

Abstract
Inhibitors targeting the influenza A virus M2 (A/M2) proton channel have lost their effectiveness due to widespread resistance. As a first step in the development of new inhibitors that address this problem, we have screened several focused collections of small molecules using two-electrode voltage patch clamp assays (TEVC) on Xenopus laevis oocytes. Diverse head groups and scaffolds of A/M2 inhibitors have been explored. It has been found that not only amine but also hydroxyl, aminooxyl, guanidine, and amidine compounds are active against the A/M2 proton channel. Moreover, the channel is able to accommodate a wide range of structural variation in the apolar scaffold. This study offers information to guide the next generation of A/M2 proton channel inhibitor design.
Keywords: Influenza A/M2, A/M2 channel inhibitor, two-electrode voltage clamp assay (TEVC)
Influenza A viruses present a severe human health threat due to the ease of transmission via the upper respiratory system. Influenza A virus constantly undergoes antigenic drift and antigenic shift, which allow them to escape from existing immunity.1 Currently, there are two classes of small molecule anti-influenza virus drugs:2 zanamivir and oseltamivir, which target neuraminidase, and A/M2 channel inhibitors, which include amantadine and rimantadine. Widespread resistance to both oseltamivir (the only orally bioavailable neuraminidase inhibitor) as well as A/M2 inhibitors presents a major health risk.
The A/M2 protein forms a homotetrameric proton selective channel3,4 that is essential for viral replication.5,6 Amantadine targets the A/M2 channel by blocking its pore.7−9 The number of drug-resistant variants of A/M2 is limited by the very conserved nature of the binding site within the channel. V27A, L26F, and S31N are the only widely occurring drug-resistant variants seen in transmissible strains of the virus.10−12
Extensive medicinal chemistry efforts have been devoted to identifying A/M2 channel inhibitors with higher potency and reduced side effects.2 However, very few chemotypes other than the adamantane scaffold have been studied for A/M2 channel inhibition.2,13 One exception is a spiro-piperidine 1, which emerged from our previous structure−activity relationship (SAR) study of 2-[3-azaspiro(5,5)undecanol]-2-imidazoline (BL-1743).14 Further structure modification yielded another class of spiran amines 2 that are not only active against wide type (WT) A/M2 but also A/M2-V27A mutant channel.15 Encouraged by these initial results, we decided to explore both headgroup and scaffold diversity to develop new generations of A/M2 inhibitors, which might similarly be modified to improve potency toward drug-resistant mutants in subsequent studies. Inhibitors tested in this study were either synthesized or purchased through commercial vendors.
The inhibitors were tested via a two-electrode patch clamp assay (TEVC) using Xenopus laevis frog oocytes microinjectected with RNA expressing the A/M2 protein as in a previous report.16 The potency of the inhibitors was expressed as the percentage inhibition of A/M2 current observed after 2 min of incubation with 100 μM compounds, and IC50 values were collected for selected potent compounds.
A common feature of all known M2 channel inhibitors is the presence of a polar headgroup, generally a primary or secondary amine, connected to a hydrophobic scaffold. Interestingly, the crystal and solid state NMR structures of amantadine complexed A/M2 did not identify any specific interactions between the amino group and the protein;9,17 instead, our most recent high-resolution X-ray structure showed a cluster of well-organized water molecules that might form hydrogen bonds with His37.18 The water cluster is highly polarized to stabilize an incoming hydronium ion, which might be mimicked by the ammonium group of the inhibitor. This observation led us to ask whether the primary amine of amantadine might be substituted by other polar groups without significantly affecting the activity. To explore whether the amine can be substituted by any other polar head groups, we examined the following panel of compounds based on an adamantane scaffold (Table 1).
Table 1. Activities of Adamantane Analogues with Diverse Polar Head Groups on WT A/M2 Inhibition.
