

Abstract
A review. Transition metal catalyzed decarboxylative allylations, benzylations, and interceptive allylations are reviewed.
Keywords: Decarboxylative, allylation, benzylation, cross-coupling
1 Introduction to Decarboxylative-Coupling
Catalytic cross-coupling reactions have had profound impact on the synthesis of pharmaceuticals, biologically active natural products, and materials.1 Such reactions typically involve the oxidative addition of an aryl or alkyl halide to a low-valent metal, followed by transmetalation and reductive elimination of the desired product (Scheme 1).2 The transmetalation steps in cross-coupling reactions often use relatively expensive, toxic, or highly basic reagents that must be prepared from other functional precursors. In addition, the reagents required for transmetalation necessarily produce stoichiometric quantities of hazardous byproducts that can complicate product purification. With this in mind, it has been recognized that it is highly desirable to develop new strategies for the generation of organometallic intermediates that utilize inexpensive substrates, proceed under mild conditions, and are environmentally benign. One such strategy is decarboxylative coupling. Decarboxylative coupling reactions utilize decarboxylative metalation to generate organometallic intermediates that are coupled via reductive elimination (Scheme 1). As compared to traditional cross-coupling methods, decarboxylative coupling has several potential advantages: 1) carboxylic acid derivatives are ubiquitous and inexpensive reactants, 2) decarboxylation can drive the formation of reactive intermediates under neutral conditions, and 3) the only stoichiometric byproduct is CO2, which is non-flammable, non-toxic, and easily removed from the reaction medium. Moreover, decarboxylation allows the site-specific generation and coupling of reactive intermediates, in contrast to reactions that generate reactive intermediates by C-H activation where regioselective formation of specific intermediates can be difficult.3
Scheme 1.

Standard Cross-coupling vs. Decarboxylative Coupling
In this review, we will focus on discussion of homogeneous catalysis of decarboxylative allylation and benzylation reactions, a subject that highlights the breadth of nucleophilic species that can be generated by decarboxylation. In addition, studies of decarboxylative allylations have shown that there are several mechanisms for decarboxylative coupling that do not necessarily follow the simplified rubric shown in Scheme 1. While several accounts have been published on this topic in the last several years,4 none has done so in the comprehensive manner of this review which covers relevant publications through August 2010.
2 Decarboxylative Allylation of Enolates
2.1 Introduction to Decarboxylative Allylation
The Tsuji-Trost reaction is a reaction that has garnered much attention due to its ability to couple allyl electrophiles with nucleophiles in a chemo-, regio-, and stereoselective fashion.5,6 In a typical Tsuji-Trost reaction, an allyl acetate or carbonate reacts with a palladium catalyst by displacement of the leaving group to give a π-allyl palladium intermediate which can undergo substitution by a nucleophile. Frequently, the nucleophiles have been limited to “soft” nucleophiles, like malonates, whose corresponding pKa’s are <20. However, successful allylation of monostabilized enolates has been achieved using preformed tin,7 boron,8 magnesium,9 and lithium enolates,10 as well as silyl enol ethers.11 While these methods have demonstrated the ability to form a new carbon–carbon bond selectively, they all suffer from the need to make a preformed organometallic which typically requires subjecting the substrate to highly basic conditions and results in a stoichiometric amount of metal salt waste. An ideal alternative synthesis would be one in which the same reaction can be accomplished yet produces only easily removed waste and does not require preformed nucleophiles, thus allowing a greater synthetic efficiency. Such a strategy requires an alternative method for the in situ generation of enolates. This review will focus on synthetic strategies that involve the direct generation of enolates and other nucleophiles via decarboxylation. Indeed, the in situ generation of nucleophiles via decarboxylation distinguishes decarboxylative allylation (DcA) reactions as an important subset of Tsuji-Trost reactions.
In 1950, Nesmayanov showed that metal enolates can be readily accessed under neutral conditions and without additives by the decarboxylation of metal β-ketocarboxylates (Scheme 2).12 While Nesmayanov utilized this decarboxylative metalation in stoichiometric transformations, he set the stage for later catalytic transformations. In the early 60’s, divalent metals like Ni(II) and Mn(II) were shown to decarboxylate malonic acids and were proposed to form intermediate metal enolates.13 While the knowledge of these transformations was applied to understanding enzymatic decarboxylations, the synthetic potential of the intermediates was not realized.
Scheme 2.

Formation of Mercury Enolate via Decarboxylation
Then in 1980, Tsuji14 and Saegusa15 almost simultaneously reported the decarboxylative allylation of β-keto allyl esters (eq 1). In this method the loss of CO2 replaces the need to selectively prepare preformed enolate equivalents. A further potential benefit of the decarboxylative allylation (DcA) is the ability to generate both nucleophile and electrophile in situ. Thus, greater functional group compatibility can be expected since the high energy intermediates are formed in catalytic concentration and the pH is formally neutral. Consequently, decarboxylative allylation is a valuable addition to the toolbox of the organic chemist. In the following section of the review, we cover the developments whose chemical lineage can be traced back to these seminal works.
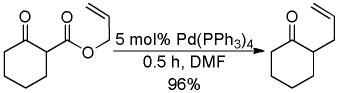 |
(1) |
2.1.1 Decarboxylative Allylation: Scope and Chemoselectivity
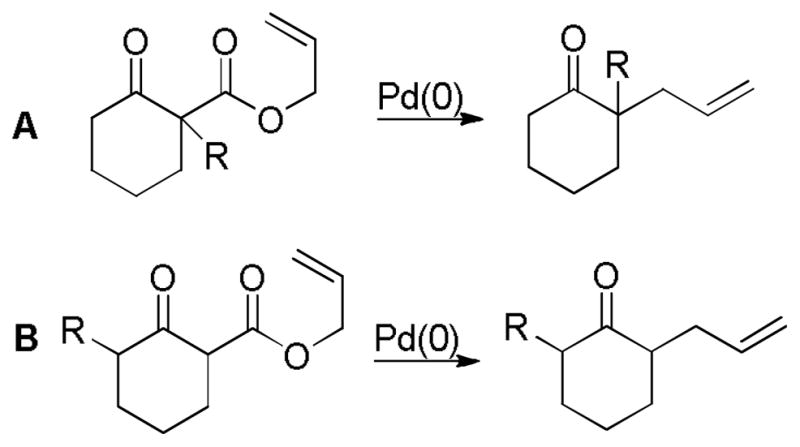
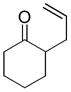
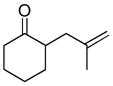
In the first disclosure of a decarboxylative allylation (DcA) reaction by Tsuji,14 allyl esters of acetoacetic acid were subjected to a catalytic amount of Pd(OAc)2 and PPh3 (Method A, Chart 1), providing γ,δ-unsaturated methyl ketones in high yield. Alternatively, Saegusa demonstrated that a variety of acyclic and cyclic ketoesters would undergo decarboxylative coupling using 5 mol % Pd(PPh3)4 as the catalyst (Method B, Chart 1).15 While these reports did not detail the functional group compatibility of decarboxylative allylation (DcA) reactions, they did demonstrate that the reaction could tolerate β-hydrogens on the allyl fragment, however the yield is significantly reduced when the product is derived from the geranyl ester (4, Chart 1). This illustrates a common challenge in decarboxylative allylation; substitution and elimination are often competitive. A recent report illustrates that substitution is favored when the α-position of the β-keto ester is unsubstituted while elimination is favored when the α-position is substituted (Scheme 3).16 This may reflect different mechanisms of allylation for the two substrates as discussed vida infra.
Chart 1.

Tsuji-Saegusa Decarboxylative Allylation
Scheme 3.
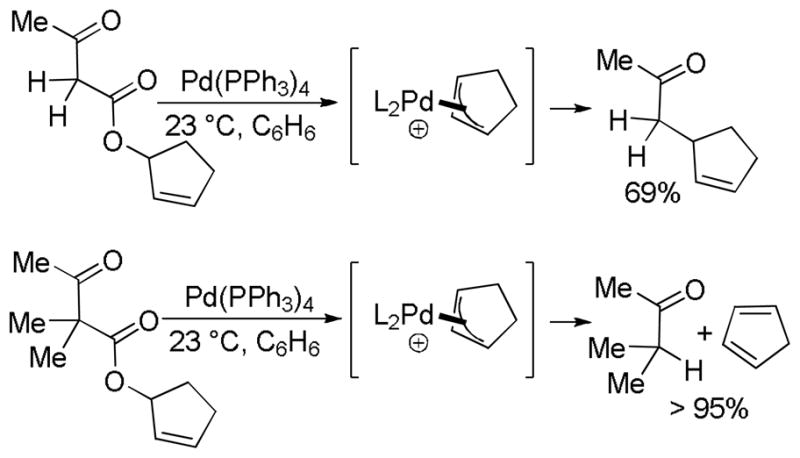
Effect of Substitution on Allylation vs. Elimination
While β-ketoester substrates that contain an α-hydrogen are less prone to competing elimination, they do suffer competing diallylation. For example, allyl acetoacetate undergoes decarboxylative coupling to give a poor yield of the desired monoallylation product due to competing diallylation (eq 2). The diallylation can be thought to result from a combination of Tsuji-Trost allylation of the ketoester followed by decarboxylative allylation. The problematic diallylation is reduced when the substrate is an aryl or cyclic ketone (2,3,5, Chart 1). Alternatively, diallylation can also be mitigated by additional substitution on the allyl electrophile (1, Chart 1).
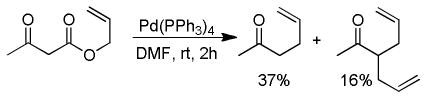 |
(2) |
2.1.2 Ester Enolates
Tsuji and co-workers showed that it was also possible to perform the decarboxylative allylation of malonate derivatives (eq 3);17 however, the reactions were much slower than their ketone counterparts and required heating at or above 100 °C. In doing so, Tsuji also reported the concomitant formation of a byproduct resulting from protonation of the ester enolate; such protonation products are commonly observed byproducts of DcA reactions. Finally, the researchers found that the DcA of α-monoalkylated substrates worked similar to that of the α,α-dialkyl derivative; however they took place at slightly lower temperature (eq 4).
 |
(3) |
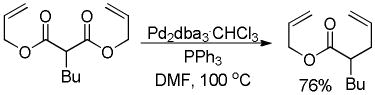 |
(4) |
More recently, Ohata et al. investigated the DcA reactions of α-aryl malonic acid derivatives (Chart 2).18 Unlike Tsuji’s report,17 the DcA of α-phenyl substituted malonic ester derivatives took place readily at room temperature. This highlights the dependence of the rate of DcA reactions on the stability of the incipient enolate; making the enolate benzylic lowers its pKa by ca. 6–7 pKa units, allowing facile decarboxylation. Such observations also suggest that decarboxylation is the rate-limiting step in the DcA of allyl malonic esters. The authors further reported that both monodentate and bidentate phosphine ligands worked well for catalysis, however, no reaction was observed with more electron-deficient phosphite ligands.
Chart 2.

DcA of α-Phenyl Diallyl Malonates
In his synthesis of precursors to carbocyclic nucleoside analogs, Miller showed that allyl trifluoroethyl malonic esters undergo facile decarboxylative allylation even when the α-carbon is not benzylic (eq 5).19 Similarly, Tunge showed that aryl esters undergo decarboxylative allylation under mild conditions (eq 6).16 Once again, the favorable reaction of these substrates can be attributed to the more facile formation of the enolates of these esters, which are less basic than those of alkyl malonates.
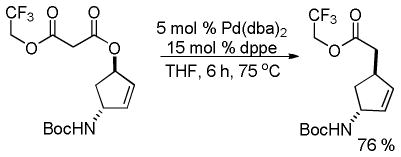 |
(5) |
 |
(6) |
2.1.3 Regioselectivity of Addition to the Allyl Electrophile
Like other related palladium-catalyzed Tsuji-Trost reactions, decarboxylative allylation (DcA) is regioselective, typically affording the linear product as the major regioisomer regardless of the regiochemistry of the reactant (Chart 1). This is easily understood if the reaction proceeds through a common Pd-π-allyl intermediate which reacts selectively with nucleophiles at the less hindered allyl terminus (Scheme 4).
Scheme 4.

Regioconvergent DcA: Evidence for Pd-π-Allyl Intermediates
A significant advantage of a decarboxylative method over more traditional deprotonation and electrophilic trapping of ketone enolates is the ability to regiospecifically generate enolates. Decarboxylation allows the site-specific generation of an enolate at the α-position that bears the CO2 (Scheme 5). Importantly, the nucleophile that is kinetically formed by decarboxylation does not isomerize to the more thermodynamically stable enolate, rather it is rapidly trapped by the allyl electrophile. Thus, the regiospecific generation of enolates via decarboxylation provides a method to access enolates that are difficult to access using acid-base chemistry. In cases such as A (Scheme 5), this regiospecificity is likely the result of the short lifetime of the enolates such that isomerization is not kinetically competitive with allylation of the enolate. In cases such as B, the regiospecificity may reflect the selective allylation of the stabilized β-ketoester enolate prior to decarboxylation (see section 4.2.2).
Scheme 5.
Regiospecific DcA
Shimizu utilized the regiospecificity of decarboxylative coupling to allylate an α-fluoro ketone which proceeds via a fluorinated enolate (eq 7);20 such fluorinated enolates are difficult to generate and alkylate using standard methods.21 Shimizu likewise demonstrated that trifluoromethyl β-keto esters could undergo DcA to give trifluoromethyl ketones (eq 8).22 The tolerance of β-hydrogens on the allyl fragment is noteworthy and may reflect the reduced basicity of the nucleophile which bears the electron-withdrawing CF3 group.
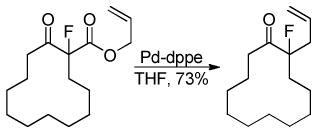 |
(7) |
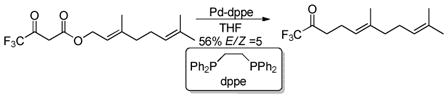 |
(8) |
2.1.4 Regioselectivity in Allylation of Dienolates
Allyl-allyl cross-coupling has historically been accomplished via transmetallation of allyl magnesiates or stannanes with Pd-π-allyl complexes formed by oxidative addition of allyl acetates.23 This type of coupling suffers from low yields and stoichiometric metal salt waste. Tunge et al. recognized that decarboxylation might replace the transmetallation step to allow a new type of catalytic allyl-allyl coupling.24 The decarboxylation of vinyl substituted β-keto esters generates the dienolate which can undergo allylation at either the α- or the γ-carbon to afford regioisomeric products (Chart 3). Indeed, when β-keto ester 6 was subjected to a catalytic amount of Pd(PPh3)4 the authors observed only the formation of the α-allylated product (7) and none of regioisomeric γ-allylated product 8. The reaction establishes that the kinetic product is the α-allylation product and this regioselectivity appears to be quite general (Chart 3). Regiocontrol of the allylation event appears to be dictated primarily by electronics since the bond formation occurs exclusively at the more hindered α-position rather than the less hindered γ-position.
Chart 3.

Hexadiene Synthesis via DcA
Importantly, the γ-allylation isomers can be readily accessed by Cope rearrangement of the kinetic products to the conjugated thermodynamic products (eq 9). Thus, controlled access to either the α- or γ-allylated regioisomers is possible.
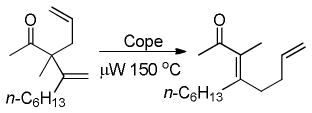 |
(9) |
In an intramolecular decarboxylative allylation, Hiyashi took advantage of the slow cyclization of the zwitterionic intermediate (A, Scheme 6) to promote a γ-selective allylation.25 The allylation also results in dearomatization of the thiophene, however the products could be rearomatized by simply allowing them to stir with alumina. While the reaction did not extend to simple α-phenyl lactones, it did work well for electron rich aromatics (Chart 4). The ease of reaction apparently correlates with the ability to dearomatize the nucleophilic arene.
Scheme 6.

Kinetic γ-Allylation of Valerolactones
Chart 4.

