 5'-end-labeled RNA was treated with RNase T1 in the presence of the concentration of CsrA shown at the top of each lane. Partial alkaline hydrolysis (OH) and RNase T1 digestion (T1) ladders, as well as a control lane in the absence of RNase T1 treatment (C), are shown. RNase T1 ladders were generated under denaturing conditions so that every G residue in the transcript could be visualized. Positions of the CsrA binding sites (BS), Shine-Dalgarno (SD) sequence and the translation initiation codon (Met) are indicated in the figure and in the hilD sequence at the bottom of figure 9E. Numbering is with respect to the start of transcription. (F) Northern blot analysis of hilD mRNA stability. WT and ΔcsrA strains were grown in LB to mid-exponential phase of growth (OD600 ~0.6), at which time transcription initiation was blocked by the addition of rifampicin. Samples were harvested at the indicated times, total RNA was prepared, and a 20 µg aliquot from each sample was analyzed by Northern blotting as described in Experimental procedures. The left panel shows the autoradiogram of the blot with the specific hilD probe, and the ethidium bromide stained rRNA bands, showed as a loading control. The right panel shows the semi-logarithmic plot of the hilD mRNA decay in the two strains.
Footprint experiments were then performed to identify the CsrA binding site(s) in the hilD and ssrAB transcripts using RNase T1 as a probe (cleaves following single-stranded G residues). CsrA protected five G residues in the hilD transcript that overlapped the hilD SD sequence and translation initiation codon (Fig. 9E). As both of these regions contain the highly conserved GGA motif (Fig. 9E), it is apparent that CsrA binds to two sites in this transcript. These results indicate that CsrA represses hilD expression, presumably by blocking 30S ribosomal subunit binding. CsrA also protected several G residues in the ssrAB transcript that overlapped its SD sequence (data not shown). Thus, it appears that CsrA is also capable of directly repressing ssrAB translation by binding to a single site in the ssrAB transcript, although the reduced affinity of CsrA for this RNA suggests that repression would be less extensive than for hilD.
By blocking ribosome binding and translation, CsrA-mediated translational repression often leads to accelerated degradation of target transcripts (Babitzke and Romeo, 1997; Lucchetti-Miganeh et al., 2008, Timmermans and Van Melderen, 2010). Therefore, we tested whether bound CsrA accelerated hilD mRNA decay. The half-life of hilD mRNA was determined in WT and csrA strains. To facilitate detection of hilD mRNA, the two strains were transformed with plasmid pT3-HilD1, which carries the hilD gene under the control of a constitutive promoter, to increase the cellular concentration of the hilD transcript. These strains were grown to mid-exponential phase in LB; then, transcription was inhibited by adding rifampicin. Culture samples were collected at different time-points after addition of the drug for RNA extraction and analysis by Northern blotting. The hilD mRNA was detected as two unique bands, which showed similar intensity in the WT and ΔcsrA mutant strains immediately after the treatment with rifampicin (Fig. 9F). However, the hilD mRNA decayed faster in the WT strain (0.79 ± 0.05 min half-life) compared to the ΔcsrA mutant (1.54 ± 0.05 min half-life) (Fig. 9F).
Taken together, it can be concluded that CsrA-mediated translational repression leads to more rapid degradation of hilD mRNA. In addition, by reducing HilD synthesis, CsrA would also affect HilD positive autoregulation (Olekhnovich and Kadner, 2002; Ellermeier et al., 2005), thus reducing hilD transcription. Furthermore, our results suggest that CsrA indirectly represses hilA and ssrAB transcription under growth conditions that favor HilD-dependent activation of the SPI-1 and SPI-2 regulons.
Discussion
In recent years it has become more evident that the mechanisms regulating virulence gene expression in Salmonella are more complex than previously thought. A myriad of regulatory factors and environmental cues have been shown to be part of the molecular events leading to the appropriate spatiotemporal expression of the genes that integrate the SPI-1 and SPI-2 regulons (Altier, 2005; Jones, 2005; Ellermeier and Slauch, 2007; Fass and Groisman, 2009). In this regard, we recently provided evidence showing that HilD is a key regulatory protein that establishes transcriptional cross-talk between the SPI-1 and SPI-2 regulons, by sequentially activating expression of the SPI-1 and SPI-2 genes when Salmonella grows in LB medium (Bustamante et al., 2008). HilD directly binds to the regulatory regions of hilA and the ssrAB operon, thereby interfering with H-NS-mediated repression of their promoters. HilA and the SsrA/B two component system, in turn, activate the SPI-1 and SPI-2 regulons, respectively (Schechter et al., 2003; Olekhnovich and Kadner, 2006; Bustamante et al., 2008). Interestingly, HilD is not required for expression of the ssrAB operon when Salmonella grows in minimal media containing low concentrations of magnesium and phosphate (Bustamante et al., 2008). Under these growth conditions ssrAB expression instead depends on other regulators such as OmpR, PhoP and SlyA (Lee et al., 2000; Feng et al., 2003; 2004; Bijlsma and Groisman, 2005; Linehan et al., 2005; Fass and Groisman, 2009).
Based on previous knowledge (Johnston et al., 1996; Ahmer et al., 1999; Goodier and Ahmer, 2001; Altier et al., 2000a; 2000b; Lawhon et al., 2003; Teplitski et al., 2003; Fortune et al., 2006; Teplitski et al., 2006) and new information generated from this study, we present the integration of the regulatory cascade that controls expression of the SPI-1 and SPI-2 virulence regulons via the SirA/BarA and Csr global regulatory systems and the Salmonella-specific regulator HilD.
