

Abstract
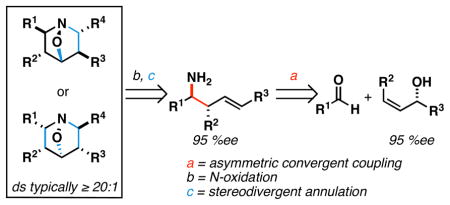
A convergent and stereodivergent pathway to highly substituted 1-aza-7-oxabicyclo[2.2.1]heptanes is described. This process begins with a coupling reaction between an allylic alcohol, aldehyde, and LiHMDS for the production of stereodefined primary homoallylic amines. Subsequent N-oxidation and condensation with formaldehyde or a glyoxylate, defines a convenient entry to densely functionalized homoallylic nitrones whose intramolecular annulation can be controlled to deliver one of two distinct heterocyclic skeletons, each with ≥20:1 stereoselection. The control of stereochemistry in these reactions is proposed to result from both the control of nitrone geometry, and a selective partitioning of reaction pathway between direct [3+2] cycloaddition, or tandem [3,3]-rearrangement/[3+2] cycloaddition.
Alkaloids are a broad class of natural products that possess a diverse array of biological properties. While examples include a vast array of molecular architectures, structurally complex polycyclic alkaloids have played a prominent role in the development of modern organic chemistry, providing inspiration for the development of synthetic methods suitable for the preparation of their intricate stereodefined skeletons. Densely functionalized piperidines are a common motif in complex alkaloids (Figure 1) and, despite the variety of chemical methods available to access this type of nitrogen-containing heterocycle, the asymmetric synthesis of highly substituted piperidines remains a significant challenge in chemical synthesis.1 Here, we describe a convergent, asymmetric, and stereodivergent entry into tri- and tetrasubstituted 1-aza-7-oxabicyclo[2.2.1]heptanes (6 and 7), precursors to complex 4-hydroxypiperidines, that derives from the union of an allylic alcohol (3) with two carbonyl electrophiles (1 and 5) and LiHMDS (2)(Figure 2). These studies are based on two central advances, 1) the establishment of a three-component coupling reaction for the stereoselective synthesis of primary homoallylic amines, and 2) control of path selectivity in annulation chemistry of highly substituted homoallylic nitrones.
Figure 1.

Piperidine-containing polycyclic alkaloids.
Figure 2.

Convergent route to 1-aza-7-oxabicyclo[2.2.1]heptanes.
Since LeBel and Lumma’s early reports,2 and Oppolzer’s subsequent investigation of regio- and stereoselectivity,3 intramolecular cycloaddition of homoallylic nitrones has been accepted as a general strategy for the synthesis of substituted 1-aza-7-oxabicyclo[2.2.1]heptanes and, on reductive cleavage of the σN–O bond, 4-hydroxypiperidines.4 The vast majority of applications in this regard describe intramolecular reactions of sparsely functionalized homoallylic nitrones, with few reports describing the utility of this cycloaddition for the stereoselective synthesis of densely functionalized of 1-aza-7-oxabicyclo[2.2.1]heptanes. This apparent lack of utility was thought to be due, in part, to: 1) difficulties associated with accessing the requisite highly substituted and stereodefined homoallylic nitrone starting materials, and 2) lack of stereochemical control in the cycloaddition reaction.5
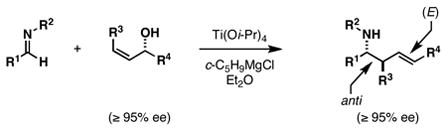
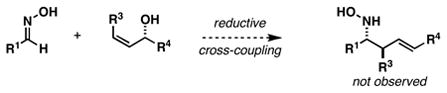
Our investigations in this area began by developing a synthetic method suitable for the preparation of highly substituted and stereodefined homoallylic hydroxylamines. Previous studies in our laboratory have established an asymmetric route to stereodefined homoallylic secondary amines based on the union of preformed imines with chiral allylic alcohols (eq 1).6 While a sequence of deprotection (removal of R2) and N-oxidation could render this process useful to intersect with the annulation chemistry of homoallylic nitrones, we opted to pursue a more direct means to accomplish this goal; namely coupling of allylic alcohols with preformed oximes (eq 2). Unfortunately, all attempts to accomplish such a bond construction were met with failure.