Decarboxylative Cyclization of Valerolactones
2.2 Intermolecular Coupling of β-Keto Acids
2.2.1 Coupling with Allyl Acetates
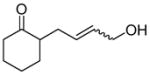
One drawback to the aforementioned decarboxylative coupling methodologies can be the need to preform the ester from the corresponding acid and allyl alcohol. However, Saegusa has shown that the acylation step is not necessary when β-keto acids are utilized as reactants.26 Specifically, he demonstrated the intermolecular coupling of allyl acetates and β-keto acids or β-keto carboxylates (Table 1). The largest limitation to this reaction appears to be the requirement of a substrate β-ketoacid that bears an α-proton; we will return to this subject in the discussion of the mechanisms of decarboxylative allylation. One remarkable difference between what Saegusa observed and the corresponding Tsuji-Trost reaction,27 in which the geometry of the trisubstituted olefin is preserved, is high selectivity for the E-olefin (entries 3–5). Substitution at the 2-position of the allyl (entry 2) and β-hydrogens (entries 3–5) were tolerated while maintaining selective monoallylation (entry 6).
Table 1.
DcA of β-Keto Acids
| entry | β-keto acid | allyl acetate | producta | yield |
|---|---|---|---|---|
| 1 |
 9 |
|
|
93% |
| 2 | 9 |
|
|
71% |
| 3 | 9 |
E-10 |
 11 |
78% |
| 4 | 9 | Z-10 | 11 | 64% |
| 5 | 9 |
|
11 | 83% |
| 6 |
|
|
|
89% |
Pd(PPh3)4, 23 °C, THF or C6H6, 0.5–20 h
2.2.2 Coupling with Vinyl Epoxides
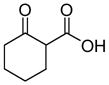
In 1986, Saegusa similarly demonstrated the ability to utilize vinyl epoxides and β-keto acids to facilitate DcA.28 Once again, the substrate scope suggests the requirement of a β-keto acid that bears an α-hydrogen for successful coupling. As will be discussed later (Section 4.2), this has important mechanistic implications. The reaction is remarkable, but in general gives modest yields and some epoxides seem prone to elimination rather than C–C bond formation (entry 5, Table 2).
Table 2.
Intermolecular DcA of β-Keto Acids and Vinyl Epoxides
| entry | β-keto acid | vinyl epoxide | producta | yield |
|---|---|---|---|---|
| 1 |
|
|
|
86% E/Z = 4.5 |
| 2 |
|
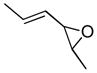
|
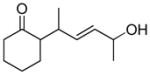
|
59% |
| 3 |
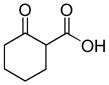
|
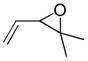
|
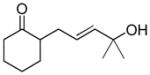
|
54% |
| 4 |
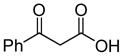
|
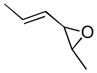
|
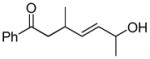
|
67% |
| 5 |
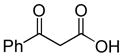
|
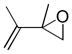
|
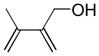
|
27% |
Pd(PPh3)4, 23 °C, THF or C6H6, 0.5–20 h
2.3 Decarboxylative Allylation via Enol Carbonates
A few years after his initial report on decarboxylative coupling of β-ketoesters, Tsuji demonstrated the ability of allyl vinyl carbonates to undergo a decarboxylative allylation to afford identical products to those observed from the DcA of the β-keto esters.29 Some key results are shown in Chart 5. Once again, the reaction is regiospecific, so either regioisomer of the product can be obtained exclusively by use of the appropriate enol carbonate (Scheme 7). However, the regiospecific DcA of enol carbonates requires that one is able to regioselectively generate the enol carbonate precursor via a classic base-induced enolization. Thus, while enol carbonates and β-ketoesters both allow regiospecific allylation, there is some synthetic advantage to the use of β-ketoesters.
Chart 5.

DcA of Allyl Enol Carbonates
Scheme 7.
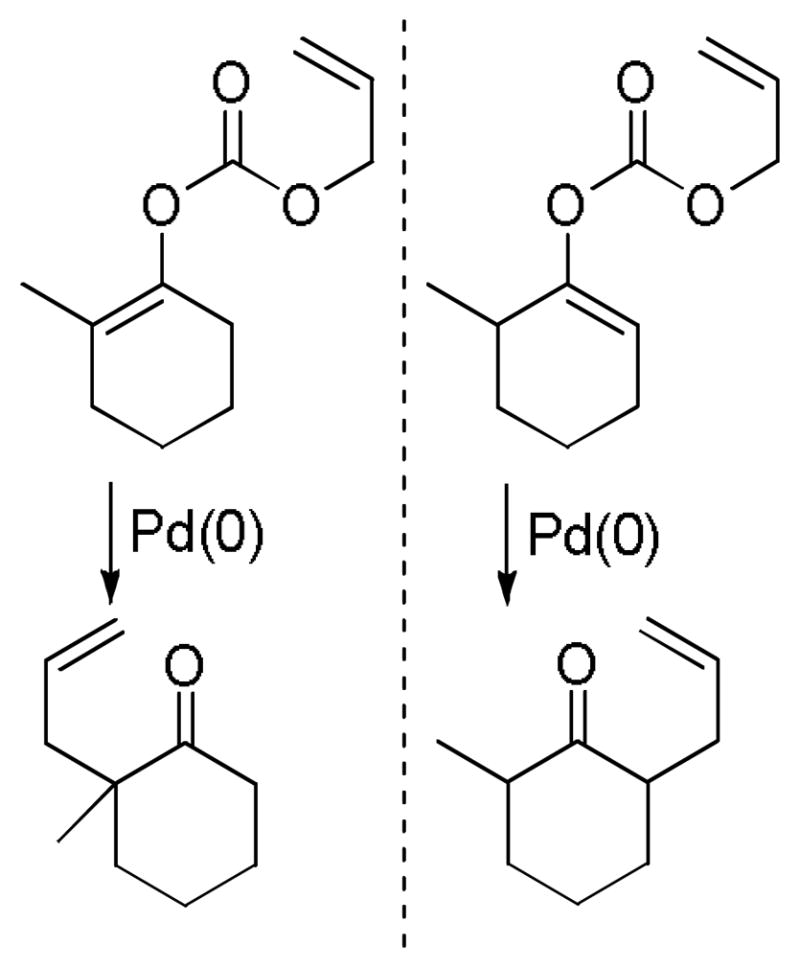
Regiospecificity in the DcA of Allyl Enol Carbonates
Enol carbonate precursors do have some advantages as well. For example, the use of enol carbonates allows the formation of homoallylic aldehydes (Chart 5); aldehyde products have not been prepared from the corresponding β-oxo esters, presumably due to the instability of reactants. Furthermore, Tsuji showed that α-allylation of the dienolate occurs preferentially over γ-allylation. Thus, dienolates generated from either β-ketoesters or enol carbonates undergo kinetic α-allylation rather than γ-allylation and the mild conditions prevent isomerization of the double bond to give the α,β-unsaturated enone.
2.4 Catalysis with Molybdenum, Nickel, and Rhodium
While palladium-based catalysts have been utilized for DcA reactions more widely than any other transition metal catalyst, several other metals have proven to be active catalysts for the decarboxylative allylation of enolates. For example, Tsuji demonstrated that Mo, Ni, and Rh catalysts were capable of facilitating DcA reactions using enol carbonates or β-ketoesters (eqs 10–12).30 To date, the scope of these transformations has not been adequately examined. Nonetheless, these few examples do provide proof that that DcA is not uniquely catalyzed by Pd. The regioselectivities of the reactions with Mo, Ni, and Rh are not well-documented; however the limited information suggests that, in the case of the enol carbonates (eq 10), the intermediate enolate does not isomerize to the more stable enolate under the reaction conditions. Since only unsubstituted allyl esters were allowed to react with Ni and Rh, the regioselectivity about the allyl electrophile cannot be determined. Interestingly, when a crotyl ester was used with the Mo(CO)6 catalyst, the linear product was the major product (eq 12); this result lies in contrast to some Mo-catalyzed allylations of stabilized malonate enolates which favor production of the branched product.31
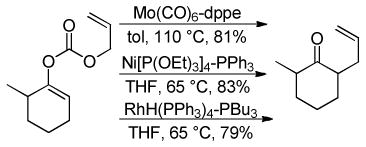 |
(10) |
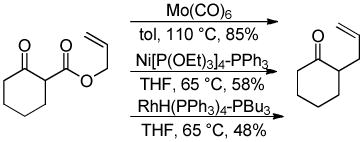 |
(11) |
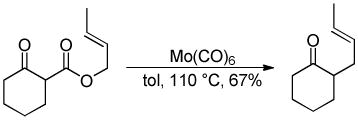 |
(12) |
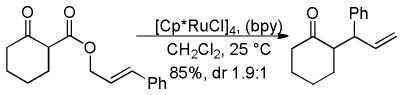
2.5 Catalysis with Ruthenium
2.5.1 Regioselectivity
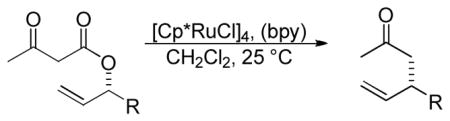
Recently, ruthenium catalysts have also been found to be active catalysts for the DcA of allyl β-ketoesters.4a,32 Specifically, it was found that [Cp*RuCl]4 and bipyridine formed an effective catalyst for decarboxylative allylation (Scheme 8). The bipyridine (bpy) was found to be an essential ligand as it helped form the active monomeric catalyst from the tetrameric precatalyst. Based on NMR spectra of a 1:1 complex of bpy:Ru, and the known coordination of ruthenium,33 the active catalyst was proposed to be a 16-electron Cp*Ru(bpy)+ complex. The reaction is regioselective for reaction of the enolate at the more substituted allyl terminus, as both the linear ester and the branched ester lead to the branched product (Scheme 8). This is easily explained by the existence of a Ru-π-allyl species which preferentially undergoes carbon-carbon bond formation at the more hindered position. This electronically-driven regioselectivity is typical for a reaction between Ru-π-allyl complexes and stabilized nucleophiles.34 While it was initially thought that decarboxylation preceded carbon-carbon bond formation, it is more likely that carbon-carbon bond formation precedes decarboxylation (see section 4.2).
Scheme 8.

Regioselectivity of the Ru-Catalyzed DcA of β-Ketoesters
2.5.2 Scope of the Ru-Catalyzed DcA
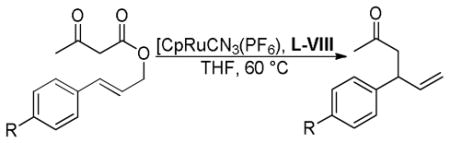
The ruthenium-catalyzed DcA is sensitive to the electronics of the aryl substituent R1, such that electron rich allyl esters undergo DcA faster than electron poor substrates (Table 3). In addition, the rate of reaction is sensitive to the sterics of the aryl substitution (entries 1 vs. 2). Furthermore, the reaction of 1,3-disubstituted substrates occurs but results in lower yield (entry 7). While the majority of substrates that were utilized did not contain α-substitution, the coupling does tolerate substitution at the α-position; however, the resulting product was formed with a low dr (eq 13).
Table 3.
Ru-Catalyzed DcA of β-Keto Esters
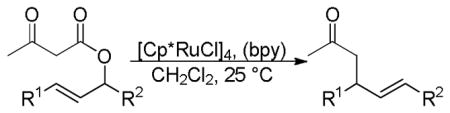 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | time (h) | yield (%) |
| 1 | p-tolyl | H | 2 h | 96 |
| 2 | o-tolyl | H | 120 h | 81 |
| 3 | p-C6H4OMe | H | 0.25 h | 93 |
| 4 | o-C6H4OMe | H | 0.25 h | 91 |
| 5 | p-C6H4Cl | H | 4 h | 96 |
| 6 | p-C6H4CF3 | H | 40 h | 90 |
| 7 | Ph | Ph | 1 h | 67 |
 |
(13) |
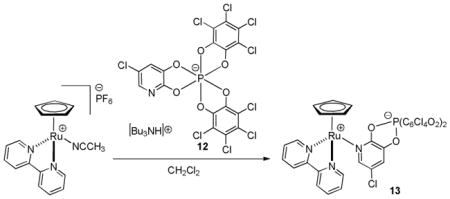
2.5.3 A Recyclable Ruthenium Catalyst
Lacour has made some significant strides in the area of developing recyclable catalysts for DcA reactions.35 One problem with the active ruthenium catalysts that are typically used for DcA reactions is their sensitivity to air and moisture. Lacour found that exchanging the PF6− counterion with the TRISPHAT-N anion 12 gave catalyst 13 which was air stable and even isolable via column chromatography (eq 14).
 |
(14) |
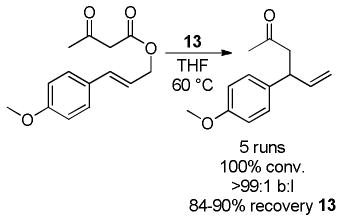
The catalytic compentency of the zwitetterionic Ru-species was tested by subjecting an allyl β-keto ester to DcA reaction conditions (eq 15); at the completion of the reaction the catalyst was recovered and reused. After each run 84–90% of the catalyst was recovered. Importantly, the recovered catalyst gave identical results to the freshly prepared catalyst in subsequent DcA reactions. While catalyst 13 was somewhat less active than the precursor PF6-complex (eq 14), it was found to be superior at higher temperatures. This observation can be attributed to the increased stability of the TRISPHAT-N catalyst complex and its decreased propensity to undergo decomposition.
 |
(15) |
3 Asymmetric DcA of Enolates
3.1 Control of Stereochemistry at the β-Carbon
3.1.1 Enantioselective DcA of β-Ketoesters
While the first disclosures of the formation of homoallylic ketones via Pd-catalyzed DcA appeared in 1980,14–15 the first enantioselective variants did not appear until 2004 when Tunge and Burger were able to use the Trost ligand L-I to effect the enantioselective DcAs of allyl β-ketoesters (Chart 6). In doing so, the authors were able to efficiently control the stereochemistry at the β-position of the homoallylic ketone product.36 As with similar asymmetric allylic alkylations,1d,2i,2k the racemic starting ester affords a meso Pd-allyl complex upon ionization. The Trost ligand introduces a chiral environment and ultimately favors attack of the nucleophile at one of the prochiral allyl termini. Ultimately, the yields of the asymmetric DcA ranged from 69–94% and ee’s ranged from 80–99%, with ee’s increasing with ring size 7>6>5. The enantioselectivity was slightly influenced by the substitution pattern at the remote α-position (14) and was quite sensitive to substitution at the reacting α-position (15, Chart 6). Unfortunately, the reaction proceeded with very little diastereoselectivity (15, dr = 1.5:1).
Chart 6.

The First Enantioselective DcA of Ketones
In addition, by comparison with the same product derived from the Tsuji-Trost reaction with the same ligand, the authors determined the absolute configuration of the DcA product to be R when the Trost ligand L-I was used (Scheme 9). Notably, the enantioselectivity of the DcA is higher than for the two-step Tsuji-Trost/decarboxylation path. The authors attributed this increase in enantioselectivity to the intermediacy of an enolate with a soft metal counterion.37
Scheme 9.

Comparison of DcA and Traditional 2-Step Method
3.1.2 Stereospecific DcA of β-Ketoesters
More recently, Spilling and Yan hoped to take advantage of the double-inversion mechanism for decarboxylative allylation to develop an enantiospecific DcA that would provide access to chiral nonracemic vinyl phosphonates (Table 4).38 As anticipated, the reaction was highly stereospecific, with perfect transfer of the reactant stereochemistry to the product. Unfortunately, competing elimination was problematic for allyl substrates that contained β-hydrogens. While Tsuji has shown that the addition of a base can help to alleviate problematic elimination,18 the authors were able to avoid elimination using a more classical intermolecular Tsuji-Trost allylation of a stabilized enolate.
Table 4.
Stereospecific DcA
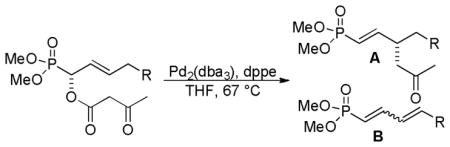 | |||
|---|---|---|---|
| entry | R | yield A | yield B |
| 1 | n-C4H8 | 61 | 27 |
| 2 | H | 71 | 2 |
| 3 | CH(CH3)2 | 35 | 31 |
3.2 Control of α-Stereochemistry
3.2.1 Enantioselective DcA of Allyl Vinyl Carbonates
While Tunge and Burger demonstrated the ability to control the stereochemistry at the β-position of the ketone, Stoltz,39 and later Trost,40 demonstrated the ability to control the stereochemistry at the α-carbon via enantioselective decarboxylative allylation of enol carbonates (Chart 7). In these reactions, the disadvantage associated with the preparation of the precursor enol carbonates is mitigated by the ability to form highly enantioenriched alkylated ketones. Both investigators demonstrated the ability to achieve carbon—carbon bond formation in high yield and in good to excellent enantioselectivity. Stoltz’s method (Conditions A) was used to quaternarize the alpha position while Trost (Conditions B) described formation of both quaternary and tertiary α-stereocenters with no apparent racemization of the tertiary products (17 and 18, Chart 7). Interestingly, Trost notes that when 16 is formed from the lithium enolate, using the same ligand, the opposite stereoisomer is formed. Remarkably, Trost’s method tolerates terminal substitution on the allyl as well as β-hydrogens (19 and 20); as mentioned above, such substrates are often prone to undergo competing elimination. In addition it appears that carbon-carbon bond forming step is slower than π–σ–π allyl isomerization; product 19 is obtained regardless of whether the reactant is the cis- or trans-crotyl ester.
Chart 7.