Firstly, we demonstrated that SirA positively controls expression of the SPI-1 and SPI-2 genes through HilD. A previous study reported that SirA-His6 bound to a region located downstream of the hilA promoter and the regulatory region of hilC (Teplitski et al., 2003), suggesting a direct SirA regulation of these SPI-1 genes. In contrast, we found that MBP-SirA specifically bound to the regulatory regions of csrB and csrC, but not to the regulatory regions of hilA and hilC containing the fragments used by Teplitski et al., 2003 (Figs. 4 and S3). Differences in these results could be attributed to different DNA binding specificities between SirA-His6 and MBP-SirA, or other technical differences; however, the genetic evidence presented here supports the notion that SirA does not directly activate hilA. Briefly, we demonstrated that SirA cannot activate expression of SsrB and HilA proteins in the absence of HilD (Figs. 2, S1 and S2), in agreement with results from a previous study (Ellermeier et al., 2005). In contrast, HilD activated expression of ssrAB and hilA in the absence of SirA, indicating that HilD does not require the presence of SirA to directly activate expression of the SPI-1 and SPI-2 regulatory genes. In agreement with our results, mathematical models considering that SirA regulates SPI-1 genes through HilD are able to predict results comparable to experimental data. However, they fail to predict the level of participation of intermediate components of the SPI-1 regulatory cascade, when considering a direct regulation of SirA on hilA or hilC (Ganesh et al., 2009). Furthermore, CsrB was able to restore the expression of the SPI-1 and SPI-2 genes in the absence of SirA, but not in the absence of HilD (Fig. 6), indicating that SirA acts through CsrB to regulate these genes, and that HilD is downstream of SirA and CsrB in the regulatory cascade controlling them. This is consistent with previous studies indicating that SirA orthologs regulate gene expression through the corresponding CsrB and CsrC orthologs (Babitzke and Romeo, 2007; Lucchetti-Miganeh et al., 2008; Brencic et al., 2009; Timmermans and Van Melderen, 2010). In this regard, we demonstrated that SirA specifically binds to the csrC regulatory region (Fig. 4), an interaction previously demonstrated only for csrB (Teplitski et al., 2003; Teplitski et al., 2006). Moreover, the demonstration that CsrA binds to the SD sequence and translation initiation codon of hilD mRNA, but not to hilA mRNA (Fig. 9), strongly correlates with the genetic and biochemical data indicating that regulation of the SPI-1 and SPI-2 regulons require direct activation of csrB and csrC by SirA, which in turn overcomes CsrA-dependent repression of HilD synthesis. HilD is then responsible for the direct activation of hilA and ssrAB and, indirectly, of the SPI-1 and SPI-2 regulons.
Additionally, we found that CsrA binds to ssrAB mRNA, but with a much lower affinity than to hilD mRNA (Kd = 340 nM vs 5 nM) (Fig. 9). Whether this weak CsrA-ssrAB RNA interaction is capable of regulating ssrAB expression remains to be determined. However, as expression of ssrAB requires HilD when Salmonella grows in LB medium, CsrA-mediated regulation of HilD synthesis appears to be the primary mechanism by which CsrA controls ssrAB expression. Moreover, SPI-2 expression was not significantly affected in the ΔsirA mutant growing in N-MM (Fig. 1), which favors HilD-independent activation of ssrAB.
A model for the regulatory cascade that controls expression of the SPI-1 and SPI-2 regulons, which summarizes our results and those from other groups, is shown in Figure 10. This regulatory cascade is under the influence of catabolite repression, through the positive control of SirA expression exerted by the cAMP receptor protein CRP (Teplitski et al., 2006). The regulatory cascade can also be activated by short-chain fatty acids, such as acetate, formate and propionate, which stimulate the sensor kinase activity of BarA, and thus phosphorylation of SirA orthologs (Chavez et al., 2010). Alternatively, acetyl-phosphate seems to directly phosphorylate the response regulator SirA or the sensor kinase BarA (Altier et al., 2000b; Lawhon et al., 2002; Teplitski et al., 2003; 2006). Acetyl-phosphate can be generated from acetate, which is commonly found in the distal ileum of the mammalian intestines. Within the bacteria, acetate is converted to acetyl-phosphate by acetate kinase (ackA), and, presumably, can also be produced endogenously from acetyl-CoA by phosphotransacetylase (pta) (Lawhon et al., 2002; Teplitski et al., 2006). Phosphorylated SirA activates expression of CsrB and CsrC by binding to the csrB and csrC regulatory regions. These RNA molecules, in turn, bind to and sequester CsrA (Babitzke and Romeo, 2007; Lucchetti-Miganeh et al., 2008; Timmermans and Van Melderen, 2010), thus counteracting CsrA-mediated translational repression of hilD. The appropriate concentration and activity of HilD is maintained by additional negative and positive regulatory mechanisms, including degradation of HilD mediated by the Lon protease (Boddicker and Jones, 2004; Takaya et al., 2005), HilD inactivation by direct interaction with HilE (Baxter et al., 2003), positive auto-regulation of HilD, as well as by a feed forward positive regulatory loop involving the HilD, HilC and RtsA AraC-like regulators (Olekhnovich and Kadner, 2002; Ellermeier et al., 2005). The regulatory cascade diverges when HilD, in a growth phase-dependent manner, activates expression of hilA and the ssrAB operon, by counteracting the repression exerted by H-NS on their promoters (Bustamante et al., 2008, Schechter et al., 2003; Olekhnovich and Kadner, 2006). Finally, HilA and the SsrA/B system positively control expression of the SPI-1 and SPI-2 regulons, respectively (Altier, 2005; Jones, 2005; Ellermeier and Slauch, 2007; Fass and Groisman, 2009; Tomljenovic-Berube et al., 2010).
Fig. 10.

Schematic representation of the SirA-CsrB/C-CsrA-HilD regulatory cascade controlling the expression of SPI-1 and SPI-2 genes in S. Typhimurium. See text for details.
The SirA-HilD divergent regulatory cascade could be important to coordinate the events leading to intestinal infection by Salmonella. Different lines of evidence support the notion that both the SPI-1 and SPI-2 genes are required for the production of enteritis in animal models (Bispham et al., 2001; Santos et al., 2003; Brown et al., 2005; Coburn et al., 2005; Coombes et al., 2005; Hapfelmeier et al., 2005; Jones et al., 2007; Haraga et al., 2008). In addition, it was shown that SPI-2 genes are expressed in the intestine prior to epithelial cell invasion (Brown et al., 2005). Moreover, previous studies indicate that SirA, CsrB/CsrC, CsrA and HilD are required by Salmonella to display phenotypic features involved in intestinal infection (Jones, 2005; Altier, 2005; Lucchetti-Miganeh et al., 2008). Once Salmonella is inside host cells or growing in N-MM, SPI-2 genes, which are essential for the systemic phase of the infection (Cirillo et al., 1998; Hensel et al., 1998; Hansen-Wester and Hensel, 2001), are expressed by a SirA-Csr-HilD-independent mechanism involving other regulators such as OmpR, PhoP and SlyA (Fass and Groisman, 2009); whereas expression of the SPI-1 genes is rapidly downregulated following SPI-1-mediated invasion or when growing in N-MM (Eriksson et al., 2003; Drecktrah et al., 2006; Bustamante et al., 2008). In this regard, it has also been shown that SPI-1 is not required for Salmonella replication inside host cells (Drecktrah et al., 2006). These results suggest that the SirA-Csr-HilD regulatory cascade is not functional during the systemic phase of the infection. Therefore, although SirA has been shown to be required for Salmonella intracellular replication (Chan et al., 2005), this does not seem to be related to the SirA-mediated regulation of the SPI-2 genes intracellularly, but instead of other target genes, in agreement with its global regulatory nature observed in Salmonella and other bacteria (Ahmer et al., 1999, Goodier and Ahmer, 2001).