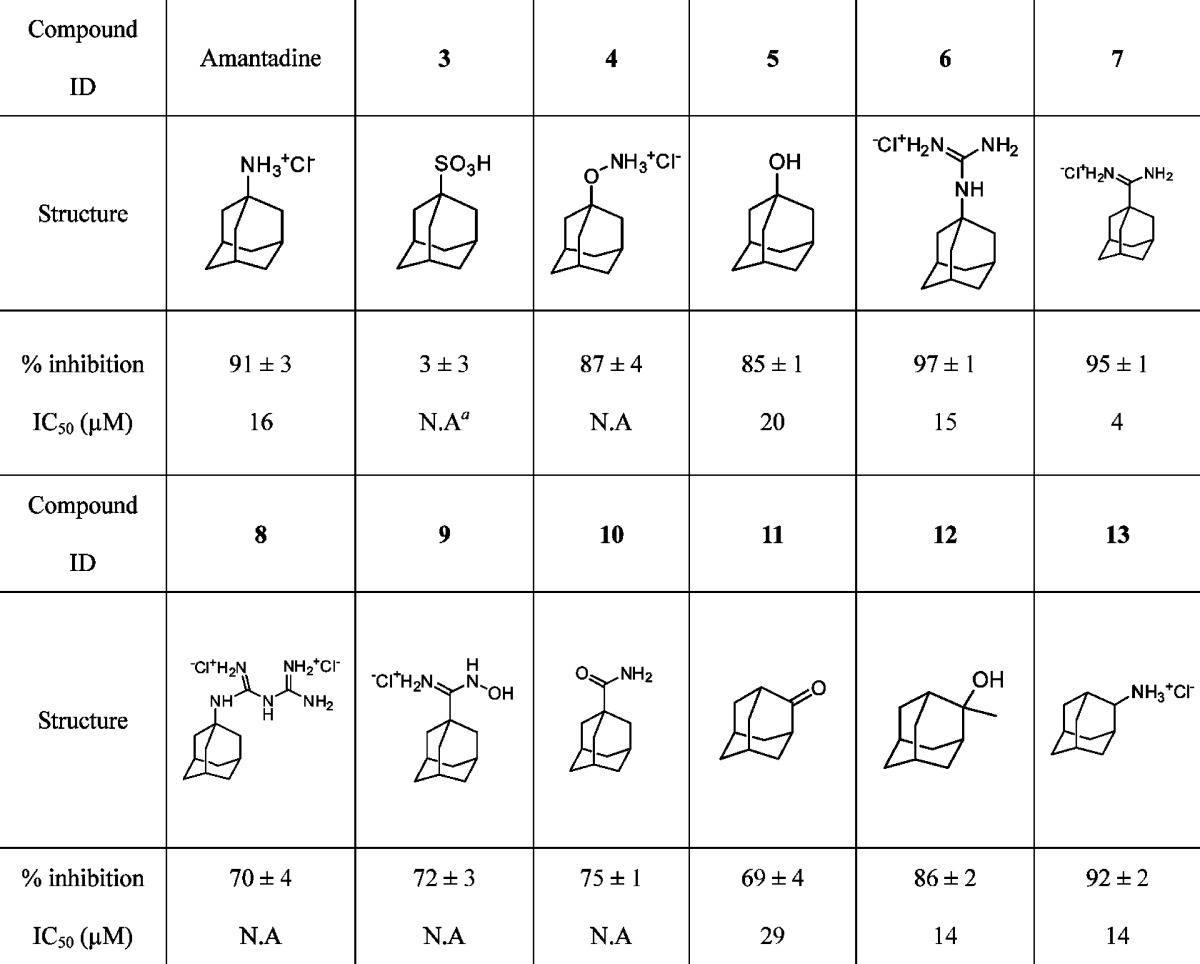 |
NA, not available.
Amantadine showed 91 ± 3% inhibition (IC50 = 16 μM) against the WT A/M2 channel and was used as a benchmark for comparison. The negatively charged 1-adamantyl sulfonic acid 3 was inactive, consistent with M2's function of stabilizing a positively charged permeant ion. Amino-oxyl 4 and 1-adamantol 5 were nearly as active as the corresponding amine, giving a similar degree of inhibition at 100 μM. Guanidine 6 (IC50 = 15 μM) had a similar activity to amantadine. In contrast, the 1-adamantyl biguanidine 8 was much less active than 1-adamantyl guanidine 6, possibly due to the decrease of hydrophobicity or the increased bulk of the polar group. Interestingly, when the amine was substituted by amidine 7, the potency increased nearly 4-fold with an IC50 of 4 μM. N′-Hydroxyadamantane-1-carboximidamide 9 was also less active than amantadine when comparing percentage inhibition at 100 μM. Neutral compounds such as amide 10 and 2-adamantanone 11 both showed decreased activities relative to the corresponding amine. 2-Methyl-2-adamantanol 12 (IC50 = 14 μM) had a higher activity than both 1-adamantol 5 (IC50 = 21 μM) and amantadine (IC50 = 16 μM). This finding was consistent with earlier studies showing the in vitro activity of 2-methyl-2-adamantol in influenza virus-induced cytopathic effect (CPE) assay.19 2-Aminoadamantane 13 was shown to have a similar activity as amantadine. In fact, a series of 2-substitited adamantane molecules are known to be highly active against influenza A virus in in vitro viral CPE assay.20
Additionally, for the spiran class of inhibitors, converting either the primary amine of 2 or the secondary amine of 1 to the corresponding hydrazine in 14 and 15, respectively, decreased potency. This is possibly due to a decrease in the basicity of the terminal amine (Table 2). In comparison, both the glutarimide 16 and the aminoglurarimide 17 were only weakly active against A/M2, probably due to the introduction of hydrogen bond acceptors and the strong decrease in basicity. In conclusion, these results are consistent with previous studies showing that alcohols can substitute for amines in M2 inhibitors19 and expand the range of groups that can be considered for design.