Enantioselective DcA of Allyl Vinyl Carbonates
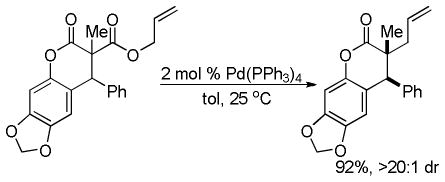
3.2. Enantioselective DcA of β-Ketoesters
The year following that of their initial publication, Stoltz and co-workers demonstrated that the DcA of the isomeric β-keto esters is also highly enantioselective (eq 16).41 Furthermore, the consistent results, whether starting from enol carbonate or isomeric β-ketoester, lend credence to the proposal of a common intermediate.
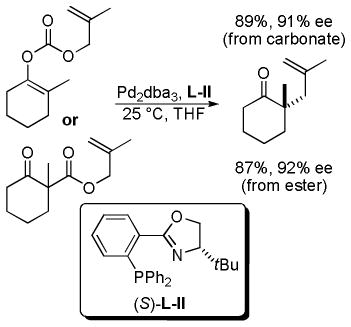 |
(16) |
While the Trost ligands and tert-butyl PHOX ligands are the most generally applicable ligands for the decarboxylative coupling of enol carbonates, ligand modifications can have a significant influence on the enantioselectivity of DcA reactions. For example, Stoltz recently reported the superiority of an electron deficient tert-butyl PHOX derivative, (S)-L-IV, for the asymmetric synthesis of a protected diketone (Scheme 10),42a while the Trost ligand (L-I) provided the product with very low ee. The authors further demonstrated that superior enantioselectivities are obtained when using hexane/toluene solvent mixtures. The authors suggest that both the low polarity solvent and the ligand serve to increase the affinity between the metal center and the enolate and, as a consequence, the ee is improved.
Scheme 10.

Superior ee’s with an Electron Deficient PHOX Ligand
3.2.3 DcA of Vinylogous Ester Derivatives
Trost and co-workers recognized that an asymmetric DcA of cyclic vinylogous esters followed by a Stork-Danheiser addition of an organometallic reagent would give access to α,β-unsaturated enones with a γ-stereocenter, an important motif in the synthesis of a variety of terpenes and alkaloids (eq 17).43
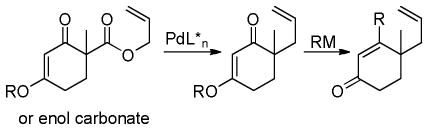 |
(17) |
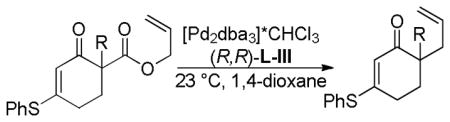
Treatment of the β-keto ester reactant 21 with palladium catalyst and the anthracenyl Trost ligand, (R,R)-L-III, provided the product in high ee, however the reaction was slow and formed product in low yield (Scheme 11). While the carbon backbone of the nucleophilic enolate generated from 21 is similar to cyclic β-keto esters that readily react, the electronics of the enolate are more akin to the corresponding malonates which typically are sluggish reactants for DcA reactions (vida supra).
Scheme 11.
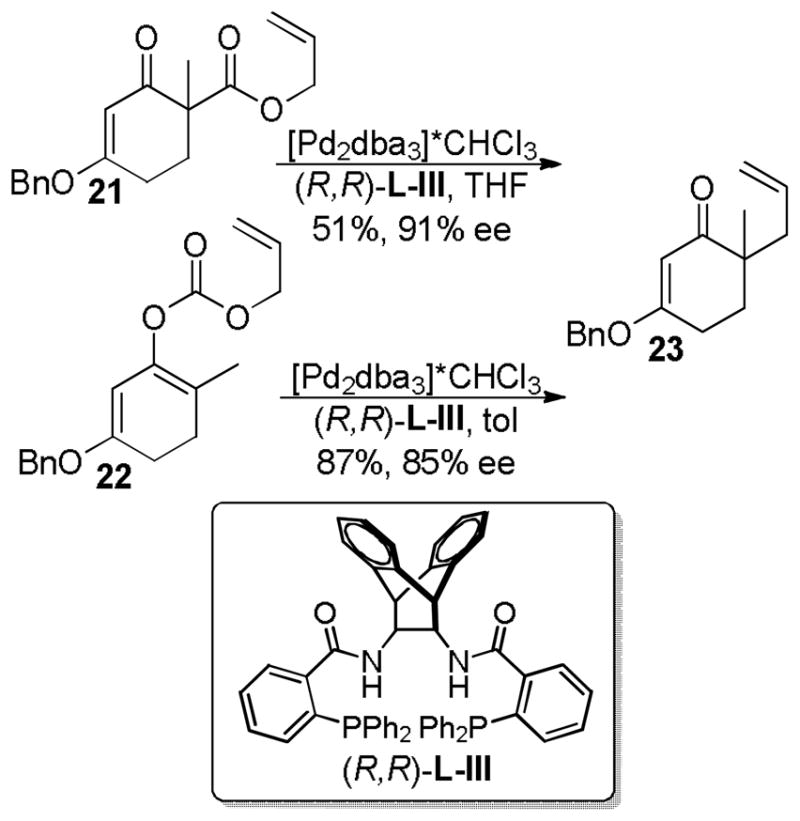
DcA of Vinylogous Ester Enolates
With the goal of developing an asymmetric DcA of cyclic vinylogous esters, allyl enol carbonate 22 was synthesized and subjected to Pd and (R,R)-L-III in toluene and afforded the desired ketone in 87% yield and 85% ee (Scheme 11). The reaction was more facile than that of the β-ketoester, presumably due to the faster decarboxylation of enolcarbonates as compared to malonates. Unfortunately, while the DcA reaction worked well, the synthesis of the requisite enol carbonate (22) was challenging, giving primarily C-carboxylation instead of O-carboxylation.
Trost et al. envisioned that replacing the vinyl ether of the vinylogous ester with a thioether would lead to a β-keto ester that would undergo more facile decarboxylation. Serendipitously, when the oxygen was replaced with a sulfur atom, trapping the enolate gave clean O-carboxylation. Fortunately, these carbonates readily underwent asymmetric DcA (Table 5). Six-membered cyclic enolates gave the highest levels of enantioenrichment, although 5- and 7-membered cyclic enolates also work well (entries 1 vs 2 and 3 vs 4). Interestingly, the allylation was facile and highly selective even when there was a quaternary carbon adjacent to the site of allylation (entry 6). Finally, the Pd-catalyzed asymmetric DcA of vinylogous thioesters also worked for the corresponding isomeric β-keto ester reactants (Table 6), in contrast to the low reactivity of the related oxygen analogs (21, Scheme 11).
Table 5.
Asymmetric DcA of Allyl Vinyl Carbonates Generated from Vinylogous Thioesters
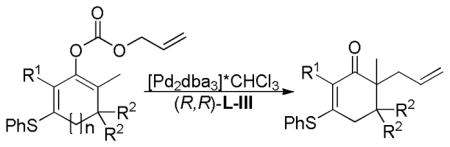 | |||||||
|---|---|---|---|---|---|---|---|
| entry | n | R1 | R2 | solvent | temp (°C) | yield (%) | ee (%) |
| 1 | 1 | H | H | THF | −20 | 100 | 98 |
| 2 | 2 | H | H | THF | 0–4 | 100 | 94 |
| 3 | 1 | Me | H | THF | 0–4 | 100 | 99 |
| 4 | 0 | Me | H | THF | 0–4 | 96 | 80 |
| 5 | 1 | Ph | H | Dioxane | 23 | 100 | 97 |
| 6 | 1 | H | Me | THF | 0–4 | 91 | 79 |
Table 6.
Asymmetric DcA of β-Ketoesters Generated from Vinylogous Thioesters
 | ||||
|---|---|---|---|---|
| entry | R | time (h) | yield (%) | ee (%) |
| 1 | Me | 16 | 75 | 100 |
| 2 | Bn | 16 | 78 | 92 |
| 3 |
|
2 | 98 | 95 |
| 4 | CH2CO2Et | 1 | 80 | 92 |
| 5 | CH2CH2CO2Et | 4 | 90 | 73 |
| 6 | CH2CH2CH2CO2Et | 2 | 86 | 94 |
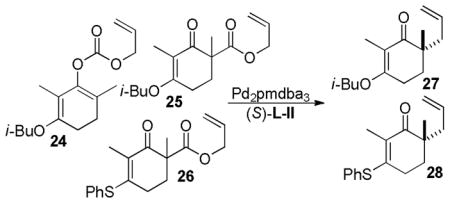
In their effort toward the synthesis of (+)-cassiol, the Stoltz group compared three related vinylogous ester reactants for their ability to form the desired enantioenriched vinylogous esters using the tert-butyl PHOX ligand (L-II) (Table 7).43,44 While the ee of 27 was good when it was derived from the enol carbonate (24), the substrate was found to be unstable in air, complicating its use. When the β-keto ester analog (25) was used, the reaction was sluggish at 50 °C and required elevated temperatures to achieve high conversions (entry 2). Unfortunately, the temperatures needed for high conversion led to lower enantioselectivity (entry 3). Finally, the vinylogous thioester (26, entries 4–7) allowed the reaction to occur at milder temperatures with high enantioselectivity. The authors further reported that the enantioselectivity obtained was rather insensitive to the solvents used (aromatic and ethereal), although the yield of product 28 dropped substantially when benzene was used as the solvent (entry 5).
Table 7.
Asymmetric DcA of Vinylogous Ester Derivatives with PHOX-Ligand
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | solvent | T (°C) | product | yield (%) | ee (%) |
| 1 | 24 | tol | 25 | 27 | 22–61 | 84–88 |
| 2 | 25 | tol | 50 | 27 | 19 | 79 |
| 3 | 25 | tol | 80 | 27 | 86 | 75 |
| 4 | 26 | tol | 50 | 28 | 86 | 92 |
| 5 | 26 | PhH | 50 | 28 | 61 | 92 |
| 6 | 26 | THF | 50 | 28 | 88 | 92 |
| 7 | 26 | dioxane | 50 | 28 | 90 | 91 |
3.2.4 AREA Reaction
Schulz and Blechert developed a DcA reaction that they called an AREA reaction (asymmetric ring expanding allylation).42b In this reaction, allyl carbonates derived from 1,3-hydroxy fused bicyclic systems such as 29 undergo enantioselective Pd-catalyzed ring-opening, ring-expanding allylation to form tertiary and quaternary stereocenters resulting in selectively allylated cyclic 1,4-diketones (30, Chart 8). The reaction is presumed to take place via ionization and decarboxylation to generate the expected alkoxide ion. The intermediate alkoxide undergoes a retro-aldol reaction to form an enolate (A) which undergoes allylation. Importantly, the requisite substrates are readily made from the cyclic β-diketone via a photo induced [2+2] reaction. In general, the AREA reaction works quite well for making quaternary stereocenters, but the enantioselectivity of the reaction is slightly lower when tertiary stereocenters are formed. Furthermore, substrates that are substituted in the 2-position of the allyl electrophile also provide products with diminished ee’s. Ultimately, this methodology is unique in its use of a fragmentation to generate the reactive nucleophile. Such a strategy allows access to enantioenriched medium-ring ketones that are not easily accessed by other methods.
Chart 8.

Select Examples of the AREA Reaction
3.2.5 Asymmetric DcA of α-Fluoro-β-ketoesters
Having been inspired by the seminal work of Shimizu,20 the research groups of Nakamura,45 Tunge,46 and Stoltz41 recognized the potential to control the stereochemistry of α-fluoroketones using DcA reactions. Controlling the α-stereochemistry through a carbon–carbon bond forming reaction could accomplish the equivalent of an asymmetric fluorination of a ketone,47 a challenging transformation. Nakamura detailed the use of the tert-butyl PHOX ligand (L-II), while Tunge compared the PHOX ligand with another P,N-ligand, (S)-QUINAP (L-VI, Chart 9). (S)-QUINAP (Conditions B) provided excellent yields of the allylated fluoroketones, however the tert-butyl PHOX ligand produced higher enantioselectivities in most cases. It is also noteworthy that the two ligands provided access to products of the opposite absolute configuration, with (S)-QUINAP giving rise to the (S)-ketone and (S)-tert-butyl PHOX giving rise to the (R)-ketone.
Chart 9.

Enantioselective DcA of α-Fluoro-β-ketoesters
Paquin has investigated the asymmetric decarboxylative allylation of the related fluorinated enol carbonates.48a Interestingly, the authors reported that the ee’s of the product fluoroketones were highly dependant on the Pd:L ratio. For example, a “traditional” use of a 1.25:1 ligand:Pd ratio provided the product in low ee (eq 18). Alternatively, use of substoichiometric amounts of ligand (L:Pd = 1:4) produced the product with high enantioselectivity. This same effect was not observed in the decarboxylative coupling of fluorinated β-ketoesters, nor was it observed in the allylations of a fluorinated silyl enol ether or an alkyl substituted enol carbonate. Thus, the unusual ligand effect is unique to fluorinated enol carbonates. While it is difficult to explain this behavior, these results do show that the reactions of enol carbonates and β-ketoesters do not always exhibit the same selectivities.
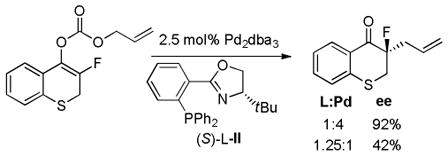 |
(18) |
With regard to the decarboxylative allylation of fluorinated β-ketoesters (Chart 9), it is noteworthy that acyclic products are significantly less enantioenriched than their cyclic counterparts. This limitation is common among decarboxylative methods that generate enolates from acyclic ketones. The ee is proportional to both the facial selectivity as well as the E/Z ratio of enolates formed, the lower enantioenrichments observed for acyclic ketones may be attributed to the formation of an imperfect mixture of enolate geometries from such substrates.
3.3 Acyclic Stereocontrol
The low enantioselectivity of DcA reactions of acyclicβ-keto esters is a general problem and is not limited to α-fluoro substrates. The apparent mixture of enolate geometries generated via decarboxylation is a general problem for DcA reactions, however a few creative solutions to this problem have emerged. Among the simplest solutions is the appropriate placement of functional groups in the ester to favor a single enolate geometry. Another, perhaps more general solution is the use of allyl enol carbonate reactants with predefined enolate geometry.
3.3.1 α-Acetamido-β-Ketoesters
Kuwano and Murakami have demonstrated that acyclic α-acetamido-β-keto esters can lead to enantioenriched γ,δ-unsaturated ketones (eq 19).48b The authors did not investigate scope of the DcA, nor did they explicitly comment on why the enantioselectivity is higher for α-acetamido β-ketoesters than for α-alkyl and α-fluoro ketoesters. That said, it is possible that the amido group in combination with the naphthol additive helps to favor a single enolate geometry via hydrogen bonding; the enantioselectivity was much lower (51% ee) without the naphthol additive.
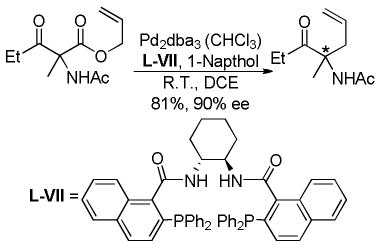 |
(19) |
3.3.2 Acyclic Allyl Enol Carbonates
Trost has offered a more general solution to the problem of acyclic control of α-stereochemistry.40b Use of preformed enol carbonates gives a handle on the geometry of the enolate that is not present when starting from the acyclic β-keto esters. Indeed, an E-enol carbonate (E-31) smoothly underwent DcA to afford the homoallylic ketone in high yield and excellent enantioenrichment (Scheme 12). When the isomeric Z-enol carbonate was allowed to react under the same conditions, the opposite enantiomer of the product (32) was obtained. However, the reaction of the Z-enol carbonate proceeds more slowly and with lower enantioselectivity than the analogous reaction with the E-enol carbonate. This indicates that the E-isomer is matched with the catalyst while the Z-isomer is mismatched. This is not general for all acyclic carbonates, but seems to be the case with carbonates derived from dialkyl ketones.
Scheme 12.
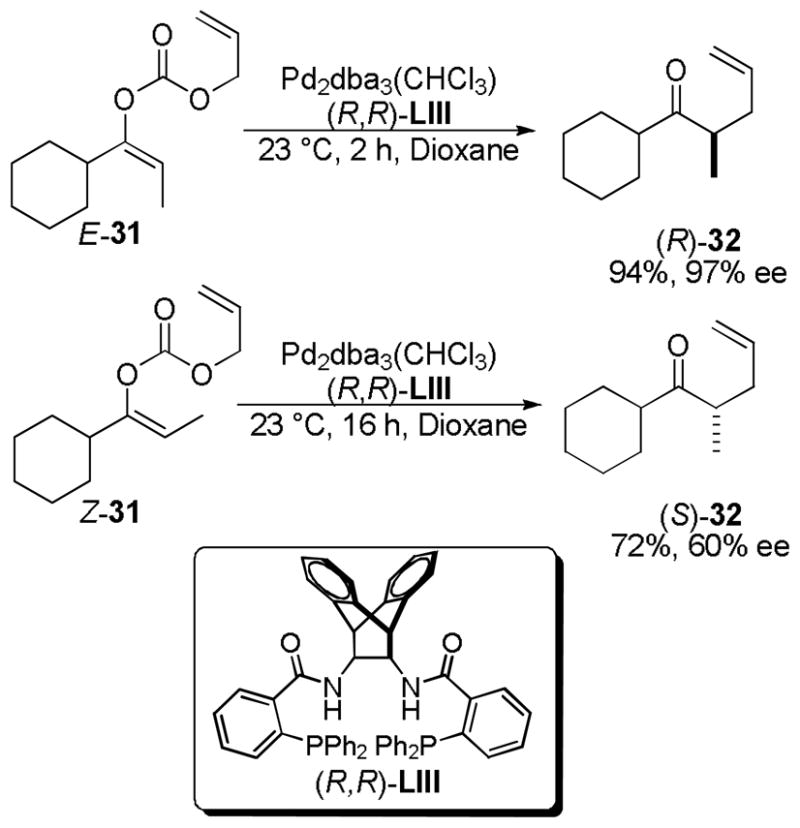
Matched/Mismatched Asymmetric DcA
While the Z-enol carbonates of alkyl ketones provide relatively low enantioselectivites, Z-enol carbonates that are derived from a phenyl ketone are excellent substrates for asymmetric DcA reactions (Table 8). The reaction is general for a variety of α-alkyl substituents (entries 2–4), but the enantioselectivity is somewhat sensitive to the steric nature of the α-substituent; when an isopropyl group was used, the reaction became very sluggish and the selectivity dropped dramatically (entry 5, Table 8).
Table 8.
Enantioselective DcA of Z-Enol Carbonates
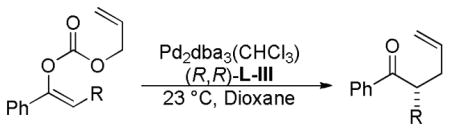 | ||||
|---|---|---|---|---|
| entry | R | time (h) | yield (%) | ee (%) |
| 1 | Me | 3 | 96 | 94 |
| 2 | Et | 2 | 94 | 94 |
| 3 | n-Pent | 16 | 93 | 92 |
| 4 | Bn | 1 | 75 | 88 |
| 5 | i-Pr | 24 | 30 | 32 |
Interestingly, the reaction with the Z-enol carbonates was quite tolerant of substitution of the phenyl ring (Chart 10). The major exception was the mesityl derived substrate which was unreactive (34). The chemoselectivity was also noteworthy, allowing the DcA to take place while a pendant aryl bromide was unscathed (33).
Chart 10.