Our results together with those from other groups, illustrate how multiple global and Salmonella-specific regulators have been integrated to form a complex regulatory circuitry that allow the appropriate spatiotemporal expression of genes that have been acquired by horizontal transfer events. In addition, they further expand our understanding about the molecular mechanisms coordinating the expression of the SPI-1 and SPI-2 genes.
Experimental procedures
Bacterial strains and growth conditions
Bacterial strains, plasmids and primers used in this work are listed in Tables 1, 2 and S1, respectively.
Table 2.
Plasmids used in this study
| Plasmid | Description | Reference or source |
|---|---|---|
| pKK232-8 | pBR322 derivative containing a promoterless chloramphenicol acetyltransferase (cat) gene, ApR | (Brosius, 1984) |
| philA-cat1 | pKK232-8 derivative containing a hilA-cat transcriptional fusion from nucleotides −410 to +446 | (Bustamante et al., 2008) |
| philD-cat1 | pKK232-8 derivative containing a hilD-cat transcriptional fusion from nucleotides −364 to +88 | (Bustamante et al., 2008) |
| pssaG-cat1 | pKK232-8 derivative containing a ssaG-cat transcriptional fusion from nucleotides −232 to +361 | (Bustamante et al., 2008) |
| pssrAB-cat1 | pKK232-8 derivative containing a ssrB-cat transcriptional fusion from nucleotides −303 to +3054 | (Bustamante et al., 2008) |
| psirA-cat1 | pKK232-8 derivative containing a sirA-cat transcriptional fusion from nucleotides −563 to +98 | This study |
| philC-cat1 | pKK232-8 derivative containing a hilC-cat transcriptional fusion from nucleotides −373 to +79 | This study |
| prtsA-cat1 | pKK232-8 derivative containing a rtsA-cat transcriptional fusion from nucleotides −465 to +97 | This study |
| pcsrB-cat1 | pKK232-8 derivative containing a csrB-cat transcriptional fusion from nucleotides −372 to +18 | This study |
| pcsrC-cat1 | pKK232-8 derivative containing a csrC-cat transcriptional fusion from nucleotides −351 to +63 | This study |
| pKD46 | pINT-ts derivative containing red recombinase system under an arabinose-inducible promoter, ApR | (Datsenko and Wanner, 2000) |
| pKD4 | pANTsγ derivative template plasmid containing the kanamycin cassette for λ Red recombination, ApR | (Datsenko and Wanner, 2000) |
| pCP20 | Plasmid expression FLP recombinase from a temperature-inducible promoter, ApR | (Datsenko and Wanner, 2000) |
| pSUB11 | pGP704 derivative template plasmid for FLAG epitope tagging | (Uzzau et al., 2001) |
| pBADMycHisA | Expression vector for constructing C-terminal MycHis fusions, ara promoter, ApR | Invitrogen |
| pBADMycHisC | Expression vector for constructing C-terminal MycHis fusions, ara promoter, ApR | Invitrogen |
| pBAD-HilD1 | pBADMycHis derivative expressing HilD-MycHis from the ara promoter | This study |
| pBAD-SirA1 | pBADMycHis derivative expressing SirA-MycHis from the ara promoter | This study |
| pMPM-K3 | p15A derivative low-copy-number cloning vector, lac promoter, KanR | (Mayer, 1995) |
| pK3-SirA1 | pMPM-K3 derivative expressing SirA from the lac promoter | This study |
| pK3-CsrB1 | pMPM-K3 derivative expressing CsrB from the lac promoter | This study |
| pK3-CsrA1 | pMPM-K3 derivative expressing CsrA from the lac promoter | This study |
| pMPM-T3 | p15A derivative low-copy-number cloning vector, lac promoter, TcR | (Mayer, 1995) |
| pT3-HilD1 | pMPM-T3 derivative expressing HilD from the lac promoter | (Bustamante et al., 2008) |
| pT3-CsrA1 | pMPM-T3 derivative expressing CsrA from the lac promoter | This study |
| pMAL-c2X | Vector for constructing MBP fusions, ApR | New England Biolabs |
| pMAL-SirA1 | pMAL-c2X derivative expressing MBP-SirA | This study |
| pCSB12 | pET21a+ derivative expressing CsrA-H6 | (Dubey et al.,2005) |
The coordinates for the cat fusions are indicated with respect to the transcriptional start site for each gene.
Bacterial cultures were grown at 37°C in LB medium, containing 1% tryptone, 0.5% yeast agar and 1% NaCl, at pH 7.5; or in N-minimal medium (N-MM) containing 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 1 mM KH2PO4, 100 mM Tris-HCl (pH 7.5), 10 µM MgCl2, 0.5% glycerol and 0.1% casamino acids. When necessary, media were supplemented with the following antibiotics: ampicillin (200 µg/ml), streptomycin (100 µg/ml), tetracycline (10 µg/ml) or kanamycin (20 µg/ml). Bacterial cultures grown overnight in LB medium were concentrated and resuspended in fresh LB or N-MM to an OD600 of 1. Then, 250-ml flasks containing 50 ml of LB or N-MM were inoculated with 1 ml of the bacterial suspensions and incubated at 37°C in a shaking water bath at 200 rpm (Gyromax 902; Amerex Instruments). Culture samples for chloramphenicol acetyl transferase (CAT) activity determination or for western blot analysis, were taken after 5 h or 10 h of growth in LB under the conditions described above. These time points were previously optimized for the analysis of SPI-1 and SPI-2 regulation, respectively (Bustamante et al., 2008).
CAT assays
The CAT assays and protein quantification to calculate CAT-specific activities were performed as described previously (Puente et al., 1996).