Table 2. Activities of Spiran Amines/Hydrazines on WT A/M2 Inhibition.
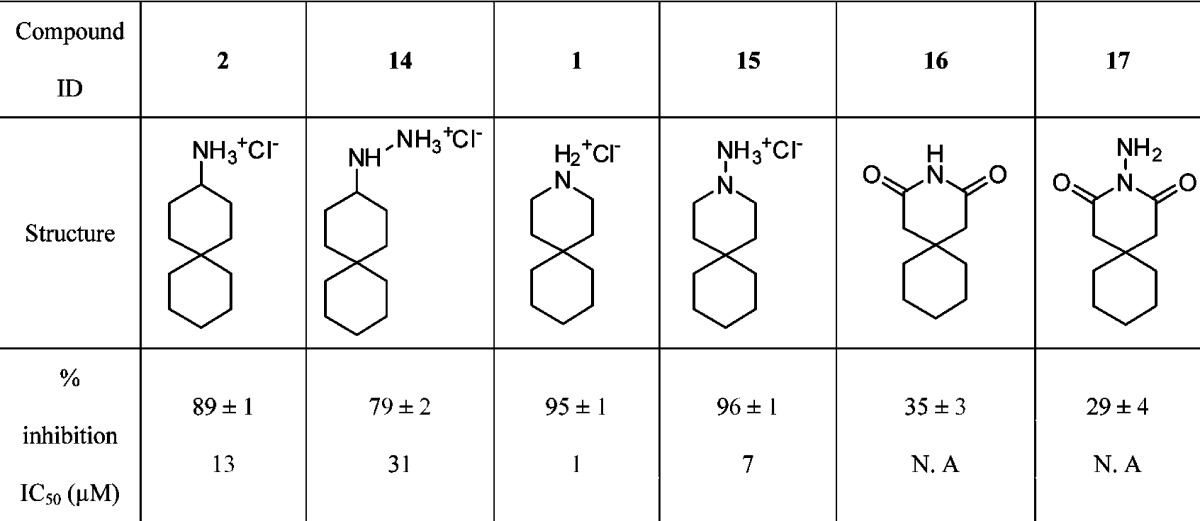 |
Pentacyclo[5.4.0.0.2,60.3,1005,9]undecane is a particularly good synthetic precursor for multifunctional inhibitors, as the diketone can be easily converted to a diol, hydroxyl amine, or hydroxyl ketone.21 Our recent high-resolution X-ray crystal structure of A/M2 revealed two layers of well-organized water molecules positioned above the proton-accepting H37 residues.18 We rationalized that an additional polar headgroup might potentially hydrogen bond to or displace these water molecules.
The desired compounds were synthesized by standard procedures from the starting diketone (Scheme 1). In this series, monoamine 20 showed the most potent inhibition against the A/M2 channel (IC50 = 8 μM), followed by amino alcohol 21 (IC50 = 24 μM). Diol 18 was also active, but the percentage inhibition at 100 μM was smaller than that of the amino alcohol. Monoalcohol 19 showed only moderate inhibition against the A/M2 channel. The lower activities of compounds with an additional polar headgroup might be due to the increased polarity, leading to a greater dehydration penalty upon entering the A/M2 channel pore.
Scheme 1. Synthesis Scheme of A/M2 Inhibitors with More than One Polar Head Group.