Enantioselective DcA of Z-Enol Carbonates
Finally, variation of the hybridization of the enol carbonate substituent revealed substantial differences in reactivity and selectivity. Specifically, the Z-enol carbonates underwent efficient asymmetric DcA reactions for sp2-hybridized ketone substituents (35, Chart 10), but led to poor ee’s if the substituent was sp3-hybridized (36); however, the DcA is well-suited for the analogous E-enol carbonate (36′). An ynone provided product in good yield and moderate enantiomeric excess (37).
3.3.3 DcA of 2-Acyl Imidazole Derivatives
More recently, Trost has circumvented the sluggish transformations of ester enolate derivatives by utilizing 2-acyl imidazoles as surrogates for ester enolates.49 The increased electrophilicity of the 2-acyl ketone, compared to the carbonyl of the ester, makes the DcA of the corresponding enol carbonates more facile. Indeed, such reactions took place at ambient temperatures, affording products in high yields and ees (Chart 11). Notably, the DcA with cyclic allyl esters allowed the formation of adjacent carbon stereocenters with high enantio- and diastereoselectivity.
Chart 11.

Enantioselective DcA of 2-Acyl Imidazoles
Having established that the 2-acyl imidazole derivatives undergo highly enantioselective DcA, the authors next demonstrated that they could easily access acids, esters, amides, and ketones in high ee’s from the acyl imidazole products. Using methyl triflate to activate the imidazole toward nucleophilic acyl substitution, the allylated ketone was converted into cetiedil, a compound that is used clinically for the treatment of vascular disease in its racemic form (eq 20).
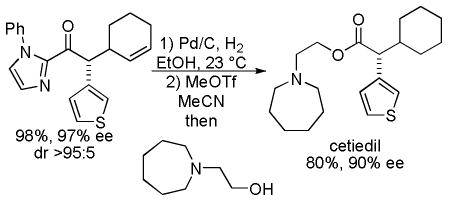 |
(20) |
3.3.4 Protected α-Hydroxy Allyl Vinyl Carbonates
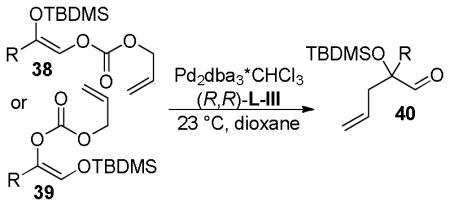
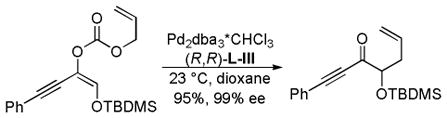
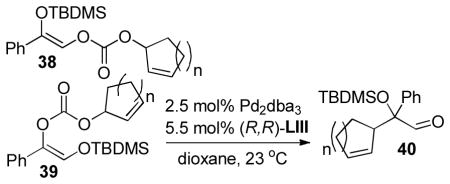
In 2007, Trost and co-workers published an interesting twist on DcA reactions that involved regioconvergent, enantioselective generation of α-tertiary hydroxy aldehydes from protected α-hydroxy enol carbonates (path A, Scheme 13).50 This report was shortly followed by a related demonstration of the dramatic ability of the ligand to alter the outcome of the DcA, such that the reaction was regiospecific rather than regioconvergent (paths B and C, Scheme 13).51 Specifically, use of the naphthyl-Trost ligand (R,R)-L-VII and a pivoyl protecting group led to a regiospecific DcA. Thus, either α-hydroxy aldehydes (40) or protected α-hydroxy ketones (41) were accessible in high yield and enantiomeric excess via regiocontrolled synthesis of enol carbonates 38 and 39.
Scheme 13.

Diverse Reactivity of Protected Hydroxy Ketones
In the initial report on regioconvergent DcA reactions, Trost et al. disclosed conditions for the formation of protected α-hydroxy homoallylic aldehydes from either of the two isomeric protected α-hydroxy enol carbonates (38 and 39, path A, Scheme 13).50 The fact that two regioisomeric reactants led to a single product (40) implied that the reaction was a convergent process where equilibration of the intermediate enolates A and B occurred more rapidly than C–C bond formation (k1+k−1 > k2,k3; Scheme 14). Furthermore, the formation of 40 from either A or B indicates that allylation preferably occurs through intermediate A (Scheme 14).
Scheme 14.

Regioconvergent, Enantioselective DcA of α-Hydroxy Aldehydes
Studying the effect of the hydroxy protecting group on the regioconvergent DcA showed that silyl groups tend to allow more isomerization, while carbonyl-based protecting groups slow the transfer process (Table 9). The effect of the ligand was perhaps more surprising. When dppe was used as opposed to the Trost ligand [(R,R)-L-III], the ketone 41 was the major product, regardless of which starting isomer was used (Table 9). These results suggest that the ligand plays an intimate role in controlling the relative rates of allylation (k2, k3, Scheme 14) and/or the favored enolate isomer (A or B, Scheme 14).
Table 9.
Influence of the Ligand on the Regioselectivity
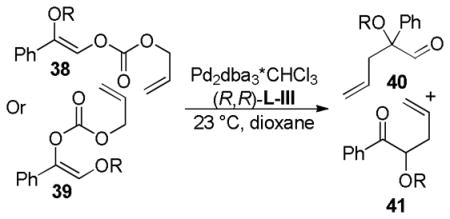 | ||||
|---|---|---|---|---|
| entry | SM | R | yield 40 | yield 41 |
| 1 | 38 | TBDMS | 93 | 0 |
| 2 | 39 | TBDMS | 86 | 0 |
| 3 | 38 | Benzoyl | 93 | 0 |
| 4 | 39 | Benzoyl | 11 | 75 (dppe) |
| 5 | 38 | Acetyl | 40 | 60 |
Using the TBDMS protecting group to facilitate enolate isomerization, the scope of the regioconvergent DcA was investigated (Table 10). As expected, use of either isomeric starting materials 38 or 39 led to the aldehydes 40 in similar yields and ee’s. The asymmetric allylation worked well with substrates derived from aromatic ketones (entries 1–7, Table 10) as well as enones and ynones (entries 8,9).
Table 10.
Scope of Protected α-Hydroxy Aldehydes
 | ||||
|---|---|---|---|---|
| entry | SM | R | yield (%) | ee (%) |
| 1 | 38 | Ph | 93 | 92 |
| 2 | 39 | Ph | 89 | 91 |
| 3 | 38 | p-MeOC6H4 | 94 | 92 |
| 4 | 39 | p-MeOC6H4 | 86 | 92 |
| 5 | 38 | o-NO2C6H4 | 69 | 79 |
| 6 | 39 | o-NO2C6H4 | 69 | 72 |
| 7 | 38 | 2-Furyl | 81 | 93 |
| 8 | 38 | 1-cyclohexenyl | 93 | 98 |
| 9 | 38 | PhCC | 76 | 89 |
As expected for a reaction involving intramolecular transfer of the protecting group, the geometry of the enol carbonate is important (eq 21). For example, when the protected α-hydroxy substitutent is trans to the enolate oxygen, allylation is faster than silyl transfer and the allylated ketone is the only product formed.
 |
(21) |
In addition to allylations with simple allyl electrophiles, a number of cyclic allyl carbonates were also tested for their ability to undergo asymmetric regioconvergent DcA reactions (Table 11). The reaction appears to be remarkably tolerant of allyls that can undergo elimination. The products were all formed with high enantioselectivity, however, the cyclopentenyl allyl electrophile provided the product with poor dr (2.5:1). In contrast, the cyclohexenyl and cycloheptenyl carbonates formed product with good dr (entries 2–5). While both isomeric carbonates (38 and 39) underwent the DcA reaction, the authors did note that the reactions that utilized the internal carbonate (39) were sluggish.
Table 11.
DcA of Protected α-Hydroxy Allyl Vinyl Carbonates
 | |||||
|---|---|---|---|---|---|
| entry | SM | n | yield (%) | dr | ee (major) |
| 1 | 39 | 1 | quant. | 2.5:1 | 92% |
| 2 | 38 | 2 | quant. | 11:1 | >99% |
| 3 | 39 | 2 | quant. | 11:1 | >99% |
| 4 | 38 | 3 | quant. | 50:1 | >99% |
| 5 | 39 | 3 | 30% | 50:1 | >99% |
Given the ability of the dppe ligand to alter the regiochemical outcome of the reaction of protected α-hydroxy enol carbonates, it is not surprising that Trost followed up his initial report with a study that detailed conditions that prevented enolate isomerization and allowed synthesis of the isomeric ketone products (Chart 12).51 Screening of reaction conditions showed that: 1) (R,R)-L-III and the iPr-PHOX ligand (L-V) tended to favor the formation of aldehyde via isomerization and 2) use of esters as protecting groups tended to slow transfer of the protecting group relative to silyl groups. Thus, optimal conditions for regiospecificity and enantioselectivity utilized acetyl or pivoyl protecting groups, naphthyl Trost ligand [(R,R)-L-VII], and DME as the solvent.
Chart 12.

Scope of DcA of α-Protected Hydroxy Allyl Vinyl Carbonates
In general the DcA of the protected α-hydroxyl allyl vinyl carbonates proceeded with good to excellent yields and ee’s (Chart 12). However, substrates with an sp3-hybridized carbon in the R1 position provide lower yields and ee’s. Aryl ketones are particularly well suited for the DcA reaction, regardless of whether an acetyl or pivoyl protecting group was used. Cyclic allyl electrophiles are also well suited for the allylation of aryl ketones and provided products with high enantio- and diastereoselectivity. Furthermore, acrylate protecting groups could be used without significantly affecting the yield or ee.
3.4 Asymmetric DcA Using Metals other than Palladium
3.4.1 Ru-Catalyzed Stereospecific DcA
After establishing that Ru could facilitate the DcA of allyl β-ketoesters,32 Burger and Tunge turned their attention to the development of an asymmetric synthesis of branched γ,δ-unsaturated ketones.52 Believing that it might be possible to take advantage of a slow π–σ–π epimerization of the Ru-allyl intermediate,53 chiral nonracemic β-keto esters were subjected to the reaction conditions for decarboxylative allylation. Indeed, good levels of stereochemical fidelity (conservation of enantioenrichment = cee = 100 × ee product/ee reactant) were observed, giving access to enantioenriched homoallylic ketones that contain a β-stereocenter (Table 12). The reaction does, however, proceed with some racemization. The authors noted that the use of TMEDA as a ligand, as opposed to bipyridine, led to increase amounts of racemization.
Table 12.
Ru-Catalyzed Stereospecific DcA of β-Ketoesters
 | ||||
|---|---|---|---|---|
| entry | R | time (h) | cee % | yield (%) |
| 1 | Ph | 1.5 h | 83 | 86 |
| 2 | p-tolyl | 2 h | 87 | 81 |
| 3 | p-C6H4OMe | 0.25 h | 93 | 83 |
| 4 | p-C6H4Cl | 0.5 h | 94 | 56 |
| 5 | p-C6H4Cl | 4 h | 86 | 70 |
| 6 | p-C6H4NO2 | 3 h | 98 | 49 |
Importantly, it was recognized that the cee was dependent on the conversion to product for slow-reacting substrates (entries 4 vs. 5). Closer investigation showed that after three hours reaction time, the p-nitrophenyl derivative (entry 6) had reached complete conversion, forming the desired product 44 in high ee as well as the linear ester 43. Prolonged reaction resulted in the DcA of achiral ester 43, which necessarily produced racemic product (Scheme 15). Thus, higher conversion to product resulted in lower enantiospecificity. Ultimately, the imperfect stereofidelity of the reaction was attributed to formation and DcA of the achiral linear isomer 43 and not π–σ–π isomerization.
Scheme 15.