Construction of mutant strains and strains expressing FLAG-tagged proteins
Non-polar gene-deletion mutant strains (Table 1) were generated by the Red recombinase system, as reported previously (Datsenko and Wanner, 2000). The genes sirA, barA, csrB, csrC or csrA were replaced with a selectable kanamycin resistance cassette in the S. Typhimurium SL1344 strain generating strains ΔsirA::kan (JPTM23), ΔbarA::kan (JPTM39), ΔcsrB::kan (JPTM40), ΔcsrC::kan (JPTM41) and ΔcsrA::kan (JPTM44), respectively. The gene uvrY was replaced with the kanamycin resistance cassette in E. coli MC4100 to generate the ΔuvrY::kan (JPMC46) strain. When required, the kanamycin resistance cassette was excised from the respective mutant strain, by using helper plasmid pCP20, expressing the FLP recombinase, as described previously (Datsenko and Wanner, 2000), generating S. Typhimurium mutant strains ΔhilA (JPTM24), ΔhilD (JPTM25), ΔhilC (JPTM26), ΔsirA (JPTM27), ΔompR (JPTM28), ΔssrB (JPTM29) and ΔcsrB (JPTM42) and E. coli ΔuvrY (JPMC47). The ΔcsrB ΔcsrC::kan double mutant (JPTM43) was generated by introducing the ΔcsrC::kan deletion into the ΔcsrB mutant (JPTM42) by P22 transduction. The sirA::Tn10d ΔcsrB ΔcsrC::kan triple mutant (JPTM45) was generated by introducing the sirA::Tn10d deletion into the ΔcsrB ΔcsrC::kan double mutant (JPTM43) by P22 transduction.
The chromosomal ssrB gene was FLAG-tagged in S. Typhimurium SL1344, using a modification of the λRed recombinase system for gene replacement, as described previously (Uzzau et al., 2001), generating strain JPTM30. The ssrB::3XFLAG-kan allele from strain JPTM30 was transferred to strains ΔhilA (JPTM24), ΔhilD (JPTM25), ΔhilC (JPTM26), ΔsirA (JPTM27) and ΔompR (JPTM28) by P22 transduction, generating strains JPTM31, JPTM32, JPTM33, JPTM34 and JPTM35, respectively. The hilA::3XFLAG-kan allele from strain JPTM36 was transferred to strains ΔhilD (JPTM25) and ΔsirA (JPTM27) by P22 transduction, generating strains JPTM37 and JPTM38, respectively. All mutant strains were verified by PCR amplification and sequencing.
SDS/PAGE and Western blot assays
Whole-cell extracts were prepared from bacterial samples collected at the indicated time points from LB cultures. Ten micrograms of each extract were subjected to electrophoresis in SDS-12% polyacrylamide gels, and then transferred to 0.45-µm pore size nitrocellulose membranes (Bio-Rad), using a semidry transfer apparatus (Bio-Rad). The membranes containing the transferred proteins were blocked in 5% nonfat milk for 1 h. Immunoblots were performed with anti-FLAG M2 (Sigma) or anti-DnaK (StressGen) monoclonal antibodies, or anti-SseB polyclonal antibody (Coombes et al., 2004), at 1:1,000, 1:20,000 and 1:2,000 dilutions, respectively. Horseradish peroxidase-conjugated anti-mouse or anti-rabbit (Pierce), at a dilution of 1:10,000, were used as the secondary antibodies. Bands on the blotted membranes were developed by incubation with the Western Lightning Chemiluminescence Reagent Plus (Perkin-Elmer) and exposed to KodaK X-Omat films.
Protein secretion analysis
S. Typhimurium strains were grown for 9 h in LB. Then, 1.5 ml samples were harvested from each culture and concentrated by centrifugation for 5 min at 16,000 X g. Proteins contained in culture supernatants were precipitated by the addition of 10% (v/v) trichloroacetic acid and overnight incubation at 4°C. Precipitated proteins were concentrated by centrifugation at 16,000 X g for 30 min at 4°C. Pellets were resuspended in SDS-PAGE loading buffer containing 10% (v/v) saturated Tris base. Samples were subjected to SDS-PAGE analysis, using 12% polyacrylamide gels and stained with Coomassie brilliant blue R-250.
DNA electrophoretic mobility shift assays
The regulatory regions of hilD, hilC, csrB, csrC, hilA, ssrAB and sigD were amplified by PCR using primer pairs hilDFBamHI/hilDRHindIII, hilCFSmaI/hilCRSalI, csrB-Fw1HindIII/csrB-Rev1, csrC-BFw1/csrC-HRv1, hilA1FBamHI/hilA2RHindIII, ssaBFBglII/ssrBRS6E (Bustamante et al., 2008) and sigDBH1F/sigDH3R, respectively, and S. Typhimurium SL1344 chromosomal DNA as template. PCR products were labeled during the reaction using 32P-5’-end-labeled primers and then purified using the QIAquick PCR purification kit (Qiagen). DNA binding reactions were performed in binding buffer (10 mM Tris-HCl [pH 8], 50 mM KCl, 1 mM DTT, 0.5 mM EDTA, 5% glycerol and 10 µg ml−1 BSA) by mixing ≈100 ng of each PCR product with ≈100 ng of the sigD fragment (negative control) and increasing concentrations of purified phosphorylated or nonphosphorylated MBP-SirA, in a total volume of 20 µl. Phosphorylated MBP-SirA was generated by incubation it with 100 mM acetyl phosphate (Sigma) for 180 min at room temperature, as previously reported (Kenney et al., 1995). Protein-DNA binding reactions were incubated at room temperature for 20 min and then electrophoretically separated in 6% nondenaturing polyacrylamide gels in 0.5X Tris-borate-EDTA buffer at room temperature. The unlabeled DNA fragments were stained with ethidium bromide and visualized with an Alpha-Imager UV transilluminator (Alpha Innotech Corp.). The 32P-5’-end-labeled DNA fragments were detected by exposing to KodaK X-Omat films.
Quantitative real-time RT-PCR (q-RT-PCR) assays
Total RNA of S. Typhimurium SL1344 and its isogenic mutants was purified from samples of cultures grown in LB medium at 37°C for 5 h, using the RNeasy Mini Kit (Qiagen). Chromosomal DNA was removed by incubating 2 µg of RNA from each strain with DNase I (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized in a reaction containing 0.5 µg of each DNase-treated-RNA and 5 pmol of primers hilD-RT-R (for hilD) and dnaK-RT-R (for dnaK), using the Revert Aid H Minus First Strand cDNA Synthesis kit (Fermentas). The obtained cDNA was used as template for Real-Time PCR assays, with 5 pmol of primer pairs for hilD or dnaK (hilD-RT-F/hilD-RT-R or dnaK-RT-F/dnaK-RT-R, respectively) and the SYBR Green PCR Master Mix (Perkin-Elmer/Applied Bio-systems). Real-Time PCR reactions were performed with the ABI Prism 7000 Sequence Detection System (Applied Bio-systems) set in the standard run mode, and data collected using the Rotor Gene 6000 series software 1.7. Reaction conditions were 10 min at 95°C, and 40 cycles at 95°C for 15 s and 60°C for 60 s. The level of dnaK mRNA was used as an internal control to normalize the results obtained for the hilD mRNA. The quantification technique used to analyze data was the 2 −Δ,ΔCT method (Livak and Schmittgen, 2001). All Real-Time PCR reactions were performed in triplicate and were repeated using RNA purified from three different bacterial cultures.