 |
(1) |
 |
(2) |
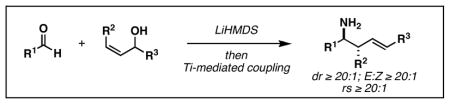
To circumvent this impediment in reactivity, we pursued an alternative coupling reaction capable of delivering a stereodefined primary homoallylic amine – a product whose subsequent N-oxidation would deliver homoallylic hydroxylamines without the need for intermediate protecting group manipulations. As depicted in Table 1, in situ generation of a TMS-imine7 proved compatible with Ti-mediated allylic alcohol-based reductive cross-coupling, defining a three-component coupling process for the synthesis of stereodefined primary homoallylic amines. Overall, exposure of an aldehyde to LiHMDS is followed by introduction of Ti(Oi-Pr)4 and c-C5H9MgCl (Et2O, −78 to −40 °C). Subsequent addition of a preformed allylic lithium alkoxide results in overall reductive cross-coupling and, on aqueous work-up, delivers primary homoallylic amines with outstanding levels of regio- and stereocontrol. This reaction proceeds effectively with a variety of aromatic and aliphatic aldehydes, and delivers stereodefined (E)-anti-homoallylic amines as single diastereomers in 53–87% yield.
Table 1.
Primary homoallylic amines from Ti-mediated coupling.
 | ||||
|---|---|---|---|---|
| entry | aldehyde | allylic alcohol | yield (%) | product |
| 1 |
8 |
9 |
80 |
 10 |
| 2 |
8 |
 11 |
76 |
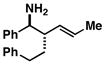 12 |
| 3 |
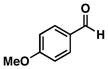 13 |
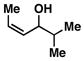 14 |
70 |
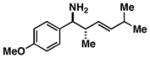 15 |
| 4 |
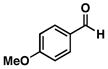 13 |
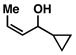 16 |
83 |
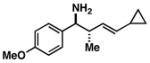 17 |
| 5 |
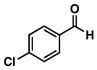 18 |
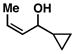 16 |
87 |
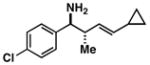 19 |
| 6 |
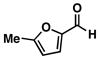 20 |
 11 |
72 |
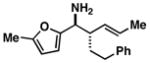 21 |
| 7 |
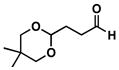 22 |
9 |
56 |
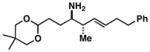 23 |
| 8 |
24 |
9 |
53 |
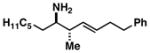 25 |
| 9 |
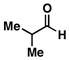 26 |
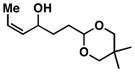 27 |
63 |
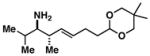 28 |
Reaction conditions: RCHO (2 eq), LiHMDS (2 equiv), THF (−78 °C for aliphatic RCHO, −10 °C for aromatic aldehyde), then Ti(Oi-Pr)4 (2–3 equiv), RLi or RMgCl (4–6 eq), then Li-alkoxide of allylic alcohol (1 equiv) (−78 °C to rt). For specific examples, see Supporting Information. Note: products are racemic.
With a robust stereoselective process for the convergent assembly of complex primary homoallylic amines secured, we turned our attention to the evaluation of intramolecular nitrone annulation chemistry as a means to generate highly substituted heterocycles. After oxidation of homoallylic amine 10 to the corresponding hydroxylamine 29,8 heating with formaldehyde at 50 °C leads to a complex product mixture (eq 3, Figure 3). While the rearranged nitrone 30 is formed in 67% yield, cycloadducts 31 and 32 are generated in 17% combined yield without substantial stereoselection (ds = 2.5:1). Interestingly, after isolation of nitrone 30, heating at 120 °C in toluene results in a highly stereoselective annulation (eq 4). Here, the isomeric cycloadduct 33 is produced in 74% yield with very high levels of stereoselection (ds ≥ 20:1).
Figure 3.

Stereochemical complexity of nitrone cycloaddition.
The difference in 2,3-stereochemistry between 33 and the cycloadducts 31 and 32 is consistent with a complex competition in reaction mechanism due to the relative rate of cycloaddition vs. sigmatropic rearrangement (Figure 4).5a,b,9 For example, initial exposure of 29 to formaldehyde at 50 °C leads primarily to the product of [3,3]-rearrangement (30), with the minor cycloadducts 31 and 32 deriving from direct [3+2]-cycloaddition of the intermediate nitrone A. After isolation of the rearranged product 30, heating in toluene at 120 °C results in a stereodivergent annulation via D to deliver 33, uncontaminated by cycloadduct 31 or 32.