Encouraged by the result that diol compounds showed good activity against A/M2, we next examined two commercially available diols (22 and 23, stereochemistry is not defined by the vendor). Both were found be less active than amantadine at 100 μM percentage inhibition. An additional hydroxyl group at the 3-position of amantadine caused a dramatic decrease in A/M2 inhibition (24). Another disubstituted oxyadamantane analogue 25 was also not active. Taken together, this might be due to either steric mismatch or decreased hydrophobicity. The ring-contracted bisnoradamantane diol 26 was only moderately active, highlighting that hydrophobic contacts are important for tight binding.
Earlier work by Clercq et al. examined additional 2-amino substitution on the potency of rimantadine against influenza A virus, finding two compounds with 1,2-diaminoethyl groups to be just as potent as rimantadine.20,22 Taken together with our data, we conclude that additional polar groups can be tolerated but do not enhance the potency.
Compound 27 was designed by Chou et al.23 to potentially target drug-resistant A/M2 mutants based on the solution NMR structure of WT A/M2 (PDB code: 2RLF).24 The design principle aimed to introduce an additional hydroxyl group at the 2-position of rimantadine to connect the two adjacent helices of A/M2, assuming the interfaces of the adjacent helices at residue Asp44 are the pharmacologically relevant drug binding sites; however, this inhibitor was found to be equally potent as amantadine (IC50 = 16 μM) toward WT A/M2 but had no inhibitory effect on either S31N or V27A mutants (Table 3).
Table 3. Activities of A/M2 Inhibitor with More than One Polar Head Group on A/M2 Inhibition.
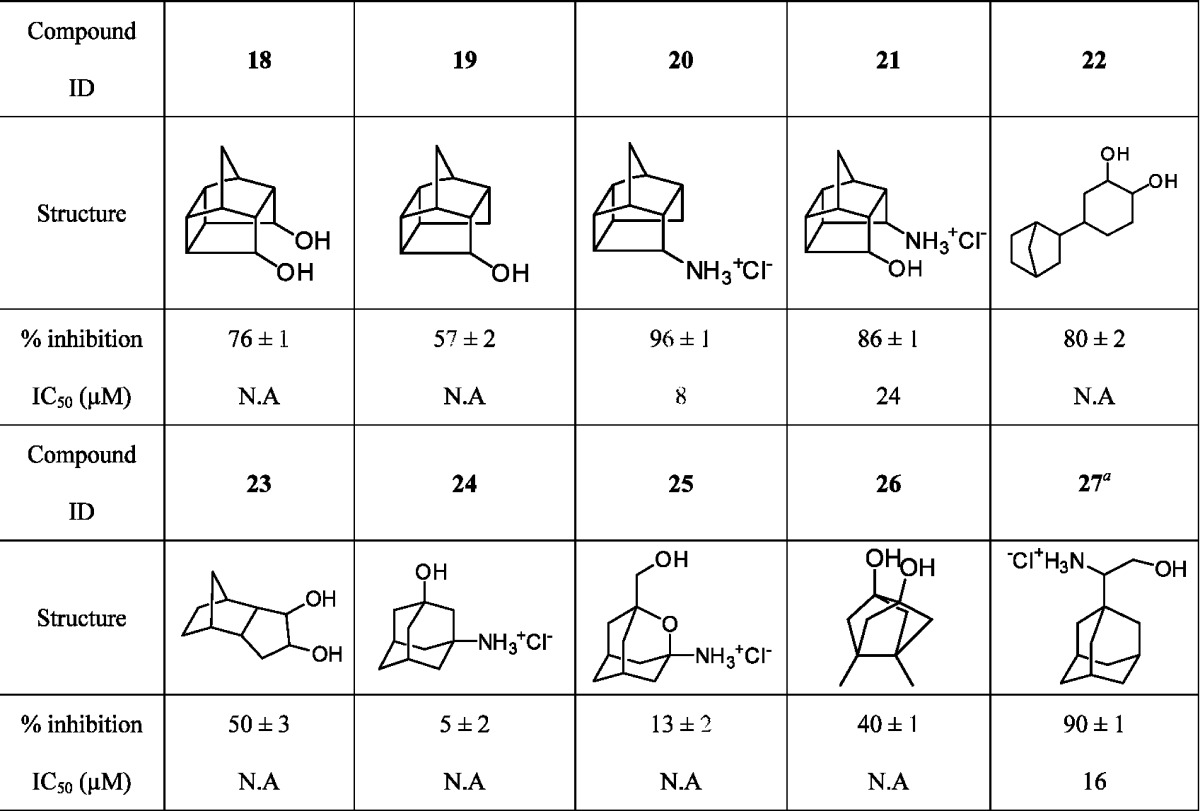 |
The percentage inhibition of 27 against the V27A and S31N mutants of A/M2 at a 100 μM concentration was 4 ± 1 and 15 ± 2%, respectively.