RNA electrophoretic mobility shift assay
Electrophoretic mobility shift assays followed published procedures (Baker et al., 2007; Yakhnin et al., 2007). His-tagged CsrA (CsrA-H6) from E. coli was purified as described previously (Dubey et al., 2005). Note that the amino acid sequence of CsrA from S. enterica and E. coli are identical. RNA was synthesized in vitro using the RNA Maxx Transcription kit (Stratagene). PCR fragments used as templates in transcription reactions contained a T7 promoter and hilA, hilD, ssrAB or csrB sequences extending from +1 to +365, +1 to +89, +1 to +183 or +1 to +371 relative to the start of transcription, respectively. Gel-purified RNA was 5′-end labeled with [γ-32P]-ATP. RNA suspended in TE buffer was heated to 90 °C for 1 min followed by slow cooling to room temperature. Binding reactions (10 µl) contained 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 100 mM KCl, 200 ng/µl yeast RNA, 0.2 mg/ml BSA, 7.5% glycerol, 20 mM DTT, 10–500 pM RNA, CsrA-H6 (various concentrations), and 0.1 mg/ml xylene cyanol. Competition assay mixtures also contained unlabeled competitor RNA. Reaction mixtures were incubated for 30 min at 37 °C to allow CsrA-RNA complex formation. Samples were then fractionated through native 10% polyacrylamide gels using 0.5X TBE as the gel running buffer. Radioactive bands were visualized with a phosphorimager and quantified using ImageQuant 5.2 software. Apparent equilibrium binding constants (Kd) of CsrA-RNA interaction were calculated as described previously (Yakhnin et al., 2000).
RNA footprint assays
RNA footpringt assays followed published procedures (Baker et al., 2007; Yakhnin et al., 2007). Binding reactions (10 µl) containing various concentrations of CsrA-H6 and 5 nM hilD or ssrAB RNA were otherwise identical to those described for the electrophoretic mobility shift assays. After the initial binding reaction, 0.03 U RNase T1 (Fermentas) was added to the reaction mixtures, and incubation was continued for 15 min at 37 °C. Reactions were terminated by the addition of 10 µl of gel loading buffer (0.37% EDTA, pH 8.0, 0.3% bromophenol blue and xylene cyanol in 95% formamide) and placed on ice. Partial alkaline hydrolysis and RNase T1 digestion ladders of each transcript were prepared as described previously (Bevilacqua and Bevilacqua, 1998). Samples were fractionated through standard 6% polyacrylamide sequencing gels. Radioactive bands were visualized with a phosphorimager.
RNA extraction, Northern blot and half-life determination
To determine hilD mRNA half-lives, S. Typhimurium WT and ΔcsrA mutant strains containing plasmid pT3-HilD1 were grown in LB to an OD600 ~0.6, at which time 200 µg/ml rifampicin was added to the cultures to prevent transcription initiation. Total RNA was purified from samples taken at 0, 0.5, 1, 2 and 4 min after rifampicin addition by the hot phenol extraction method, as described previously (Georgellis et al., 1992).
Northern blot analysis was performed by fractionation of the purified RNA samples (20 µg) on a 1% agarose-formaldehyde gel, which were then transferred to a nitrocellulose membrane by capillary transfer. Membranes were crosslinked using a crosslinking device (Stratalinker, Stratagene), and prehybridized for 3 h at 42°C in a buffer containing 5 X Denhardt’s solution, 5X SSC, 0.2% SDS, 50% formamide, and 250µg of sheared salmon sperm DNA per ml. Subsequently, a radio-labeled hilD specific DNA probe, denatured at 90° C for 5 min, was added to the prehybridization buffer and the membranes were incubated at 42° C overnight. The hilD specific probe was obtained by digesting plasmid pT3-HilD1 with SalI and BamHI, separation of the fragments on agarose gels and purification of the hilD specific band by using the Qiagen Agarose Purification Kit. Probe labeling was performed by using [α-32P]dCTP and the Rediprime II Kit (Invitrogen), according to the manufacturer’s instructions. Membranes were washed twice with 50 ml of SSC 2X and SDS 0.1% at 42° C, and twice with SSC 0.2X and SDS 0.1% at 42° C. Images were obtained using the phosphoimager screens and analyzed using the Typhoon image scanner (Amersham). Determination of the hilD mRNA half-life was performed as previously described (von Gabain et al., 1983).
Construction of plasmids and Expression and purification of MBP-SirA sections are described in Experimental procedures in Supporting Information.
Supplementary Material
Acknowledgments
We thank A. Vázquez and F.J. Santana for technical assistance, E. Calva for critical reading of the manuscript, S. Miller and F. Heffron for providing Salmonella strains, and B.B. Finlay for providing the anti-SseB polyclonal antibody. This work was supported by grants from Dirección General de Asuntos del Personal Académico (DGAPA) (IN227306-3 and IN210309-3 to V.H.B., IN224107-3 to J.L.P., and IN219709-3 to D.G), from Consejo Nacional de Ciencia y Tecnología (CONACYT) (83277 to V.H.B. and 6079 to J.L.P.), and from the National Institutes of Health (Public Health Service grant GM059969 to P.B.), and from the International Centre for Genetic Engineering and Biotechnology (ICGEB) (CRP/MEX08-02) (to D.G.). L.C.M is supported by a pre-doctoral fellowship from CONACYT (No. 169380).