Figure 4.

Direct [3+2] vs. tandem [3,3]/[3+2].
This tandem [3,3]-sigmatropic rearrangement/[3+2] cycloaddition is a useful stereoselective process for the production of densely functionalized 2-aryl-substituted 1-aza-7-oxabicyclo[2.2.1]-heptanes (Table 2). Entries 1 and 2 demonstrate that the stereoselection of this process is highest with electron deficient aromatics α- to the intermediate nitrone (dr ≥20:1). Entries 3 and 4 depict annulation events that deliver products of identical stereochemistry as that seen in entries 1 and 2, albeit with compromised levels of stereocontrol (dr = 6:1). Finally, entry 5 provides a glimpse of the potential utility of this process for the synthesis of fully-substituted heterocyclic systems. In this more complex example, reaction with an aliphatic aldehyde produces the tetrasubstituted heterocycle 42 in 80% yield (dr = 4:1).10
Table 2.
Stereoselective synthesis of (2,3-syn-3,4-syn-4,5-syn-5,6-anti)-1-aza-7-oxabicyclo[2.2.1]heptanes.
 | |||||
|---|---|---|---|---|---|
| entry | homoallylic hydroxylamine | R3CHO | yield (%) | dr | product |
| 1 |
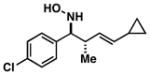 34 |
|
61 | ≥20:1 |
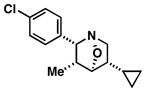 35 |
| 2 |
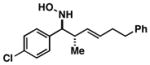 36 |
|
60 | ≥20:1 |
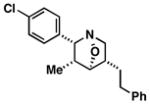 37 |
| 3 |
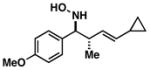 38 |
|
66 | 6:1a |
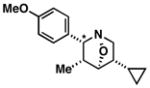 39 |
| 4 |
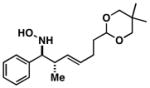 40 |
|
61 | 6:1a |
 41 |
| 5 |
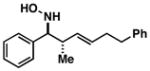 29 |
|
80 | 4:1 |
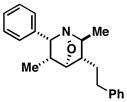 42 |
= minor product is epimeric at the highlighted (*) position.
Note: Initial sigmatropic rearrangement for reactions summarized in entries 1 and 2 occur in <2 h, whereas substrates depicted in entries 3 and 4 required up to 48 h. Similarly, the subsequent [3+2]annulation in entries 3 and 4 require prolonged heating in comparison to entries 1 and 2 (48 h at 120 °C vs. 12 h at 120 °C). Note: products are racemic.
The stereochemical control of the annulation events depicted in Table 2 derives from the enhanced propensity of the initially formed nitrones to undergo [3,3]-sigmatropic rearrangement, with subsequent [3+2] cycloaddition occurring by way of the transposed nitrone (via D; Figure 4). With this model in mind, we speculate that more electron rich nitrones in intermediate D have the potential to isomerize to the (E)-O nitrone—a process that we reason is in part responsible for the decreased levels of stereoselection observed in entries 3 and 4 of Table 2. In cases where the initial [3,3]-rearrangement (B → C; Figure 4) is not driven by thermodynamic stabilization of the resulting conjugated nitrone, attempted annulation reactions with formaldehyde or acetaldehyde result in a complex mixture of reaction products.
In an attempt to broaden the scope of this convergent route to complex heterocycles we aimed to define an alternative means to control the mechanistic course of these annulation processes. Specifically, we sought to identify conditions suitable to promote direct [3+2] annulation of the initially formed nitrone as a path to heterocycles of a distinct substitution and stereochemistry. Without a mechanistically lucid means by which to pursue this goal, we opted to drastically change the electronic and stereochemical features of the initially formed nitrone A (Figure 4).