Chen et al. reported a screen of 70 primary amines including linear, aromatic, monocyclic, bicyclic, and tricyclic compounds as A/M2 inhibitors.25 Five compounds were found to have potency against A/M2 comparable to amantadine. We have examined >30 additional amines with various sized apolar groups, focusing primarily on compounds with sizes and shapes roughly conforming to the steric requirements of the binding site. Some of the structures are shown in Table 4. Consistent with previous findings; amines with aromatic substitutions (28 and 29) are significantly less active than the corresponding hydrocarbons, possibly due to the decreased hydrophobicity.
Table 4. Activities of A/M2 Inhibitor with Diverse Scaffolds on A/M2 Inhibition in TEVC.
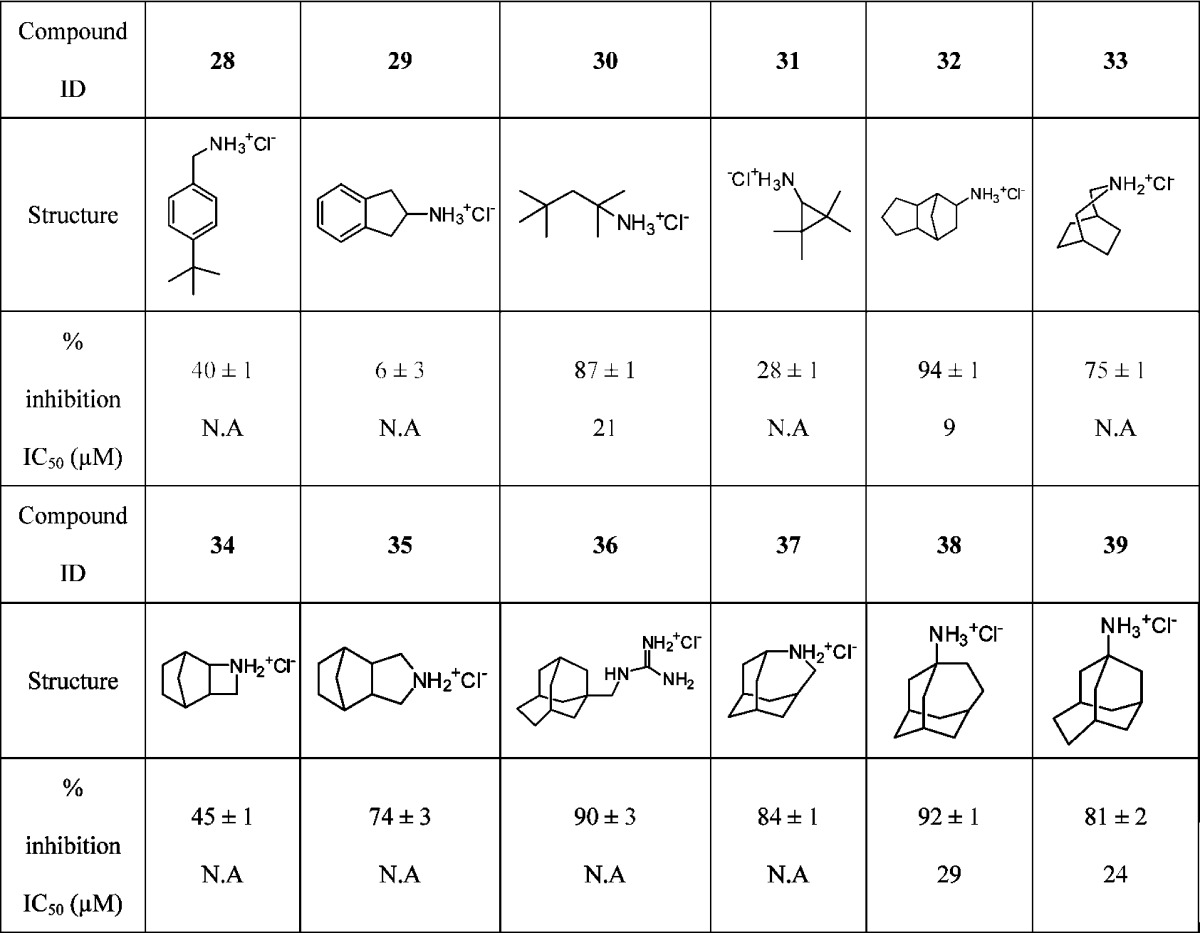 |
All linear alkyl amines tested (up to eight carbon atoms) in the previous report were found to be inactive.25 Surprisingly, we found the branched alkyl amine 30 to be nearly as active as amantadine (IC50 = 21 μM), suggesting that the A/M2 channel can accommodate a wide range of structural diversity.