References
- Ahmer BM, van Reeuwijk J, Watson PR, Wallis TS, Heffron F. Salmonella SirA is a global regulator of genes mediating enteropathogenesis. Mol Microbiol. 1999;31:971–982. doi: 10.1046/j.1365-2958.1999.01244.x. [DOI] [PubMed] [Google Scholar]
- Altier C. Genetic and environmental control of Salmonella invasion. J Microbiol. 2005;43(Spec No):85–92. [PubMed] [Google Scholar]
- Altier C, Suyemoto M, Lawhon SD. Regulation of Salmonella enterica serovar Typhimurium invasion genes by CsrA. Infect Immun. 2000a;68:6790–6797. doi: 10.1128/iai.68.12.6790-6797.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Suyemoto M, Ruiz AI, Burnham KD, Maurer R. Characterization of two novel regulatory genes affecting Salmonella invasion gene expression. Mol Microbiol. 2000b;35:635–646. doi: 10.1046/j.1365-2958.2000.01734.x. [DOI] [PubMed] [Google Scholar]
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol. 2007;10:156–163. doi: 10.1016/j.mib.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Bajaj V, Lucas RL, Hwang C, Lee CA. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol Microbiol. 1996;22:703–714. doi: 10.1046/j.1365-2958.1996.d01-1718.x. [DOI] [PubMed] [Google Scholar]
- Baker CS, Eory LA, Yakhnin H, Mercante J, Romeo T, Babitzke P. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J Bacteriol. 2007;189:5472–5481. doi: 10.1128/JB.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CS, Morozov I, Suzuki K, Romeo T, Babitzke P. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol. 2002;44:1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- Baxter MA, Fahlen TF, Wilson RL, Jones BD. HilE interacts with HilD and negatively regulates hilA transcription and expression of the Salmonella enterica serovar Typhimurium invasive phenotype. Infect Immun. 2003;71:1295–1305. doi: 10.1128/IAI.71.3.1295-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua JM, Bevilacqua PC. Thermodynamic analysis of an RNA combinatorial library contained in a short hairpin. Biochemistry. 1998;37:15877–15884. doi: 10.1021/bi981732v. [DOI] [PubMed] [Google Scholar]
- Bijlsma JJ, Groisman EA. The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica. Mol Microbiol. 2005;57:85–96. doi: 10.1111/j.1365-2958.2005.04668.x. [DOI] [PubMed] [Google Scholar]
- Bispham J, Tripathi BN, Watson PR, Wallis TS. Salmonella pathogenicity island 2 influences both systemic salmonellosis and Salmonella-induced enteritis in calves. Infect Immun. 2001;69:367–377. doi: 10.1128/IAI.69.1.367-377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddicker JD, Jones BD. Lon protease activity causes down-regulation of Salmonella pathogenicity island 1 invasion gene expression after infection of epithelial cells. Infect Immun. 2004;72:2002–2013. doi: 10.1128/IAI.72.4.2002-2013.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol. 2009;73:434–445. doi: 10.1111/j.1365-2958.2009.06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius J. Plasmid vectors for the selection of promoters. Gene. 1984;27:151–160. doi: 10.1016/0378-1119(84)90136-7. [DOI] [PubMed] [Google Scholar]
- Brown NF, Vallance BA, Coombes BK, Valdez Y, Coburn BA, Finlay BB. Salmonella pathogenicity island 2 is expressed prior to penetrating the intestine. PLoS Pathog. 2005;1:e32. doi: 10.1371/journal.ppat.0010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante VH, Martinez LC, Santana FJ, Knodler LA, Steele-Mortimer O, Puente JL. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc Natl Acad Sci U S A. 2008;105:14591–14596. doi: 10.1073/pnas.0801205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Chan K, Kim CC, Falkow S. Microarray-based detection of Salmonella enterica serovar Typhimurium transposon mutants that cannot survive in macrophages and mice. Infect Immun. 2005;73:5438–5449. doi: 10.1128/IAI.73.9.5438-5449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez RG, Alvarez AF, Romeo T, Georgellis D. The physiological stimulus for the BarA sensor kinase. J Bacteriol. 2010;192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo DM, Valdivia RH, Monack DM, Falkow S. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol Microbiol. 1998;30:175–188. doi: 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- Coburn B, Li Y, Owen D, Vallance BA, Finlay BB. Salmonella enterica serovar Typhimurium pathogenicity island 2 is necessary for complete virulence in a mouse model of infectious enterocolitis. Infect Immun. 2005;73:3219–3227. doi: 10.1128/IAI.73.6.3219-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes BK, Brown NF, Valdez Y, Brumell JH, Finlay BB. expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J Biol Chem. 2004;279:49804–49815. doi: 10.1074/jbc.M404299200. [DOI] [PubMed] [Google Scholar]
- Coombes BK, Coburn BA, Potter AA, Gomis S, Mirakhur K, Li Y, Finlay BB. Analysis of the contribution of Salmonella pathogenicity islands 1 and 2 to enteric disease progression using a novel bovine ileal loop model and a murine model of infectious enterocolitis. Infect Immun. 2005;73:7161–7169. doi: 10.1128/IAI.73.11.7161-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin KH, Miller VL. InvF is required for expression of genes encoding proteins secreted by the SPI1 type III secretion apparatus in Salmonella typhimurium. J Bacteriol. 1999;181:4949–4954. doi: 10.1128/jb.181.16.4949-4954.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker SC, Marchal K, Verhoeven TL, Engelen K, Vanderleyden J, Detweiler CS. Microarray analysis and motif detection reveal new targets of the Salmonella enterica serovar Typhimurium HilA regulatory protein, including hilA itself. J Bacteriol. 2005;187:4381–4391. doi: 10.1128/JB.187.13.4381-4391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiwick J, Nikolaus T, Erdogan S, Hensel M. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol Microbiol. 1999;31:1759–1773. doi: 10.1046/j.1365-2958.1999.01312.x. [DOI] [PubMed] [Google Scholar]
- Drecktrah D, Knodler LA, Ireland R, Steele-Mortimer O. The mechanism of Salmonella entry determines the vacuolar environment and intracellular gene expression. Traffic. 2006;7:39–51. doi: 10.1111/j.1600-0854.2005.00360.x. [DOI] [PubMed] [Google Scholar]