Mechanism of Racemization
3.4.2 Ru-Catalyzed Enantioselective DcA
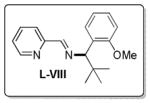
A few years later Lacour and co-workers developed an enantioselective ruthenium-catalyzed DcA that allowed the more accessible linear esters to undergo an asymmetric conversion to the branched homoallylic ketones (Table 13).54 While the DcAs of only three linear esters were investigated, the enantioselectivity appears to be dependent on the substitution of the aryl ring. The more electron rich, methoxy-substituted cinnamyl ester derivative gave the highest enantioenrichment (entry 1, Table 13), while the electron deficient p-chloro cinnamyl ester gave the lowest ee (entry 3). As previously noted by Tunge,32 the electronics of the allyl moiety also significantly affect the reaction time, so the electron deficient ester only reached 75% conversion after 120 h.
Table 13.
Ru-Catalyzed Entantioselective DcA of β-Ketoesters
 | |||||
|---|---|---|---|---|---|
| entry | R | ee % | time (h) | conv. (%) | b:l |
| 1 | OMe | 80 | 24 h | 100 | >99:1 |
| 2 | H | 74 | 24 h | 100 | 94:6 |
| 3 | Cl | 66 | 120 h | 75 | 95:5 |
| |||||
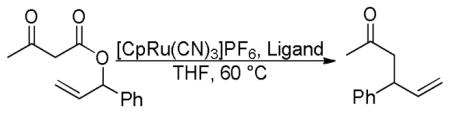
Lacour et al. also briefly investigated the effect of optically active catalysts on the stereospecific DcA that was previously demonstrated by Burger and Tunge.54,52 Interestingly, Lacour found that the conservation of enantioenrichment was dependent on the ligand. For example when bipyridine was used as the ligand, the stereochemical fidelity of the DcA was low (entries 1 and 2, Table 14), while use of the unsymmetrical achiral ligand L-IX led to product with higher cee (entries 3 and 4). When chiral ligand L-X was used with the S-ester the product with the same sign was obtained in 84% ee, however, if the R-ester was used with the same ligand, then the R-product was produced in only 68% ee (entries 5 and 6). Likewise, the chiral ligand L-VIII demonstrated similar behavior when used (entries 7 and 8). These results are most readily explained by a combination of enantiospecific and enantioselective allylations. To better explain, it helps to refer back to Scheme 15. Enantiospecific DcA of the (S)-ester will produce enantioenriched product (S)-44 as well as linear ketoester 43. If the enantioselective DcA of 43 produces the S-product (i.e. with L-X, entries 5 and 7), this reinforces the enantiospecific DcA. In contrast, the enantioselective production of (S)-44 will deteriorate the ee of the product derived from R-ester (entries 6 and 8).
Table 14.
Enantiospecific DcA of β-Ketoesters
 | ||||||
|---|---|---|---|---|---|---|
| entry | ester | ligand | ee (%) | time (h) | prod. | b:l |
| 1 | (S) | bpy | 48 | 2 h | (+), S | 94:6 |
| 2 | (R) | bpy | 46 | 2 h | (−), R | 93:7 |
| 3 | (S) | L-IX | 72 | 6 h | (+), S | 94:6 |
| 4 | (R) | L-IX | 72 | 6 h | (−), R | 94:6 |
| 5 | (S) | L-X | 84 | 10 h | (+), S | 92:8 |
| 6 | (R) | L-X | 68 | 6 h | (−), R | 92:8 |
| 7 | (S) | L-VIII | 92 | 10 h | (+), S | 93:7 |
| 8 | (R) | L-VIII | 70 | 6 h | (−), R | >99:1 |
| ||||||
3.4.3 Ir-Catalyzed Enantioselective DcA
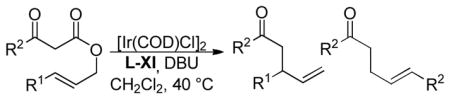
In 2007, You et al. published a report detailing the branched-selective, enantioselective DcA of β-keto esters (Table 15).55 This methodology appears to be superior to the ruthenium-catalyzed DcA in several ways. First, the ee’s are uniformly high (89–96%) and, in contrast to the ruthenium variant, appears to be insensitive to the electronics of the allyl fragment. In addition, for all aryl substituted allyls (entries 1–6, Table 15) the branched to linear selectivity was greater than 98:2. Furthermore, this method was tolerant of β-hydrogens on the allyl (entries 7 and 8) which has not been shown to be the case with any of the ruthenium-catalyzed methods. The reaction of alkyl substituted allyls did, however, take place with diminished branched to linear ratios as well as yields (entries 7 and 8). One drawback of the iridium-catalyzed DcA was the need to use added DBU as a base. Without the base, both the yield and the enantioenrichment are reduced. The role of the base was not completely understood, but the authors noted that similar results could be achieved by the use of several weak bases other than DBU. As shown by Hartwig, one role of the base is to facilitate formation of an active metallacyclic iridium complex from phosphoramidite ligand L-XI.56 Another possible role of the base is to deprotonate the ketoester, forming a stabilized enolate that can undergo iridium-catalyzed Tsuji-Trost type allylation prior to decarboxylation.56 The next section of this review will address the likelyhood of such alternate reaction mechanisms for DcA.
Table 15.
Ir-Catalyzed Enantioselective DcA of β-Ketoesters
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | ee (%) | time (h) | yield (%) | b:l |
| 1 | 4-MeOC6H4 | Ph | 95 | 12 h | 70 | >99:1 |
| 2 | Ph | Ph | 95 | 16 h | 83 | 99:1 |
| 3 | 4-CF3C6H4 | Ph | 91 | 17 h | 71 | >99:1 |
| 4 | 2-Furyl | Ph | 94 | 3 h | 73 | 98:2 |
| 5 | Ph | 2-napthyl | 93 | 16 h | 71 | 98:2 |
| 6 | Ph | 4-MeOC6H4 | 95 | 4 h | 67 | 99:1 |
| 7 | Me | Ph | 90 | 21 h | 61 | 94:6 |
| 8 | n-C5H11 | Ph | 89 | 22 h | 52 | 80:20 |
| ||||||
4 Mechanistic Aspects of the DcA of Enolates
As alluded to several times throughout this review, there are several feasible mechanisms that have been proposed for decarboxylative allylation reactions. In this section, it is our goal to summarize the known mechanistic information and to provide general conclusions. However, it is apparent that the mechanism of decarboxylation can change based on substrate and it is also expected that simple changes to the reaction conditions or ligands may also result in changes in mechanism. For those reasons, the mechanistic features that are characteristic of PPh3-ligated palladium may not be identical to those of palladium ligated by the Trost ligand, PHOX ligand, or any other ligand. Similar caution must be used in applying the mechanistic knowledge of palladium-catalyzed DcA reactions to the less studied molybdenum, nickel, ruthenium, and rhodium-catalyzed DcA reactions.
4.1 Ionization/Oxidative Addition
While there are several different mechanisms that are possible for palladium-catalyzed DcA reactions, all proposals begin with palladium-induced ionization of the allyl carboxylate or carbonate, similar to that proposed for allyl acetates in Tsuji-Trost chemistry.1d,2i,2k The ionization likely produces a π-allyl palladium carboxylate ion-pair that is in equilibrium with the neutral σ-allyl complex (Scheme 16). When Ln is a bidentate ligand, as is the case with most asymmetric allylation catalysts, the binding of the carboxylate requires the slippage of the π-allyl ligand to a σ-allyl binding mode in order to preserve the preferred 16 e-, square-planar geometry about palladium. The Stoltz group has characterized such a (σ-allyl)palladium-β-ketocarboxylate intermediate and identified it as the resting state for the catalytic cycle mediated by a Pd(PHOX) complex.57
Scheme 16.
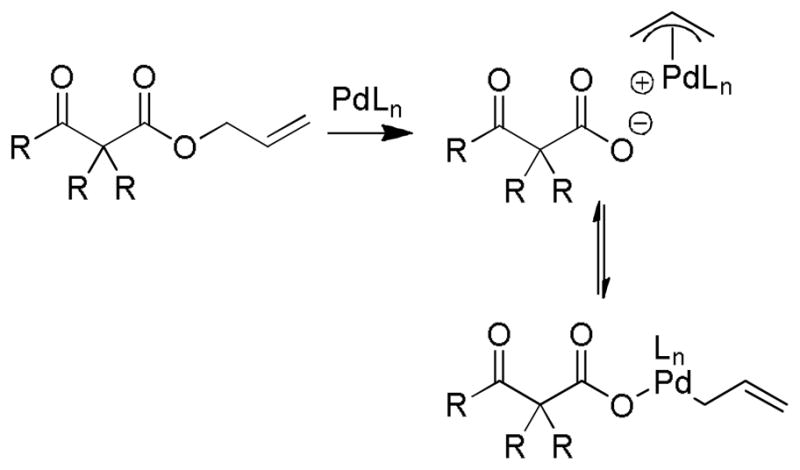
Ionization of Allyl Carboxylates
While the (allyl)palladium carboxylate intermediates have several potential binding modes, the main mechanistic classes for decarboxylative allylation are really defined by the answers to two fundamental questions: 1) Does decarboxylation precede allylation, or does allylation precede decarboxylation? and 2) Does allylation occur through an inner-sphere mechanism where the enolate is bound to palladium prior to reductive elimination or via an outer-sphere mechanism where the enolate directly attacks the allyl ligand?
4.2 Which Comes First, Allylation or Decarboxylation?
4.2.1 The Case of α,α-Disubstituted Esters
The former question can be answered definitively in several specific classes of β-oxoesters. Specifically, α,α-disubstituted-β-oxoesters must undergo decarboxylation prior to allylation (Scheme 17). In this class of compounds decarboxylation is necessary to form a reactive site for allylation. Decarboxylation of the intermediate palladium allyl carboxylate leads to a palladium enolate species. Like the carboxylates, the palladium enolate can potentially exist as an ion-pair or a neutral O- or C-bound enolate. Allylation of this enolate produces the product.
Scheme 17.
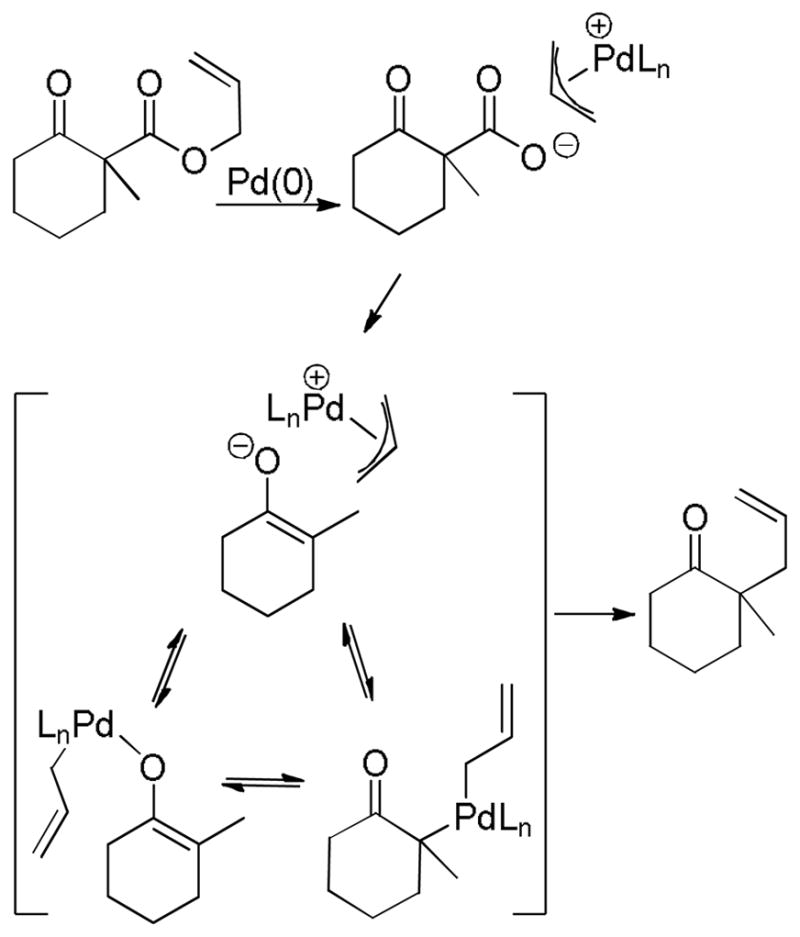
Decarboxylation Prior to Allylation
4.2.2 The Case of Substrates that Possess an α-Hydrogen
In the case of β-oxoesters that bear α-hydrogens, a new mechanistic possibility arises. In these cases, the intermediate carboxylate can undergo proton transfer to form the stabilized enolate (A, Scheme 18). This enolate can undergo Tsuji-Trost allylation followed by decarboxylation of the β-oxo acid to form product. Evidence that this path is occurring is provided by: stereochemical studies by Tunge that suggest that the α-stereochemistry of the allylation is determined by decarboxylative protonation of the β-oxo acid intermediate,16 the observation of β-oxo acid intermediates in the decarboxylative couplings of dihydrocoumarins (Scheme 19),16 and the formation of diallylated byproducts in decarboxylative allylations (Scheme 18).14 Interestingly, Tsuji showed that, when decarboxylative allylation was performed in the presence of methyl acetoacetate, the external acetoacetate was not allylated (Scheme 18).14 This suggests that the intramolecular proton transfer from the α-position (pKa ~ 14 in DMSO) to the carboxylate (pKa ~ 13 in DMSO) is faster than intermolecular proton transfer. While less is known about the mechanisms of catalysis with metals other than palladium, Tunge observed a similar result with a ruthenium catalyst, suggesting a similar mechanism.32
Scheme 18.

Allylation Before Decarboxylation
Scheme 19.

Evidence for Allylation Prior to Decarboxylation
Ultimately, while it is possible for β-oxoesters that have α-hydrogens to react via a mechanism where decarboxylation precedes allylation, the preponderance of the evidence at this point suggests that decarboxylation of such substrates primarily occurs after allylation. This proposal also readily explains why substrates that contain α-hydrogens are much less likely to undergo competing elimination reactions (see Table 1); such substrates react via less basic, stabilized enolates that are less prone to promote elimination. In addition the proposal is fitting with the results of Fiaud,58a Bäckvall,58b Saegusa,26 Spilling,38 and Tunge36 that show that such substrates undergo decarboxylative allylation through a double-inversion mechanism with outer-sphere attack of the stabilized nucleophile on the allyl ligand (Scheme 20). The somewhat lower stereospecificities reported in the examples in Scheme 20 could be attributed to competing inner-sphere reductive elimination, however, epimerization of the reactant has been implicated in similar reactions.58
Scheme 20.

Evidence for an Outer-sphere Allylation
4.2.3 The Case of Allyl Vinyl Carbonates
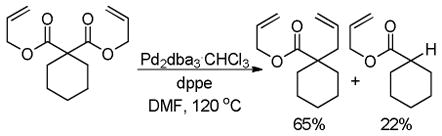
Whether enol carbonates undergo decarboxylation prior to, or after allylation remains an unanswered question. Since enol carbonates are expected to decarboxylate more readily than their β-ketocarboxylate counterparts, most researchers have assumed that decarboxylation precedes allylation. With respect to the Trost ligand-supported palladium catalyst, it has been suggested that the decarboxylation is probably rate-limiting and must occur with assistance from Pd; however, the authors offer no support for the latter conclusion and, to our knowledge, definitive experiments have not been conducted.40b One result that suggests that allylation before decarboxylation may be possible is the observation of diallylated products from the DcA of enol carbonates (Scheme 21); the observation of similar products in the DcA of β-ketoesters has been used as evidence for allylation prior to decarboxylation. Diallylated products could also arise from deprotonation and allylation of the product mono-allylated ketone, however such intermolecular proton transfers have not been previously observed, and the allyl ketone product (45) was not significantly racemized as would be expected if it were possible to deprotonate it under the reaction conditions.
Scheme 21.

Hypothetical Allylation Prior to Decarboxylation
4.3 Decarboxylation: the Birth of a Nucleophile
Decarboxylation is the key step in decarboxylative coupling and is believed to be the rate-limiting step in the decarboxylative allylation of β-oxo esters as well as enol carbonates (with Trost ligand/Pd). Unfortunately, little is known with regard to this all-important elementary step. Saegusa showed that palladium was necessary to catalyze decarboxylation of a sodium β-ketocarboxylate,15 so palladium plays a critical role in the decarboxylation. It has been suggested that the “softness” of palladium, which leads to facile ionization of Pd-carboxylate complexes, is important in facilitating decarboxylation.18 While the intimate details of decarboxylation have not been studied using Pd, we can infer possible mechanisms based on other known mechanisms for decarboxylation. For example, one is tempted to suggest decarboxylation via mechanism A (Scheme 22) that is akin to the decarboxylation of β-keto acids.59 Such a suggestion is fitting with the relative insensitivity of DcA reactions to different solvents. However, Darensbourg has investigated the catalytic decarboxylation of malonic acids with metals including Cu(I) and Zn(II) and shown that ionization of the metal carboxylate bond facilitates decarboxylation.60 His conclusion that softer metals promote faster decarboxylation is in line with Tsuji’s hypothesis. This would indicate decarboxylation by mechanism C, which closely resembles the mechanism of decarboxylation of other β-keto carboxylates.61 Moreover, Darensbourg suggests that coordination of the ketone oxygen may facilitate decarboxylation by allowing the formation of M-bound enolate as opposed to a “free” enolate (D).13,60 Applying these observations to palladium-catalyzed decarboxylation of β-oxo esters suggests that an ionic mechanism for decarboxylation is favored over the cyclic transition state favored for decarboxylation of β-keto acids. While ionic mechanisms may not seem favorable for decarboxylations in non-polar solvents, it is known that non-polar solvents increase the rates of related decarboxylations.59,62 Moreover, it is well-known that palladium allyl carboxylates can exist as ion-pairs even in non-polar solvents.1d,2i,2k
Scheme 22.