A/M2 pore lining residues drug binding site V27, A30, and G34 create a hydrophobic drug binding pocket that shows a preference for inhibitors with clogP ≥ 1.5.17 The results presented here, together with previous studies by Chen et al.25 show that a minimum of eight aliphatic carbons are required for potent inhibition. Tetramethyl cyclopropane amine 31, which has seven aliphatic carbons, showed weak inhibition (28% inhibition). Compound 32 showed higher activity than amantadine and has the same number of carbon atoms as amantadine (IC50 = 9 μM). Three bicyclic inhibitors (33, 34, and 35) were also tested and found to be less active than amantadine at 100 μM percentage inhibition. Four ring-expanded adamantane inhibitors (36, 37, 38, and 39) showed similar activity as amantadine; this result suggests that WT A/M2 is insensitive to minor scaffold modifications.
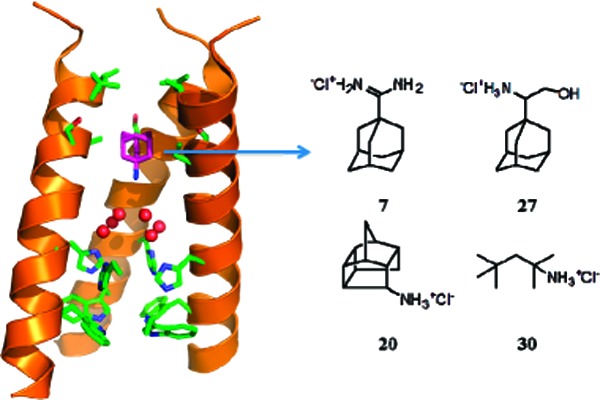
As an initial step toward the design of inhibitors targeting drug-resistant M2, it is important to first understand the mechanism by which the WT protein binds inhibitors. X-ray crystal structures17,18 and SSNMR structures9 of the channel have indicated that amantadine binds with its aliphatic region projecting toward an apolar pocket (Figure 1). These structures indicate that there is sufficient space in the pocket to accommodate larger hydrophobic groups. The SAR shown in this paper for both the polar headgroup and the apolar scaffold is consistent with such a mode of binding. In the previous literature, almost all potent A/M2 channel inhibitors are cyclic compounds with an amine headgroup.2 In this study, we have found that (1) the amine group is not essential for activity and can be substituted by hydroxyl, guanidine, amidine, and aminooxyl groups; (2) potent inhibition does not require a cyclic scaffold, so long as the shape of the molecule conforms to the cavity, such as the linear branched amine (30); (3) compound hydrophobicity is very important for the potency of the inhibitor, and the effective A/M2 inhibitor should possess clogP ≥ 1.5 or 8 aliphatic carbon atoms. While these compounds are active against the WT protein, they were found to be less potent or inactive against V27A and/or S31N mutant forms. The percentage inhibition of all compounds other than amantadine against the V27A and S31N mutants of A/M2 at a 100 μM concentration was less than 15 and 33%, respectively. (Amantadine showed 0 and 35% inhibition against V27A and S31N A/M2 mutant at a 100 μM concentration.) This is understandable, because both mutations would increase the polarity of the pore.
Figure 1.