- Dubey AK, Baker CS, Romeo T, Babitzke P. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005;11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelberg K, Galan JE. Differential regulation of Salmonella typhimurium type III secreted proteins by pathogenicity island 1 (SPI-1)-encoded transcriptional activators InvF and HilA. Infect Immun. 1999;67:4099–4105. doi: 10.1128/iai.67.8.4099-4105.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier CD, Ellermeier JR, Slauch JM. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2005;57:691–705. doi: 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- Ellermeier JR, Slauch JM. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr Opin Microbiol. 2007;10:24–29. doi: 10.1016/j.mib.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol. 2003;47:103–118. doi: 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- Fass E, Groisman EA. Control of Salmonella pathogenicity island-2 gene expression. Curr Opin Microbiol. 2009;12:199–204. doi: 10.1016/j.mib.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Oropeza R, Kenney LJ. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol Microbiol. 2003;48:1131–1143. doi: 10.1046/j.1365-2958.2003.03502.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Walthers D, Oropeza R, Kenney LJ. The response regulator SsrB activates transcription and binds to a region overlapping OmpR binding sites at Salmonella pathogenicity island 2. Mol Microbiol. 2004;54:823–835. doi: 10.1111/j.1365-2958.2004.04317.x. [DOI] [PubMed] [Google Scholar]
- Fortune DR, Suyemoto M, Altier C. Identification of CsrC and characterization of its role in epithelial cell invasion in Salmonella enterica serovar Typhimurium. Infect Immun. 2006;74:331–339. doi: 10.1128/IAI.74.1.331-339.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh AB, Rajasingh H, Mande SS. Mathematical modeling of regulation of type III secretion system in Salmonella enterica serovar Typhimurium by SirA. In Silico Biol. 2009;9:S57–S72. [PubMed] [Google Scholar]
- Georgellis D, Arvidson S, von Gabain A. Decay of ompA mRNA and processing of 9S RNA are immediately affected by shifts in growth rate, but in opposite manners. J Bacteriol. 1992;174:5382–5390. doi: 10.1128/jb.174.16.5382-5390.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach RG, Jackel D, Geymeier N, Hensel M. Salmonella pathogenicity island 4-mediated adhesion is coregulated with invasion genes in Salmonella enterica. Infect Immun. 2007;75:4697–4709. doi: 10.1128/IAI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier RI, Ahmer BM. SirA orthologs affect both motility and virulence. J Bacteriol. 2001;183:2249–2258. doi: 10.1128/JB.183.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman EA, Ochman H. How Salmonella became a pathogen. Trends Microbiol. 1997;5:343–349. doi: 10.1016/S0966-842X(97)01099-8. [DOI] [PubMed] [Google Scholar]
- Hansen-Wester I, Hensel M. Salmonella pathogenicity islands encoding type III secretion systems. Microbes Infect. 2001;3:549–559. doi: 10.1016/s1286-4579(01)01411-3. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, Heikenwalder M, et al. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar Typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J Immunol. 2005;174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- Haraga A, Ohlson MB, Miller SI. Salmonellae interplay with host cells. Nat Rev Microbiol. 2008;6:53–66. doi: 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- Hensel M. Salmonella pathogenicity island 2. Mol Microbiol. 2000;36:1015–1023. doi: 10.1046/j.1365-2958.2000.01935.x. [DOI] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, et al. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol Microbiol. 1998;30:163–174. doi: 10.1046/j.1365-2958.1998.01047.x. [DOI] [PubMed] [Google Scholar]
- Hoiseth SK, Stocker BA. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature. 1981;291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- Johnston C, Pegues DA, Hueck CJ, Lee A, Miller SI. Transcriptional activation of Salmonella typhimurium invasion genes by a member of the phosphorylated response-regulator superfamily. Mol Microbiol. 1996;22:715–727. doi: 10.1046/j.1365-2958.1996.d01-1719.x. [DOI] [PubMed] [Google Scholar]
- Jones BD. Salmonella invasion gene regulation: a story of environmental awareness. J Microbiol. 2005;43(Spec No):110–117. [PubMed] [Google Scholar]
- Jones MA, Hulme SD, Barrow PA, Wigley P. The Salmonella pathogenicity island 1 and Salmonella pathogenicity island 2 type III secretion systems play a major role in pathogenesis of systemic disease and gastrointestinal tract colonization of Salmonella enterica serovar Typhimurium in the chicken. Avian Pathol. 2007;36:199–203. doi: 10.1080/03079450701264118. [DOI] [PubMed] [Google Scholar]
- Kenney LJ, Bauer MD, Silhavy TJ. Phosphorylation-dependent conformational changes in OmpR, an osmoregulatory DNA-binding protein of Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:8866–8870. doi: 10.1073/pnas.92.19.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapouge K, Schubert M, Allain FH, Haas D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol. 2008;67:241–253. doi: 10.1111/j.1365-2958.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- Lawhon SD, Frye JG, Suyemoto M, Porwollik S, McClelland M, Altier C. Global regulation by CsrA in Salmonella typhimurium. Mol Microbiol. 2003;48:1633–1645. doi: 10.1046/j.1365-2958.2003.03535.x. [DOI] [PubMed] [Google Scholar]
- Lawhon SD, Maurer R, Suyemoto M, Altier C. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol. 2002;46:1451–1464. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- Lee AK, Detweiler CS, Falkow S. OmpR regulates the two-component system SsrA-SsrB in Salmonella pathogenicity island 2. J Bacteriol. 2000;182:771–781. doi: 10.1128/jb.182.3.771-781.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan SA, Rytkonen A, Yu XJ, Liu M, Holden DW. SlyA regulates function of Salmonella pathogenicity island 2 (SPI-2) and expression of SPI-2-associated genes. Infect Immun. 2005;73:4354–4362. doi: 10.1128/IAI.73.7.4354-4362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lostroh CP, Bajaj V, Lee CA. The cis requirements for transcriptional activation by HilA, a virulence determinant encoded on SPI-1. Mol Microbiol. 2000;37:300–315. doi: 10.1046/j.1365-2958.2000.01991.x. [DOI] [PubMed] [Google Scholar]
- Lostroh CP, Lee CA. The HilA box and sequences outside it determine the magnitude of HilA-dependent activation of PprgH from Salmonella pathogenicity island 1. J Bacteriol. 2001;183:4876–4885. doi: 10.1128/JB.183.16.4876-4885.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas RL, Lee CA. Roles of hilC and hilD in regulation of hilA expression in Salmonella enterica serovar Typhimurium. J Bacteriol. 2001;183:2733–2745. doi: 10.1128/JB.183.9.2733-2745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchetti-Miganeh C, Burrowes E, Baysse C, Ermel G. The post-transcriptional regulator CsrA plays a central role in the adaptation of bacterial pathogens to different stages of infection in animal hosts. Microbiology. 2008;154:16–29. doi: 10.1099/mic.0.2007/012286-0. [DOI] [PubMed] [Google Scholar]