Potential Mechanisms for Decarboxylation
As mentioned, the decarboxylation of enol carbonates has not been the subject of direct experimentation. However, the related decarboxylation of simple alkyl carbonates is known to occur spontaneously as well as by acid-catalysis.63 That said, most mechanistic investigations of carbonate decarboxylation reactions were conducted in water, where solvation is substantially different than in the organic solvents used in DcA reactions. A strong correlation of decarboxylation rates with pKa of the alkoxide (β = 1.4) does suggest a facile spontaneous decarboxylation of enol carbonates (G);63b the decarboxylation of phenyl carbonate is predicted to occur with a rapid rate of 2 × 103 s−1 in water at 25 °C. Alternatively, a Lewis-acid could facilitate the decarboxylation via mechanism (H), which is similar to the mechanism for Brønsted acid-catalyzed decarboxylation. The assessment of the true pathway for decarboxylation will require more detailed experimentation, perhaps in combination with computation.
4.4 Reductive Elimination: Inner-sphere vs. Outer-sphere
4.4.1 Calculated Transition States
We have already detailed that substrates that allylate prior to decarboxylation do so via an outer-sphere mechanism where the nucleophile directly attacks the allyl ligand of a π-allyl palladium complex. However, there remains a debate over whether non-stabilized palladium enolates react via an inner-sphere or outer-sphere mechanism; it is likely that both are possible. Stoltz has proposed that PHOX-ligated palladium allyl enolates react via an inner-sphere mechanism on the basis of DFT calculations (Chart 13).71 That said, the difference in transition state energies for the inner-sphere (B) and outer-sphere processes (A) is a mere 1.6 kcal/mol, and the calculations are reported to have an rms deviation of 1.2 kcal/mol. Nonetheless, the calculated mechanism for the inner-sphere process provides useful insight and suggests an interesting elimination via a 7-membered transition state (B) akin to that proposed by Echavarren.64 Notably, this 7-membered transition state (B) is 41 kcal/mol lower in energy than the “traditional” 3-center reductive elimination (C). The proposal of a 7-membered transition state for reductive elimination via a σ-allyl palladium complex suggests that the regioselectivity of allylation (linear vs. branched) may be different for decarboxylative allylations via Pd(PHOX) complexes and may favor the branched product rather than the typical linear product. To date, the regiochemical outcomes of Pd(PHOX)-catalyzed DcA reactions have not been reported.
Chart 13.

Representations of Calculated Transition States
4.4.2 Stereochemical Probes
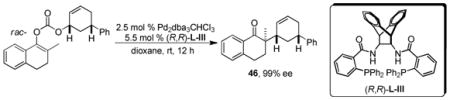
Trost was able to experimentally verify that the decarboxylative allylation, as catalyzed by Trost ligand complexes of palladium, occurs with retention of stereochemistry via a typical double-inversion mechanism.40b This indicates that the DcA occurs via an outer-sphere process similar to that observed for the allylation of lithium enolates (eq 22). Interestingly, however, the analogous synthesis of product 46 from the lithium enolate produced the opposite enantiomer. Thus, the nature of the attacking nucleophile in this DcA is substantially different from a typical enolate.
 |
(22) |
4.4.3 Crossover Reactions
Another potential way to probe whether “free” enolates or Pd-bound enolates are being formed is through a crossover experiment. In fact, the groups of Saegusa,15 Tunge,36 Trost,40b Stoltz,41 and Danishefsky65 have all performed crossover experiments aimed at determining whether the intermediate palladium enolates exchange between different palladium complexes (Scheme 23). Unfortunately, the initial ionization of the allyl carboxylate or carbonate likely creates ion pairs that are capable of complete crossover prior to decarboxylation. Thus, the observation of crossover can have ambiguous interpretations.
Scheme 23.

Ambiguous Results of a Typical Crossover Experiment
Trost has argued that crossover in DcA of enol carbonates occurs via ion-pairs and not charge-separated enolates based on the fact that acidic additives [e.g. CH2(CO2Me)2] are not extensively deprotonated under the standard conditions for DcA in dioxane (Scheme 24).40b While a charge-separated, unstabilized enolate would be expected to deprotonate such an acidic additive on thermochemical grounds, these experiments are complicated by the notoriously slow kinetics for deprotonation of carbon acids. Nevertheless, the small amount of proton transfer in dioxane coupled with the observation of more protonation in a more ionizing solvent (THF), suggests that palladium enolates exist as tightly associated species in dioxane. Trost further suggests that protonation and crossover are more likely to occur from π-allylpalladium carbonates because the more stable carbonate anion should be more charge-separated than the analogous palladium enolate.
Scheme 24.

Acidic Additives in DcA of an Enol Carbonate
4.5 Kinetics
Little experimentation on the kinetics of decarboxylative allylation has been published. Stoltz has reported that the decarboxylative allylation of enol carbonates is first-order in Pd(PHOX) catalyst and zero-order in substrate.71 The zero-order dependence on substrate is most easily attributed to the rapid formation of the resting state (allyl)Pd(carboxylate) complex under conditions of catalysis, consistent with a mechanism involving rate-limiting decarboxylation.
4.6 Mechanistic Conclusions
Ultimately, decarboxylative allylation is a field that would benefit from more in-depth mechanistic knowledge. That said, some important mechanistic features can be inferred from the vast knowledge of Tsuji-Trost allylation reactions. In addition, stereochemical studies have provided significant insight into the process. The current state of understanding of the mechanisms is summarized below (Scheme 25). Allyl β-oxo esters that contain an α-hydrogen undergo DcA reactions primarily via an allylation-decarboxylation mechanism involving outer-sphere attack of a stabilized enolate on a π-allyl palladium complex (top mechanism, Scheme 25), while allyl β-oxo esters that do not contain α-hydrogens undergo DcA reactions through a decarboxylation-allylation mechanism (middle mechanism, Scheme 25). It is reasonable to propose that allyl enol carbonates undergo DcA by decarboxylative formation of enolates followed by allylation. However, definitive rate data are needed to eliminate alternative mechanisms. The C–C bond forming allylation can occur either by outer-sphere attack of the enolate or via a 7-membered cyclic transition state for reductive elimination; outer-sphere attack is favored by the Trost ligand while the inner-sphere process may be favored by PHOX ligands, although both pathways are feasible.
Scheme 25.

Current Mechanistic Understanding
5 Applications of the DcA of Enolates
Given the ability to allylate enolates under formally neutral conditions, it is not surprising that DcA reactions have found utility in the synthesis of numerous biologically active natural products. Rather than providing an in-depth review of these syntheses, this section aims to concisely communicate how DcA reactions have facilitated some of these syntheses.
5.1 Dienolate DcA
The Upjohn company utilized DcA of a dienol carbonate in their synthesis of trospectomycin sulfate, an aminocyclitol antibiotic 48 (Scheme 26).66 In the DcA, the γ-position is selectively allylated in preference to the α-position to afford product 47 in modest yield. While simple dienolates derived from β-ketoesters give rise to selective α-allylation,24 the incorporation of the diene within a pyran moiety may alter the electronics such that γ-allylation is preferred.
Scheme 26.

Synthesis of trospectomycin sulfate
5.2 Diastereoselctive DcA
Tanaka and coworkers utilized Pd-catalyzed DcA to provide access to the prostaglandin analog (5E)-PGE2 (eq 23).67 Rather than production of the naturally occurring Z configuration of PGE2, isomerization of the Z-olefin under the reaction conditions provided the E-olefin product. The trans olefin product forms because C–C bond formation is slower than π-σ-π isomerization to the thermodynamically more stable syn-Pd-π-allyl complex, which gives the trans olefin.
 |
(23) |
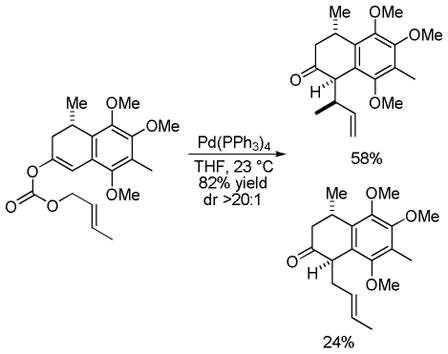
Nicolaou and coworkers performed a diastereoselective DcA of an enol carbonate in the total synthesis of colombiasin A (eq 24).68 Interestingly, this DcA favored a single diastereomer of the branched allylation product over the corresponding linear product. This observation of branched selectivity is somewhat unexpected,69 however crotylations are known to give rise to increased branched products in some Tsuji-Trost allylations.70 A more intriguing possibility is that the branched selectivity results from an inner-sphere attack of the enolate as proposed by Stoltz (B, Chart 13).71
 |
(24) |
Danishefsky and co-workers have performed a diastereoselective DcA of an allyl enol carbonate as a key step the synthesis of (±)-jiadifenin (Scheme 27).65 Importantly, Danishefsky reported that attempts to synthesize the required allylated ketone (50) by conventional enolate chemistry produced mixtures of allylated products. Believing that allylation α to the ester was a complicating side reaction, they turned to the regiospecific DcA reaction. The DcA allowed the synthesis of the requisite allylated ketone in 62–65% yield with acceptable diastereocontrol. The observed relative stereochemistry, along with its dependence on the size of the silyl protecting group, suggests that the distal α-position exhibits moderate diastereocontrol in the allylation of an intermediate enolate.
Scheme 27.

Synthesis of (±)-jiadifenin
Martin utilized DcA of a tricyclic β-keto ester as a key step in the total synthesis of (±)-lycopladine A (Scheme 28).72 Here, the authors demonstrated the use of a DcA reaction following a facile cycloaddition to an alkylidene ketoester. This highlights a particular advantage of DcA reactions of β-ketoesters; the ketoester fragment that is required for DcA often facilitates the synthesis of the requisite reactant in addition to the desired product. Decarboxylation and allylation led to the cis-ring fused allyl ketone in 80% yield. A catalytic hydroboration/oxidation sequence completed the total synthesis.
Scheme 28.

Synthesis of (±)-lycopladine A
Building off of work by Deslongchamps,73 in which the allyl ester 52 undergoes an annulation followed by decarboxylative protonation, Brückner and Tricotet investigated the ability to perform a DcA to form a new carbon-carbon bond rather than the traditional decarboxylative protonation (Scheme 29).74a When the ketal 51 reacted with 52 a mixture of diastereomeric allyl ketoesters was formed (53). When the mixture of diastereomers was subjected to the DcA conditions described by Tsuji,14 two diastereomeric products were formed in a 3:1 ratio (Scheme 29). Interestingly, when the silyl protected alcohol (55) was used, the DcA formed only a single diastereomer, albeit in modest yield. Nonetheless, the ability to control 5-contiguous stereocenters from one center is remarkable. Similarly to the lycopladine synthesis above, this annulation-allylation strategy illustrates the ability of the ketoester to facilitate a cyclization prior to DcA. Thus, the allyl ester activates the reactant (52) and delivers an olefin functional group via DcA.
Scheme 29.

Outcome of Annulation/DcA
Forsyth utilized a clever Diels-Alder/DcA reaction to set the stereochemistry of the trans-decalin required for the synthesis of salvinorin A (Scheme 30).74b While the Diels-Alder reaction led to a 1:1 mixture of the trans-decalin along with the desired cis-decalin, it did regioselectively produce the enol carbonate 57 that would be difficult to form by other means. DcA of 57 proceeded smoothly and diastereoselectively to form 58 with the two key quaternary stereocenters in place.
Scheme 30.

Diels-Alder/DcA
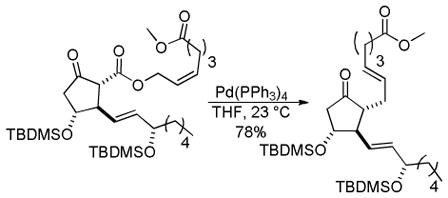
Trost and co-workers utilized the DcA of a highly stabilized allyl anion in a recent synthesis of spirotryprostatin B. Trost and Stiles took particular advantage of the distabilized allyl nucleophile, which is less prone to promote elimination, in developing a decarboxylative prenylation (Scheme 31).75 The requisite substrate for the prenylation had significant complexity, but was made as a mixture of diastereomers in just four steps. Reaction with an achiral catalyst provided the desired product in low dr, implying only a small degree of substrate control. Consequently, catalyst control of the stereochemistry was achieved by utilizing the Trost ligand to effect the formation of the desired isomer with excellent dr. It is noteworthy that decarboxylation produces a nucleophile that could undergo allylation at either of the two nucleophilic sites, however prenylation occurs preferably at the oxindole terminus with good regioselectivity (14:1).
Scheme 31.

Synthesis of spirotryprostatin B
5.3 Total Syntheses via Enantioselctive DcA
Using the asymmetric DcA reaction developed in their lab, McFadden and Stoltz76 completed the total synthesis of the (+)-R-dichronanone (Scheme 32). This synthesis, and many others that use DcA reactions, take advantage of the ability to set quaternary carbon stereocenters that are difficult to control using other methods. Thus, the α-stereocenter formed from the DcA of the allyl enol carbonate was carried through to establish the stereochemistry of the relatively congested carbocyclic core of dichronanone.
Scheme 32.

Synthesis of (+)-R-dichronanone
Enquist and Stoltz77 published a concise synthesis (−)-cyanthiwigin in which a key step was the asymmetric double decarboxylative allylation (Scheme 33). This synthesis takes full advantage of the stereoconvergent nature of the DcA and allows a 1:1 mix of diastereomeric diesters (59), prepared on a large scale through classical chemistry, to be transformed to the desired diastereomer of the enantioenriched ketone 60 (dr = 4.4:1). Furthermore, the clever use of two sequential DcA reactions allowed for statistical asymmetric amplification,78 affording a highly enantioenriched product ((R,R)-60) from a moderately enantioselective allylation. In 6 more steps the product was transformed to the natural product, (−)-cyanthiwigin, with the two quaternary carbon centers in place.
Scheme 33.

Synthesis of (−)-cyanthiwigin
5.4 Synthesis via Enantioselctive DcA of Vinylogous Ester Derivatives
Stoltz and co-workers have also utilized the decarboxylative allylation of vinylogous esters and thioesters in the synthesis of a number of natural products. For example, the asymmetric DcA of vinylogous thioester 61 using tBu-Phox ligand allowed access to the key quaternary carbon stereocenter for the synthesis of (+)-cassiol, a compound with potent antiulcerogenic activity in rats (Scheme 34).79 Optimal conditions involved the use of the p-methoxy dibenzylidene acetone (pmdba) ligated palladium precatalyst. This more electron rich dba analog binds more weakly to the palladium and provides a more active catalyst. A key feature of this synthesis was the use of Stork-Danheiser addition/hydrolysis to introduce the olefinic side chain and translate the α-quaternary center to the γ-quaternary center necessary for cassiol. From the same intermediate allylated ketone, Stoltz and coworkers completed the total synthesis of (+)-carrisone.44 Again the remaining carbons were installed via a Stork-Danheiser addition/hydrolysis protocol.
Scheme 34.