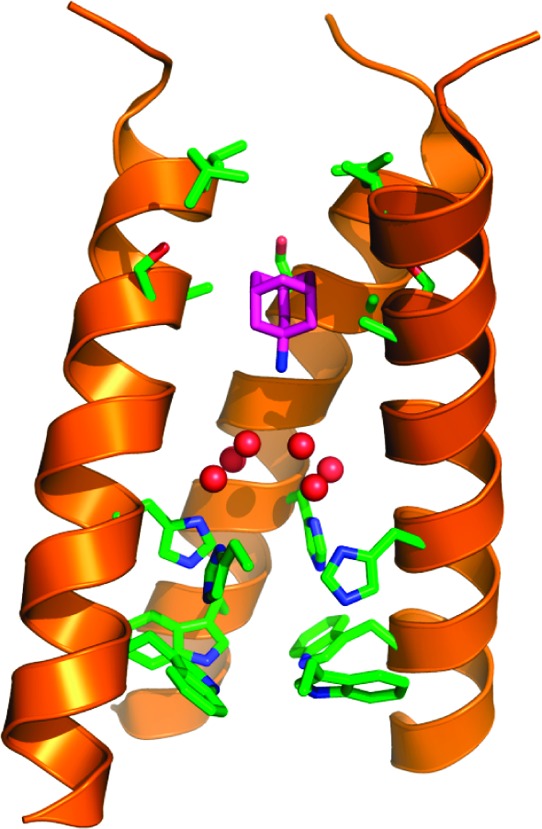
Structure of amantadine (purple carbon atoms) in complex with the transmembrane domain of M2, as determined by solid-state NMR9 (PDB: 2KQT), with the water structure seen in the high-resolution crystallographic structure (PDB: 3LBW) superimposed. Six water molecules from 3LBW, shown as red spheres, lie above the four His37 and Trp41 residues shown near the bottom of the structure (side chains shown in stick). The apolar region of the drug projects into the cavity formed by Val27, Ser31, and Ala30 (near the upper portion of the structure), while the ammonium group projects downward toward the water cluster. One helix has been removed for clarity.
A second mode of binding suggested by Chou and co-workers involves a more peripheral site on the surface of the protein.24 Chou et al. proposed that, if this mode of binding were correct, compound 27 should be a potent inhibitor of both WT protein as well as S31N, given that the peripheral binding sites were on the surface of the protein.23 On the other hand, the prediction of the pore-binding model was that this compound should bind only to the WT protein. We therefore tested this compound and found it to be a good inhibitor of WT channels; however, it did not show significant inhibition of V27A and S31N mutant forms. Thus, our results are fully consistent with the pore-binding site being the pharmacologically relevant site of inhibitor binding and do not support the peripheral mode of binding. In summary, these results are fully consistent with the expectation that drugs bind within the pore, and we will focus future work on the design of new inhibitors onto this location.
Acknowledgments
J.W. thanks Kathleen Molnar for help with HRMS data collection.
Supporting Information Available
Experimental procedures for the synthesis and characterization of A/M2 inhibitors (1H and 13C NMR, ESI-MS, and HRMS). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was funded by the NIH (GM56423 and AI74571).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug. Discovery 2006, 5, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagoja I. M.; De Clercq E. Anti-influenza virus agents: Synthesis and mode of action. Med. Res. Rev. 2008, 28, 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhmakov I. V.; Geraghty F. M.; Ogden D. C.; Hayhurst A.; Antoniou M.; Hay A. J. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J. Physiol. (London) 1996, 494, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L. H.; Holsinger L. J.; Lamb R. A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [DOI] [PubMed] [Google Scholar]
- Martin K.; Helenius A. Nuclear transport of influenza virus ribonucleoproteins: The viral matrix protein (M1) promotes export and inhibits import. Cell 1991, 67, 117–130. [DOI] [PubMed] [Google Scholar]
- Takeda M.; Pekosz A.; Shuck K.; Pinto L. H.; Lamb R. A. Influenza A virus M-2 ion channel activity is essential for efficient replication in tissue culture. J. Virol. 2002, 76, 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Takeuchi K.; Pinto L. H.; Lamb R. A. Ion channel activity of influenza A virus M2 protein: Characterization of the amantadine block. J. Virol. 1993, 67, 5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies W. L.; Hoffmann C. E.; Paulshock M.; Wood T. R.; Haff R. F.; Grunert R. R.; Watts J. C.; Hermann E. C.; Neumayer E. M.; McGahen J. W. Antiviral Activity of 1-Adamantanamine (Amantadine). Science 1964, 144, 862–863. [DOI] [PubMed] [Google Scholar]