- Lundberg U, Vinatzer U, Berdnik D, von Gabain A, Baccarini M. Growth phase-regulated induction of Salmonella-induced macrophage apoptosis correlates with transient expression of SPI-1 genes. J Bacteriol. 1999;181:3433–3437. doi: 10.1128/jb.181.11.3433-3437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus SL, Brumell JH, Pfeifer CG, Finlay BB. Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect. 2000;2:145–156. doi: 10.1016/s1286-4579(00)00273-2. [DOI] [PubMed] [Google Scholar]
- Mayer MP. A new set of useful cloning and expression vectors derived from pBlueScript. Gene. 1995;163:41–46. doi: 10.1016/0378-1119(95)00389-n. [DOI] [PubMed] [Google Scholar]
- Miao EA, Miller SI. A conserved amino acid sequence directing intracellular type III secretion by Salmonella typhimurium. Proc Natl Acad Sci U S A. 2000;97:7539–7544. doi: 10.1073/pnas.97.13.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusaki H, Takaya A, Yamamoto T, Aizawa S. Signal pathway in salt-activated expression of the Salmonella pathogenicity island 1 type III secretion system in Salmonella enterica serovar Typhimurium. J Bacteriol. 2008;190:4624–4631. doi: 10.1128/JB.01957-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Ohl ME, Miller SI. Salmonella: a model for bacterial pathogenesis. Annu Rev Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- Olekhnovich IN, Kadner RJ. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J Bacteriol. 2002;184:4148–4160. doi: 10.1128/JB.184.15.4148-4160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olekhnovich IN, Kadner RJ. Crucial roles of both flanking sequences in silencing of the hilA promoter in Salmonella enterica. J Mol Biol. 2006;357:373–386. doi: 10.1016/j.jmb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Puente JL, Bieber D, Ramer SW, Murray W, Schoolnik GK. The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol Microbiol. 1996;20:87–100. doi: 10.1111/j.1365-2958.1996.tb02491.x. [DOI] [PubMed] [Google Scholar]
- Santos RL, Tsolis RM, Baumler AJ, Adams LG. Pathogenesis of Salmonella-induced enteritis. Braz J Med Biol Res. 2003;36:3–12. doi: 10.1590/s0100-879x2003000100002. [DOI] [PubMed] [Google Scholar]
- Schechter LM, Damraue SM, Lee CA. Two AraC/XylS family members can independently counteract the effect of repressing sequences upstream of the hilA promoter. Mol Microbiol. 1999;32:629–642. doi: 10.1046/j.1365-2958.1999.01381.x. [DOI] [PubMed] [Google Scholar]
- Schechter LM, Lee CA. AraC/XylS family members, HilC and HilD, directly bind and derepress the Salmonella typhimurium hilA promoter. Mol Microbiol. 2001;40:1289–1299. doi: 10.1046/j.1365-2958.2001.02462.x. [DOI] [PubMed] [Google Scholar]
- Schechter LM, Jain S, Akbar S, Lee CA. The small nucleoid-binding proteins H-NS, HU, and Fis affect hilA expression in Salmonella enterica serovar Typhimurium. Infect Immun. 2003;71:5432–5435. doi: 10.1128/IAI.71.9.5432-5435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev. 2004;17:14–56. doi: 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors O, Georgellis D, et al. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol. 2002;184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya A, Kubota Y, Isogai E, Yamamoto T. Degradation of the HilC and HilD regulator proteins by ATP-dependent Lon protease leads to downregulation of Salmonella pathogenicity island 1 gene expression. Mol Microbiol. 2005;55:839–852. doi: 10.1111/j.1365-2958.2004.04425.x. [DOI] [PubMed] [Google Scholar]
- Teplitski M, Goodier RI, Ahmer BM. Pathways leading from BarA/SirA to motility and virulence gene expression in Salmonella. J Bacteriol. 2003;185:7257–7265. doi: 10.1128/JB.185.24.7257-7265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplitski M, Goodier RI, Ahmer BM. Catabolite repression of the SirA regulatory cascade in Salmonella enterica. Int J Med Microbiol. 2006;296:449–466. doi: 10.1016/j.ijmm.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Timmermans J, Van Melderen L. Post-transcriptional global regulation by CsrA in bacteria. Cell Mol Life Sci. 2010;67:2897–2908. doi: 10.1007/s00018-010-0381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomljenovic-Berube AM, Mulder DT, Whiteside MD, Brinkman FSL, Coombes BK. Identification of the regulatory logic controlling Salmonella pathoadaptation by the SsrA-SsrB two-component system. PLoS Genet. 2010;6:e1000875. doi: 10.1371/journal.pgen.1000875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A. 2001;98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Gabain A, Belasco JG, Schottel JL, Chang ACY, Cohen SN. Decay of mRNA in Escherichia coli: investigation of the fate of specific segments of transcripts. Proc Natl Acad Sci U S A. 1983;80:653–657. doi: 10.1073/pnas.80.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walthers D, Carroll RK, Navarre WW, Libby SJ, Fang FC, Kenney LJ. The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol Microbiol. 2007;65:477–493. doi: 10.1111/j.1365-2958.2007.05800.x. [DOI] [PubMed] [Google Scholar]
- Walthers D, Li Y, Liu Y, Anand G, Yan J, Kenney LJ. The Salmonella enterica response regulator SsrB relieves H-NS silencing by displacing H-NS bound in polymerization mode and directly activates transcription. J Biol Chem. 2010 doi: 10.1074/jbc.M110.164962. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, et al. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol. 2003;48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- Worley MJ, Ching KH, Heffron F. Salmonella SsrB activates a global regulon of horizontally acquired genes. Mol Microbiol. 2000;36:749–761. doi: 10.1046/j.1365-2958.2000.01902.x. [DOI] [PubMed] [Google Scholar]
- Yakhnin AV, Trimble JJ, Chiaro CR, Babitzke P. Effects of mutations in the L-tryptophan binding pocket of the Trp RNA-binding attenuation protein of Bacillus subtilis. J Biol Chem. 2000;275:4519–4524. doi: 10.1074/jbc.275.6.4519. [DOI] [PubMed] [Google Scholar]
- Yakhnin H, Pandit P, Petty TJ, Baker CS, Romeo T, Babitzke P. CsrA of Bacillus subtilis regulates translation initiation of the gene encoding the flagellin protein (hag) by blocking ribosome binding. Mol Microbiol. 2007;64:1605–1620. doi: 10.1111/j.1365-2958.2007.05765.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.