Syntheses of (+)-cassiol and of (+)-carrisone
Stoltz et al. also successfully employed an asymmetric DcA of an allyl enol carbonate that was derived from a cyclic vinylogous ester in the total synthesis of elatol, a member of chamigrene subclass of sesquiterpenes (Scheme 35).80 The authors found that reaction of 62 using the standard (S)-tert-butyl PHOX ligand proceeded sluggishly and they reasoned that this was due to slow alkylation. This conclusion was based on their ability to get 62 to undergo decarboxylative protonation under alternative conditions and secondly the ability for the non-chlorinated analog to undergo DcA. In support of their theory, inclusion of CF3 substituents into the ligand led to more rapid allylation and smoother DcA. This synthesis also highlights the ability to use DcA in combination with RCM to control the stereochemistry of spirocyclic carbocycles. A more detailed report has recently been published.81
Scheme 35.
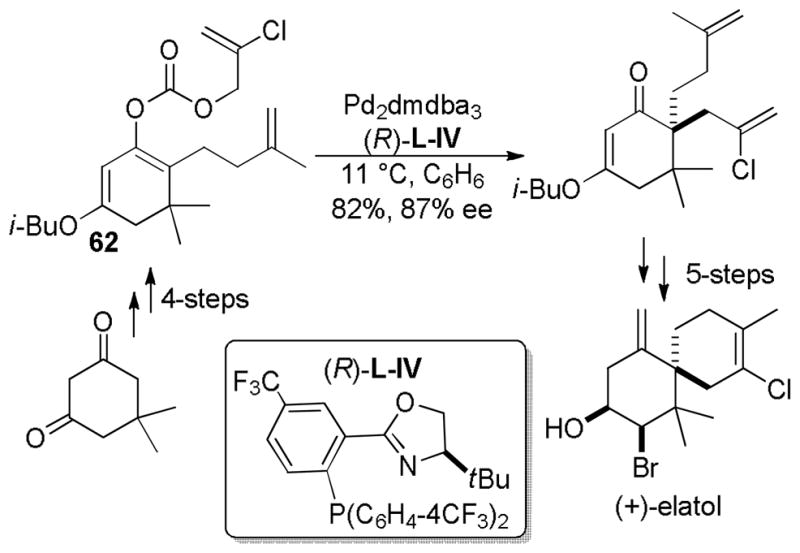
Synthesis of (+)-Elatol
6 Carbon Nucleophiles other than Enolates
While the majority of synthetic efforts have focused on the decarboxylative allylation of enolates, DcA has proven to be a rather general method for in situ formation of a number of carbon nucleophiles including: acetylides,15 α-cyano anions,15 acrylonitriles,15 coumarins,82 nitronates,18 heteroaromatic alkanes,83 nitrotolyl anions,84 α-ketimine anions,85 and α-sulfonyl anions.86 In addition to the carbon nucleophiles generated via decarboxylation, heteroatom nucleophiles have also been shown to participate in DcA reactions and will be discussed in a later section. These decarboxylative coupling methods have often proven complimentary or superior to more classical methods for generating reactive carbon nucleophiles because DcA reactions occur under mild, formally neutral conditions. Moreover, catalytic decarboxylative couplings ensure that reactive nucleophiles are generated in low concentration and in close proximity to electrophilic reaction partners. As a consequence the coupling is often faster than many other competing processes, such as proton transfer from more acidic functional groups.
6.1 2-Azallyl Anions
Homoallylic amines are an important building block for a variety of nitrogen-containing natural products. Much of the synthetic methodology for accessing homoallylic amines has focused on the addition of stoichiometric organometallic allyl nucleophiles to imines.87 Burger and Tunge sought to design a complimentary route in which an allyl electrophile is coupled to a nucleophilic α-imino anion that is derived from an α-amino acid.85
Taking note of how nature achieves the decarboxylative protonation of amino acids-namely pyridoxal-5′-phosphate (PLP)-dependent decarboxylases facilitate condensation of PLP and the amino acid to generate an iminium carboxylate (eq 25). The iminium carboxylate decarboxylates to generate an α-imino anion which ultimately protonates.88 Burger and Tunge postulated that coordination to a metal, rather than a proton, might also facilitate the decarboxylation and give rise to an anion that could be used for C–C bond formation (eq 26).
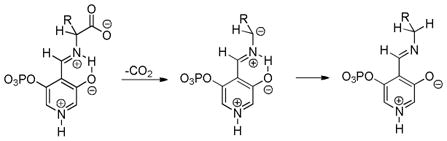 |
(25) |
 |
(26) |
To begin their investigation Burger and Tunge synthesized several ketimine protected α-amino allyl esters and subjected them to a variety of Pd(0) catalyst sources. The DcA reaction took place smoothly at 25–40 °C and gave rise to isomeric products 63 and 64 (Scheme 36). Additionally, in some cases protonation of the α-imino anion was problematic, but protonation could be mitigated by using dppb as the ligand. The formation of 64 was suggested to arise from an intermediate 2-aza-allyl anion (C) or equilibrating palladium σ-azaallyl complexes (A and B). The ratio of 63:64 was somewhat dependent on solvent, with THF providing the highest ratios of 63:64. In addition, the effect of substitution on the 2-position of the allyl was notable. While the reaction was more sluggish when the 2-position was substituted, the ratio of 63:64 was substantially improved (R = H vs Me, Scheme 36). Thus, a substituent in the 2-position led to significantly more of the desired product. One potential explanation is that C–C bond formation is primarily determined by sterics, and an increase in the steric size of the electrophilic Pd-π-allyl increases the regioselectivity. In partial support of this hypothesis, reaction of a sterically more encumbered α,α-disubstituted amino acid gave poor regioselectivity (eq 27). While not synthetically valuable, the ability of disubstituted amino acid derivatives to undergo DcA has an important implication; it suggests that decarboxylation of the amino acid precedes allylation.
Scheme 36.

DcA of Amino Acid Derivatives
 |
(27) |
A series of α-ketimine protected amino allyl esters were subjected to the DcA reaction conditions and modest to excellent yields of the product homoallylic amines were achieved (entries 1–10, Table 16). While the reactions of a variety of aryl amino acids proceeded under mild conditions (entries 2, 5, and 6), DcA of alkyl-substituted amino acid derivatives required substantially higher temperatures to achieve reaction. This likely reflects the more difficult decarboxylation of α-alkyl imino acids and implicates decarboxylation as the rate-limiting step of the reaction. In addition to their requisite reaction conditions, some α-alkyl amino acids undergo a different DcA reaction when coupled with unsubstituted allyl esters. For example, the attempted DcA of a protected tryptophan derivative gave the N-allyl aziridine rather than the expected C-allylation product (eq 28).
Table 16.
DcA of α-Ketamine Protected Amino Acid Allyl Esters
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | solvent | temp (°C) | ligand | yield (%) |
| 1 | Ph | H | THF | 25 | dppf | 67 |
| 2 | Ph | Me | THF | 40 | dppf | 81 |
| 3 | Ph | Ph | THF | 40 | dppf | 75 |
| 4 | 4-F-C6H4 | H | THF | 25 | dppb | 66 |
| 5 | 4-F-C6H4 | Me | THF | 40 | dppb | 93 |
| 6 | 4-MeO-C6H4 | Me | THF | 40 | dppb | 85 |
| 7 | CH2Ph | Me | Toluene | 110 | dppf | 63 |
| 8 | CH2Ph | Ph | Toluene | 110 | dppf | 46 |
| 9 |
|
H | Dioxane | 102 | dppb | 67 |
| 10 |
|
Me | Toluene | 110 | dppf | 26 |
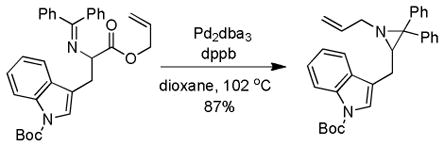 |
(28) |
Like the observation of allyl aziridine side products, the stereochemistry of amino acid DcA has important mechanistic implications. Specifically, when a non-racemic ester was allowed to undergo DcA, it produced racemic product (eq 29). Importantly, the reactant was not racemized under the reaction conditions. Not only does this show that racemization occurs via an intermediate formed in the reaction, it also supports a mechanism involving decarboxylation prior to allylation. Since the loss of stereochemistry in the DcA of an enantioenriched substrate is indicative of an achiral or rapidly racemizing intermediate, the C–C bond formation is likely to be the stereodetermining step, which should allow for enantioselective DcA. Indeed when a racemic substrate was subjected to DcA using (R)-BINAP as a ligand, a modestly enantioenriched homoallylic amine was produced (eq 30).
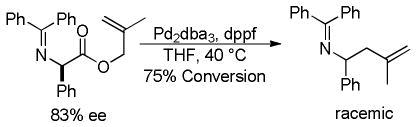 |
(29) |
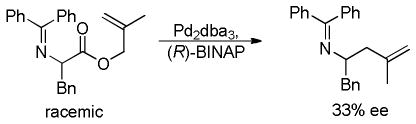 |
(30) |
The authors proposed the following mechanism (Scheme 37). Ionization of the reactant ester leads to Pd-π-allyl carboxylate A. This intermediate may undergo spontaneous decarboxylation to form an azaallyl anion. Alternatively, decarboxylation may be facilitated through an intermediate like B, in analogy to related acid-catalyzed decarboxylations.88b Intermediate C can react via two pathways; a 1,2-Pd-shift leads to D and ultimately the observed homoallylic amine product. Alternatively, electrocyclization leads to the Pd-aziridine E, ultimately forming the N-allyl aziridine. The observed N-allyl aziridines could likewise be formed by N-allylation followed by electrocyclization.
Scheme 37.
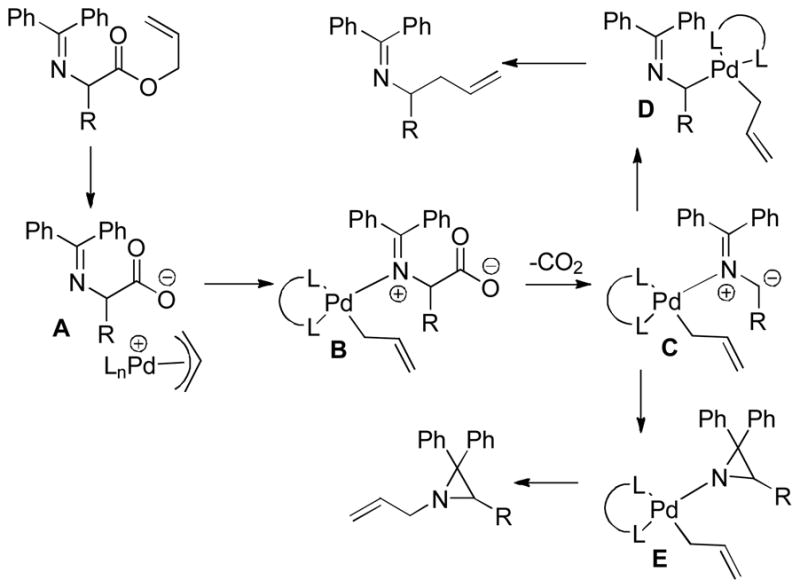
Proposed Mechanism for the DcA of α-Imino Esters
Soon after Burger and Tunge85 published their findings, Yeagley and Chruma89 published a complimentary method which generates the same intermediates through diphenylglyicinate imines (Scheme 38). By utilizing diphenylglycine as the amino acid, the authors showed that the coupling partner could be readily varied by condensation with a variety of aromatic aldehydes. This provides isomeric starting materials to those utilized by Tunge. Ultimately, by deriving the precursor from condensation with aldehydes, the Chruma method allows for more straightforward access to products that are derived from non-natural amino acids.
Scheme 38.

Regioconvergent DcA of α-Imino Allyl Esters
The dependence of the regiochemical outcome of the allylation on the electronics of the R substituent as observed by Chruma are consistent with observations made by Tunge, suggesting the formation of a common intermediate from either isomeric allyl amino acid (Scheme 38, Table 17). In addition, an increase in the steric size of the R2 substituent also makes the reaction more selective for allylation of the less crowded terminus of the 2-aza-allyl anion (Table 17, entries 1 and 2). Entries 6–8 demonstrate the advantage of Chruma’s DcA strategy; synthesis of these products using Tunge’s strategy would require the arduous synthesis of unusual, unnatural amino acids.
Table 17.
DcA of α-Imino Allyl Esters
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | time (h) | yield % | 63:64 |
| 1 |
|
H | 0.5 | 96 | 6.1:1 |
| 2 |
|
Me | 2 | 82 | >20:1 |
| 3 |
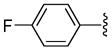
|
H | 1 | 97 | 5.5:1 |
| 4 |
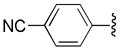
|
H | 1 | 91 | 20:1 |
| 5 |
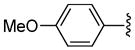
|
H | 7 | 52 | 2.2:1 |
| 6 |
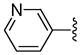
|
H | 2.5 | 47 | >20:1 |
| 7 |
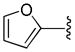
|
H | 2 | 75 | 4.1:1 |
| 8 |
|
H | 1.5 | 91 | >20:1 |
Chruma and co-workers have gone on to disclose slightly different conditions that utilize Pd(PPh3)4 as a catalyst to facilitate a tandem DcA-Heck cyclization (eq 31).90 While the reaction can be conducted in one step, the one-pot, two-step reaction provides higher yields. This chemistry highlights a common theme in decarboxylative couplings: decarboxylative allylation is facile and tolerant of aryl halide substituents. Thus, tandem decarboxylative allylation/cross-coupling reactions can allow multiple bond formations in single pot reactions.
 |
(31) |
6.2 Nitronates
While DcA reactions of amino acid derivatives provide access to protected homoallylic amines, the formation of tertiary homoallylic amines from amino acids is plagued by poor regiochemistry (eq 27). Similar homoallylic amine derivatives can potentially be accessed via the DcA of nitroacetic esters followed by reduction of the nitrogen, a reaction first demonstrated by Tsuji.18 Specifically, Tsuji reported the successful DcA of a single substrate, in which equal amounts of the isomeric C-and O-allylation products are formed (eq 32). Grenning and Tunge recognized that, if competing O-allylation could be avoided, the DcA of α-nitroacetic allyl esters might allow facile access to tertiary homoallylic amines that cannot be accessed via the DcA of amino acid esters.91
 |
(32) |
Tunge found that the DcA of allyl nitroacetic esters took place smoothly and rapidly in CH2Cl2 in the presence of 5 mol% Pd(PPh3)4 to afford the desired C-allylated product in high yield (Chart 14). The reaction works well for a range of α,α-dialkyl substrates as well as an α-phenyl α-fluoro ester. In addition, because the intermediate nitronates are relatively non-basic, the reaction tolerates some β-hydrogens on the allyl electrophile. Similar to the DcA of enolates, the reaction of α-nitro acetates is stereoconvergent with the α-stereoselectivity of the reaction being determined by the allylation. Furthermore, the anti or exo diastereoselectivity that is observed suggests that the allylation occurs via steric control (Chart 14).
Chart 14.

DcA of Allyl Nitroacetic Esters
While no O-allylation product was observed by Grenning and Tunge,91 they did observe competing formation of α,β-unsaturated aldehyde, which they propose came from the breakdown of the O-allylated product (B, Scheme 39). Consistent with a reversible O-allylation, they found that increasing the concentration of catalyst or reaction favored the C-allylated product. This higher catalyst concentration favors bimolecular reformation of the π-allyl palladium complex (A) from the O-allyl nitronate (B), allowing formation of C-allylated product 65 to occur more rapidly than unimolecular decomposition of B (Scheme 37).
Scheme 39.
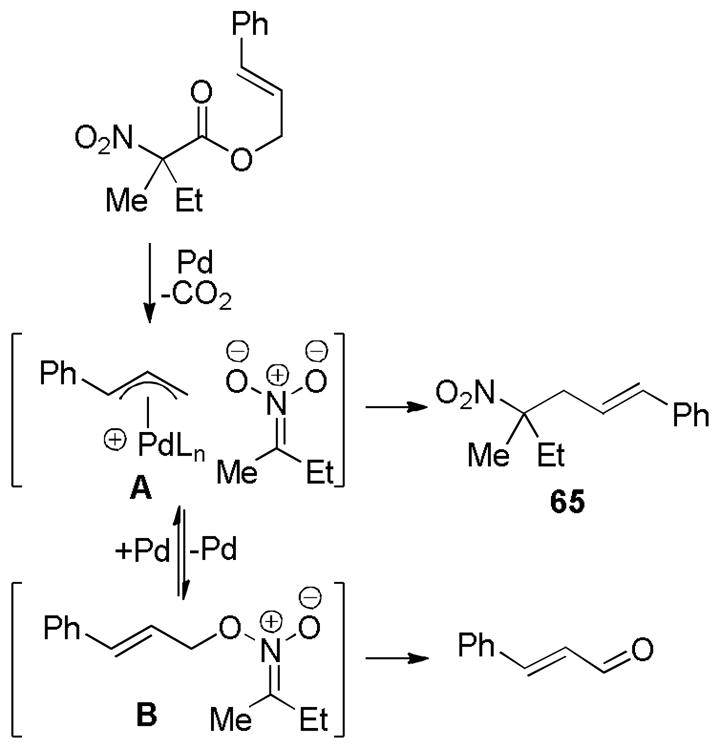
C- vs. O-allylation
6.3 Nitroaromatic Alkanes
Tunge and Waetzig showed that anilines, a common motif in the pharmaceutical industry, could be accessed via DcA of nitroarene acetic esters.84 Optimizations revealed that little reaction of an allyl ortho-nitrobenzene acetic ester took place at ambient temperature (entry 1, Table 18), but proceeded smoothly at 110 °C to give a mixture of the desired product (66) and undesired allylated aromatics, 67 (entry 2). Changing to the bidentate rac-BINAP ligand (entry 3) led exclusively to the desired product. The product ratio decreased when changing to the para-nitro analog, but allylated alkane 66 was still the dominant product (entry 4).
Table 18.
Optimization of the DcA of Nitroarene Acetic Esters
 | ||||
|---|---|---|---|---|
| entry | R | catalyst/Ln | temp (°C) | 66:67 |
| 1 | o-NO2 | Pd(PPh3)4 | 25 | NR |
| 2 | o-NO2 | Pd(PPh3)4 | 110 | 1.8:1 |
| 3 | o-NO2 | Pd2dba3/rac-BINAP | 110 | >20:1 |
| 4 | p-NO2 | Pd2dba3/rac-BINAP | 110 | 4.9:1 |
| 5 | p-NO2 | Pd2dba3/dppe | 110 | 2:1 |
| 6 | p-NO2 | Pd2dba3/dppf | 110 | 1.6:1 |
With optimal conditions in hand, Waetzig and Tunge set about studying the scope of the reaction. Pendant electron-withdrawing functional groups are well tolerated and the reaction works well even with electron donating substituents on the nitroaromatic ring (Chart 15). While the para-nitro derivatives allowed use of α,α-disubstituted substrates (68), the α-position cannot be fully substituted if the substrate is an ortho-nitro arene. This is likely due to the inability to achieve the requisite conformer for decarboxylation because of developing A-strain with the ortho-nitro substituent (eq 33).
Chart 15.