With this goal in mind, the related annulation of homoallylic hydroxylamines with ethylglyoxate was explored. Compared to nitrones resulting from condensation of hydroxylamines with formaldehyde or acetaldehyde, nitrones derived from glyoxylates are electronically and stereochemically distinct. While more electron deficient, these nitrones prefer an (E)-geometry in hydrocarbon solvents and exist in rapid equilibrium with their (Z)-isomers.11
As illustrated in Table 3, we were delighted to find that annulation with ethylglyoxylate defines a stereochemically unique path to highly substituted heterocycles – in this case, delivering 1-aza-7-oxabicyclo[2.2.1]heptane products containing a 2,3-anti-3,4-syn-4,5-anti-5,6-anti stereochemical relationship. Cycloaddition proceeds with hydroxylamines that have neighboring aliphatic- as well as aromatic- substitution, and delivers stereodefined heterocycles in 52–64% yield (entries 1–5; in all cases, no evidence could be found for the production of stereoisomeric cycloadducts).
Table 3.
Stereoselective synthesis of (2,3-anti-3,4-syn-4,5-anti-5,6-anti)-1-aza-7-oxabicyclo[2.2.1]heptanes.
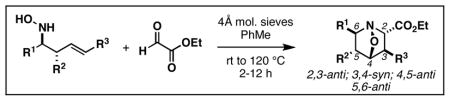 | ||||
|---|---|---|---|---|
| entry | homoallylic hydroxylamine | yield (%) | dr | product |
| 1 |
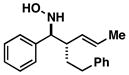 43 |
64 | ≥20:1 |
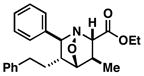 44 |
| 2 |
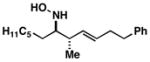 45 |
55 | ≥20:1 |
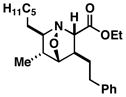 46 |
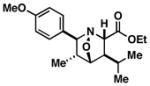
| 3 |
 47 |
64 | ≥20:1 |
 48 |
| 4 |
 49 |
52 | ≥20:1 |
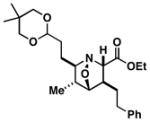 50 |
| 5 |
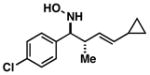 34 |
60 | ≥20:1 |
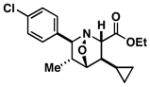 51 |
Note: products are racemic.
The stereochemistry of the heterocycles depicted in Table 3 is consistent with a reaction sequence that proceeds by direct stereoselective intramolecular [3+2] cycloaddition of the initially formed electron deficient nitrone E, uninterrupted by [3,3]-rearrangement (F → G; Figure 5). While this provides a robust means to control stereoselection in annulation events of homoallylic nitrones derived from aliphatic aldehydes (entries 2 and 4), entries 1, 3, and 5 indicate that even when substrates possess neighboring aromatic substitution that has the potential to drive the [3,3]-sigmatropic rearrangement, reactions with ethylglyoxylate proceed in a highly stereoselective manner that is apparently uncomplicated by this process.12 The facial selectivity of the cycloaddition is further supported by minimization of A-1,3 strain about the substituted nitrone E, resulting in a pseudoequatorial disposition of R1, and subsequently securing the orientation of R2 and R 3 in the transition state for cycloaddition.
Figure 5.

Stereoselective annulation with ethylglyoxylate.
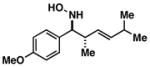
Finally, this sequence for the synthesis of complex 1-aza-7-oxabicyclo[2.2.1]heptanes is ideally suited for enantioselective synthesis. In accord with previous observations,6 Ti-mediated coupling of optically active allylic alcohols (Figure 6) proceeds with exquisite levels of stereocontrol and delivers optically active homoallylic amine products (52 and 56). As anticipated, a sequence of N-oxidation and nitrone cycloaddition by either direct [3+2] annulation or tandem [3,3]/[3+2] provides a convenient asymmetric entry to complex tri- and tetrasubstituted 1-aza-7-oxabicyclo[2.2.1]heptanes (54, 55 and 58).
Figure 6.

Asymmetric entry to 1-aza-7-oxabicyclo[2.2.1]heptanes. Reaction conditions: a) PhCHO, LiHMDS, THF (−10 °C), then Ti(Oi-Pr)4, c-C5H9MgCl, then Li-alkoxide of allylic alcohol (1 equiv) (−78 °C to rt); b) (BzO)2, K2HPO4, DMF, rt; c) NH2NH2, EtOH; d) ethylglyoxylate, PhMe, 4 Å mol. sieves, 100 °C; e) (CH2O)n, PhMe, 4 Å mol. sieves, 50 °C; f) C6H13CHO, LiHMDS, THF (−78 °C), then Ti(Oi-Pr)4, c-C5H9MgCl, then Li-alkoxide of allylic alcohol (1 equiv) (−78 °C to rt). * % ee determined by Mosher ester analysis of derivatives, see Supporting Information for details.