- Cady S. D.; Schmidt-Rohr K.; Wang J.; Soto C. S.; DeGrado W. F.; Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature 2010, 463, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright R. A.; Medina M. J.; Xu X. Y.; Perez-Oronoz G.; Wallis T. R.; Davis X. H. M.; Povinelli L.; Cox N. J.; Klimov A. I. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: A cause for concern. Lancet 2005, 366, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Saito R.; Sakai T.; Sato I.; Sano Y.; Oshitani H.; Sato M.; Suzuki H. Frequency of amantadine-resistant influenza A viruses during two seasons featuring cocirculation of H1N1 and H3N2. J. Clin. Microbiol. 2003, 41, 2164–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyde V. M.; Xu X. Y.; Bright R. A.; Shaw M.; Smith C. B.; Zhang Y.; Shu Y. L.; Gubareva L. V.; Cox N. J.; Klimov A. I. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J. Infect. Dis. 2007, 196, 249–257. [DOI] [PubMed] [Google Scholar]
- Miller J. A.; Ullah G. M.; Welsh G. M.; Hall A. C. 8-aminobicyclo[3.2.1]octanes: synthesis and anti-viral activity. Tetrahedron Lett. 2001, 42, 7503–7507. [Google Scholar]
- Wang J.; Cady S. D.; Balannik V.; Pinto L. H.; DeGrado W. F.; Hong M. Discovery of Spiro-Piperidine Inhibitors and Their Modulation of the Dynamics of the M2 Proton Channel from Influenza A Virus. J. Am. Chem. Soc. 2009, 131, 8066–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V.; Wang J.; Ohigashi Y.; Jing X. H.; Magavern E.; Lamb R. A.; DeGrado W. F.; Pinto L. H. Design and Pharmacological Characterization of Inhibitors of Amantadine-Resistant Mutants of the M2 Ion Channel of Influenza A Virus. Biochemistry 2009, 48, 11872–11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balannik V.; Lamb R. A.; Pinto L. H. The oligomeric state of the active BM2 ion channel protein of influenza B virus. J. Biol. Chem. 2008, 283, 4895–4904. [DOI] [PubMed] [Google Scholar]
- Stouffer A. L.; Acharya R.; Salom D.; Levine A. S.; Di Costanzo L.; Soto C. S.; Tereshko V.; Nanda V.; Stayrook S.; DeGrado W. F. Structural basis for the function and inhibition of an influenza virus proton channel. Nature 2008, 451, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya R.; Carnevale V.; Fiorin G.; Levine B. G.; Polishchuk A. L.; Balannik V.; Samish I.; Lamb R. A.; Pinto L. H.; DeGrado W. F.; Klein M. L. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolocouris A.; Spearpoint P.; Martin S. R.; Hay A. J.; López-Querol M.; Sureda F. X.; Padalko E.; Neyts J.; De Clercq E. Comparisons of the influenza virus A M2 channel binding affinities, anti-influenza virus potencies and NMDA antagonistic activities of 2-alkyl-2-aminoadamantanes and analogues. Bioorg. Med. Chem. Lett. 2008, 18, 6156–6160. [DOI] [PubMed] [Google Scholar]
- Tataridis D.; Fytas G.; Kolocouris A.; Fytas C.; Kolocouris N.; Foscolos G. B.; Padalko E.; Neyts J.; De Clercq E. Influence of an additional 2-amino substituent of the 1-aminoethyl pharmacophore group on the potency of rimantadine against influenza virus A. Bioorg. Med. Chem. Lett. 2007, 17, 692–696. [DOI] [PubMed] [Google Scholar]
- Romanski J.; Mloston G.; Linden A.; Heimgartner H. Synthesis and structure of spirocyclic tetrahydrothiophene derivatives bearing a 'cage' residue. Pol. J. Chem. 2005, 79, 973–979. [Google Scholar]
- Zoidis G.; Kolocouris N.; Kelly J. M.; Prathalingam S. R.; Naesens L.; De Clercq E. Design and synthesis of bioactive adamantanaminoalcohols and adamantanamines. Eur. J. Med. Chem. 2010, 45, 5022–5030. [DOI] [PubMed] [Google Scholar]
- Du Q. S.; Huang R. B.; Wang S. Q.; Chou K. C. Designing Inhibitors of M2 Proton Channel against H1N1 Swine Influenza Virus. PLos One 2010, 5, e9388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R.; Chou J. J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W.; Zeng S.; Li C.; Jie Y.; Li Z.; Chen L. Identification of Hits as Matrix-2 Protein Inhibitors through the Focused Screening of a Small Primary Amine Library. J. Med. Chem. 2010, 53, 3831–3834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.