In summary, we describe a convergent asymmetric entry to densely functionalized tri- and tetrasubstituted 1-aza-7-oxabicyclo[2.1.1]heptanes. This sequence is defined by: 1) a new diastereo- and enantioselective convergent synthesis of (E)-anti-homoallylic primary amines, 2) N-oxidation, and 3) a stereodivergent nitrone annulation process. Based on the highly stereoselective nature of this synthesis pathway, the complexity of products produced, and the frequency with which substituted piperidines appear in alkaloids, we look forward to future applications in target-oriented synthesis.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the National Institutes of Health –NIGMS (GM80266 and GM80266-04S1).
Footnotes
SUPPORTING INFORMATION PARAGRAPH:
Experimental procedures and tabulated spectroscopic data for new compounds (PDF) are available free of charge via the Internet at http://pubs.acs.org/page/jacsat/submission/authors.html.
References
- 1.For reviews, see: Buffat MGP. Tetrahedron. 2004;60:1701–1729.Weintraub PM, Sabol JS, Kane JM, Borcherding DR. Tetrahedron. 2003;59:2953–2989.Baliah V, Jeyaraman R, Chandrasekaran Chem Rev. 1983;83:379–423.
- 2.a) LeBel NA, Slusarczuk GMJ, Spurlock LA. J Am Chem Soc. 1962;84:4360–4361. [Google Scholar]; b) Lumma WC. J Am Chem Soc. 1969;91:2820–2821. [Google Scholar]
- 3.Oppolzer W, Siles S, Snowden RL, Bakker BH, Petrzilka M. Tetrahedron. 1985;41:3497–3509. [Google Scholar]
- 4.For an example, see: Hoffmann RW, Endesfelder A. Liebigs Ann Chem. 1986;11:1823–1826.
- 5.For a discussion concerning the observation of products derived from aza-Cope/intramolecular [3+2] nitrone cycloaddition, see: Wuts PGM, Jung YW. J Org Chem. 1988;53:1957–1965.Merino P, Mannucci V, Tejero T. Eur J Org Chem. 2008:3943–3959.For a review of [3+2] nitrone cycloaddition, see: Confalone PN, Huie EM. Org React. 1988;36:1–173.
- 6.(a) Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648–3652. doi: 10.1002/anie.200900236. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Umemura S, McLaughlin M, Micalizio GC. Org Lett. 2009;11:5402–5405. doi: 10.1021/ol9022134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang D, Micalizio GC. J Am Chem Soc. 2009;131:17548–17549. doi: 10.1021/ja908504z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen MZ, McLaughlin M, Takahashi M, Tarselli MA, Yang D, Umemura S, Micalizio GC. J Org Chem. 2010;75:8048–8059. doi: 10.1021/jo101535d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Hart DJ, Kanai K, Thomas DG, Yang TK. J Org Chem. 1983;48:289–294. [Google Scholar]; (b) Ha DC, Hart DJ, Yang TK. J Am Chem Soc. 1984;106:4819–4825. [Google Scholar]
- 8.Berman AM, Johnson JS. J Org Chem. 2006;71:219–224. doi: 10.1021/jo051999h. [DOI] [PubMed] [Google Scholar]
- 9.For a computational assessment related to the competition between [3,3]-rearrangement and [3+2]-cycloaddition reactions of homoallylic nitrones, see: Merino P, Tejero T, Mannucci V. Tetrahedron Lett. 2007;48:3385–3388.
- 10.See Supporting Information for details.
- 11.Inouye Y, Hara J, Kakisawa H. Chem Lett. 1980:1407–1410.Inouye Y, Takaya K, Kakisawa H. Bull Chem Soc Jpn. 1983;56:3541–3542.It has also been proposed that intermolecular [3+2] cycloaddition reactions occur faster by way of (E)-nitrones: Burdisso M, Gandolfi R, Grünanger P. J Org Chem. 1990;55:3427–3429.
- 12.These observations do not exclude control by the Curtin-Hammett principle where reversible [3,3]-rearrangement of the initially formed glyoxylate-based nitrone is irrelevant in determining product distribution.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.