DcA of Nitroarene Allyl Acetic Esters
 |
(33) |
In addition to α,α-disubstituted o-nitrobenzene acetates which were unreactive, allyl ester 69 (eq 34) was unreactive under standard conditions. However, when the catalyst was switched to Pd(PPh3)4, the substrate underwent elimination to afford the diene, showing that β-hydrogens on the allyl fragment are not tolerated (eq 34). However, if a second nitro group was included on the substrate (eq 35), several effects were noted: 1) the reaction took place smoothly at room temperature 2) Pd(PPh3)4 was a competent catalyst, and 3) the additional stabilization of the anion made the reaction more tolerant towards allyls that possessed β-hydrogens; however, prenylation was still not possible.
 |
(34) |
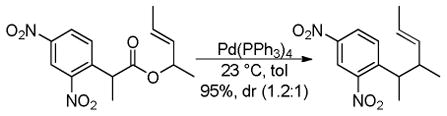 |
(35) |
6.4 Heteroaromatic Alkanes
In looking at DcA reactions of other electron deficient aryl acetic esters, Waetzig and Tunge demonstrated the ability for the allyl esters of α-heteroaromatic acetic acids to undergo DcA (Chart 16).83 Several things were unusual about this coupling; 1) the reaction was highly selective for the branched product 2) the resulting dr was unusually high for an allylation of an acyclic substrate 3) prenyl groups were reasonably well-tolerated. The ability of aromatic substrates with an N-atom at the 2-position to undergo DcA was fairly general (Chart 16). Moreover, the reaction was unusually tolerant of β-hydrogens on the allyl portion such that even reverse prenylation to form adjacent quaternerary centers was accomplished, albeit in modest yields (70). Reverse prenylation of substrates possessing only one α-substituent provided better yields of allylated products (69). Crotylation took place more readily than prenylation (71 and 72) but occurred with variable diastereoselectivity.
Chart 16.
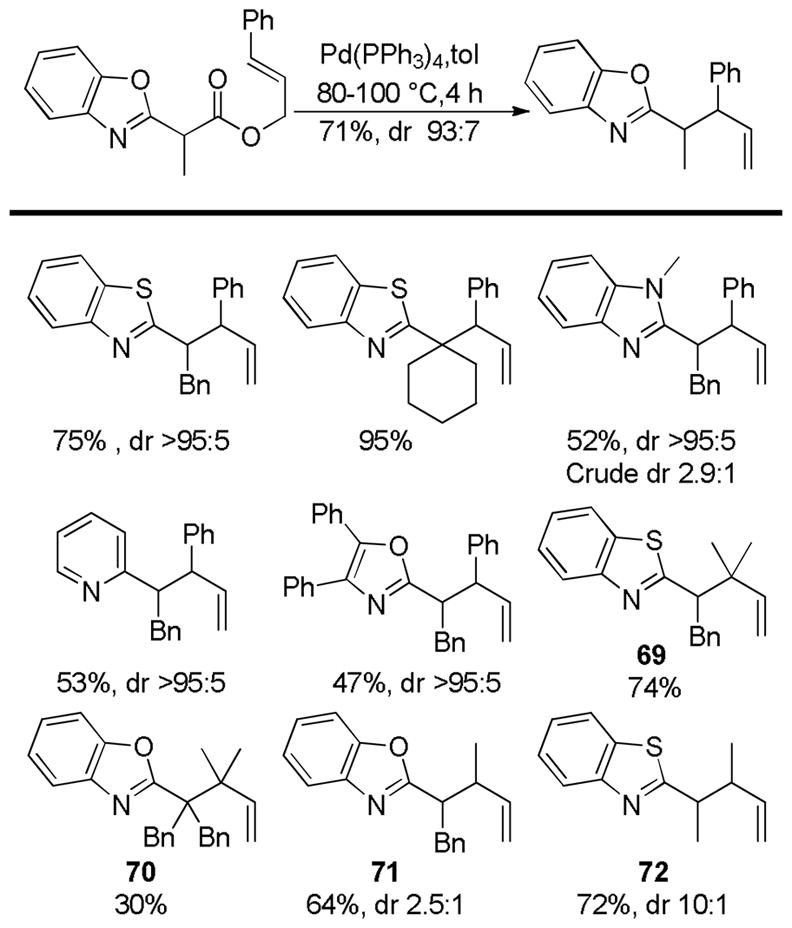
DcA of α-Heteroaromatic Acetic Allyl Esters
The ability to accomplish the decarboxylative reverse prenylation suggested that a benzylic anion is not formed; such a basic reactant would be expected to cause extensive elimination. Furthermore, a Lewis acid mediated Carroll-like rearrangement was ruled out based on the fact that the branched ester leads to the branched product (eq 36). The same product is formed from linear ester, suggesting a common Pd-π-allyl intermediate is formed in the reaction (eq 36).
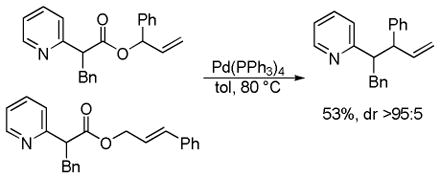 |
(36) |
Furthermore, when a 4-pyridyl analog was subjected to the catalytic conditions, it did not lead to formation of any desired product (eq 37). Thus, a nitrogen must be in the 2-position for successful reaction.
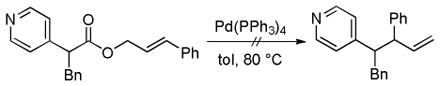 |
(37) |
Based on these results the authors proposed the following mechanism. Ionization leads to Pd-π-allyl carboxylate A (Scheme 40). N-allylation of the heteroaromatic leads to pyridinium B. This pyridinium is activated to undergo charge-neutralizing decarboxylative dearomatization to generate C, which undergoes facile aza-Cope rearrangement via a boat conformation to lead to the observed allylated alkyl pyridine.
Scheme 40.
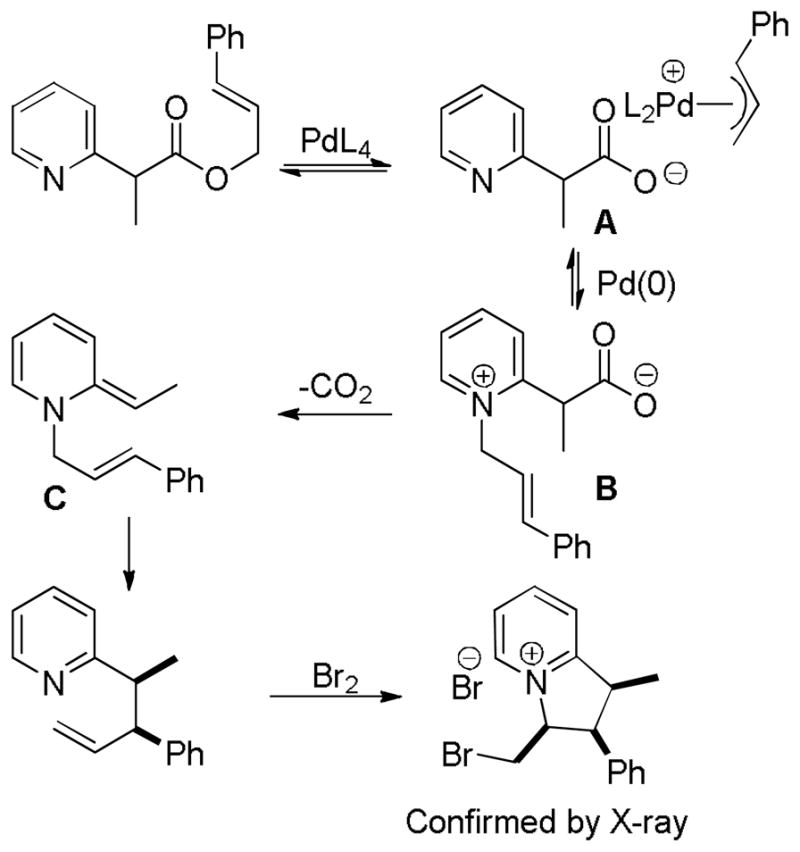
Proposed Mechansim for the DcA of Heteroaromatic Allyl Acetic Esters
6.5 α-Cyano Anions
In his seminal paper on the decarboxylative coupling of β-keto esters,15 Saegusa attempted the DcA of an allyl cyanoacetic ester. In one example, the substrate decarboxylated to provide the product in 69% yield accompanied by 16% of the diallylated product (eq 38). Tsuji later reported two examples of a similar reaction and showed that it was possible for an α-quaternary cyanoacetic acid derivative to undergo DcA, but the formation of protonation byproduct was problematic (eq 39).
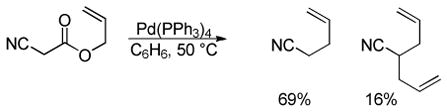 |
(38) |
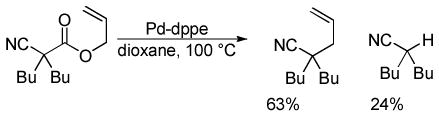 |
(39) |
In 2009, Recio and Tunge published an improved synthetic protocol for decarboxylative allylations of α-cyanoacetic esters and included a study of the scope.92 Ligand screening revealed that rac-BINAP could almost completely suppress the protonation product observed with many other ligands (Chart 17). The reaction appears to tolerate terminal substitution of the allyl electrophile with α-aryl nitriles, but α,α-dialkyl substrates promoted extensive β-hydride elimination in such cases. As with other DcA reactions, the α-phenyl substituent also has pronounced affect on the rate of the reaction; α-aryl nitriles undergo DcA at room temperature while α,α-dialkyl nitriles only react at elevated temperature (100 °C).
Chart 17.
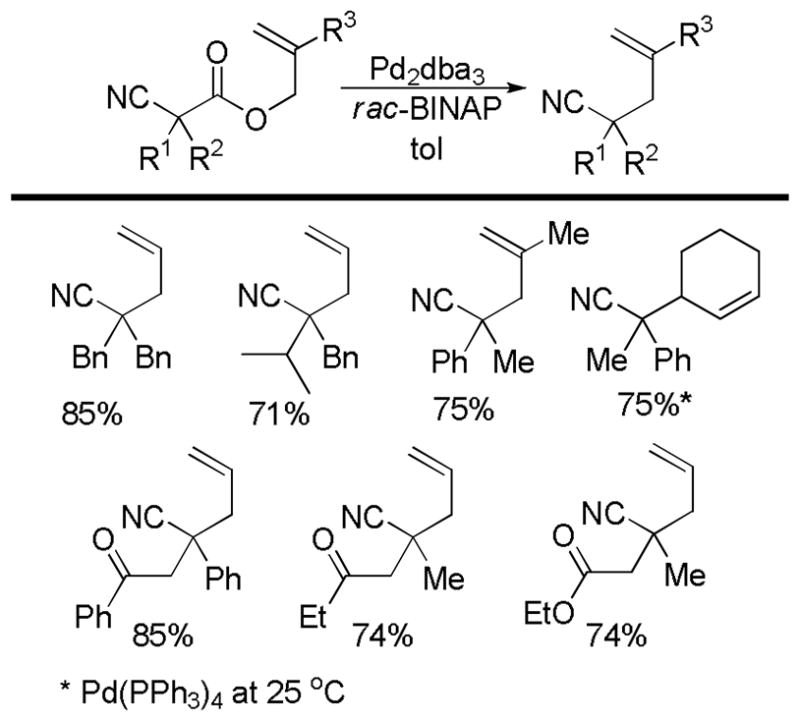
DcA of α-Cyano Allyl Acetic Esters
Like the preceding reaction involving heteroaromatic alkanes, this reaction differs from most other DcA in its preferences for formation of the branched product when α,α-dialkyl substituted cyanoacetates are utilized (Table 19). The preference for the branched product is not uniform, but rather appears to be substrate dependent (entries 1 and 2 vs. 3). If the ester possesses an α-phenyl substituent, then the ratio changes in favor of the linear product (entry 3).
Table 19.
DcA of α-Cyano Allyl Acetic Esters-Regioselectivity
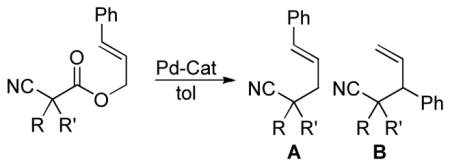 | ||
|---|---|---|
| entry | R, R′ | Yield %, A:B |
| (1) | Allyl, Allyl | 73%, 1:>9 |
| (2) | Bn, Bn | 62%, 1:5 |
| (3) | Ph, Me | 65%, 1.5:1 |
The authors explain this observation via competing kinetic N- and C- allylation of a common intermediate (Scheme 41). When the nucleophile is unstabilized and sterically large at the α-position N-allylation is preferred (entries 1 and 2, Table 19) and when the nucleophile is stabilized by an α-phenyl group, the major kinetic product is C-allylation. In this case, the low regioselectivity suggests there is only a small energy difference between the competing pathways. After kinetic N-allylation, [3,3]-sigmatropic rearrangement is expected to occur to afford the branched product; related rearrangements are known to take place rapidly at room temperature.93 Such regioselectivity could also be explained via competing inner-sphere and outer-sphere allylation mechanisms as discussed for enolate intermediates (Section 4.4.1). With this mechanism in mind, the less stabilized α,α-dialkyl anions would be prone to react via an inner-sphere mechanism giving the branched product, while the benzylically stabilized anion could prefer to react via an outer-sphere mechanism.
Scheme 41.
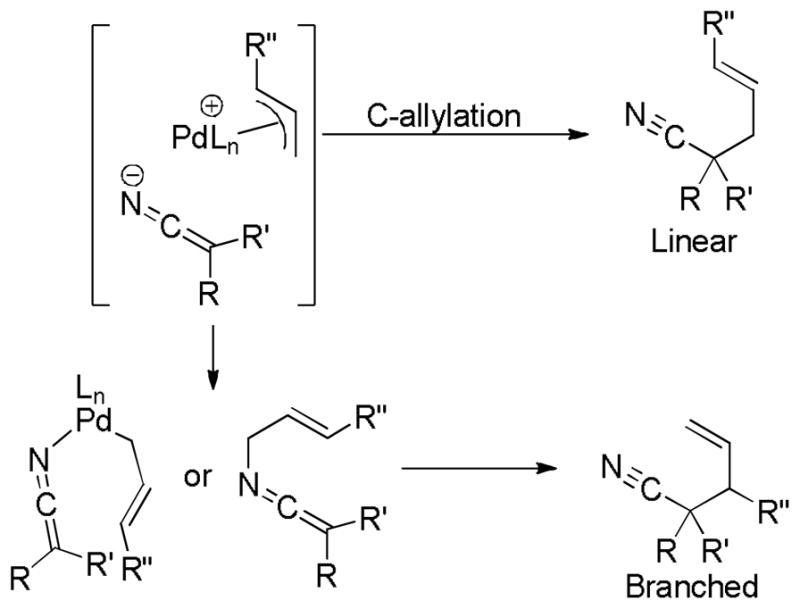
Rationale for Linear and Branched Products
Lastly, the decarboxylative generation of nitrile-stabilized anions is regiospecific, allowing the site-specific generation and reaction of a nucleophile at the position that bears CO2. In this way decarboxylation is capable of circumventing pKa issues that hinder formation of anions derived from less acidic positions using traditional acid-base chemistry. For example, the DcA of 73 is regiospecific (Scheme 42), with no isomerization of the intermediate nucleophile to the more stable anion, even though the malonate hydrogen is ~1015 more acidic than a tertiary nitrile (Ha vs. Hb, 74). In this way, decarboxylative metalation could be a useful synthetic tool for kinetic formation of anions that are not accessible by standard acid-base chemistry.
Scheme 42.

Access to Thermodynamically Unfavorable Nucleophiles
6.6 Vinylogous Malononitriles
Tunge et al. have also demonstrated that the DcA of vinylogous malononitriles is facile and kinetically favors α-allylation (Chart 18).24 Allylation occured preferentially α-to the nitriles, suggesting that the regiochemistry is electronically controlled. The yields were generally good to excellent and the regioselectivities were high; however, allylation with a 2-substituted allyl electrophile provided the product with low regioselectivity (75). Finally, the fact that product 76 could be obtained in high yield suggested that decarboxylation must precede C–C bond formation (Chart 18).
Chart 18.
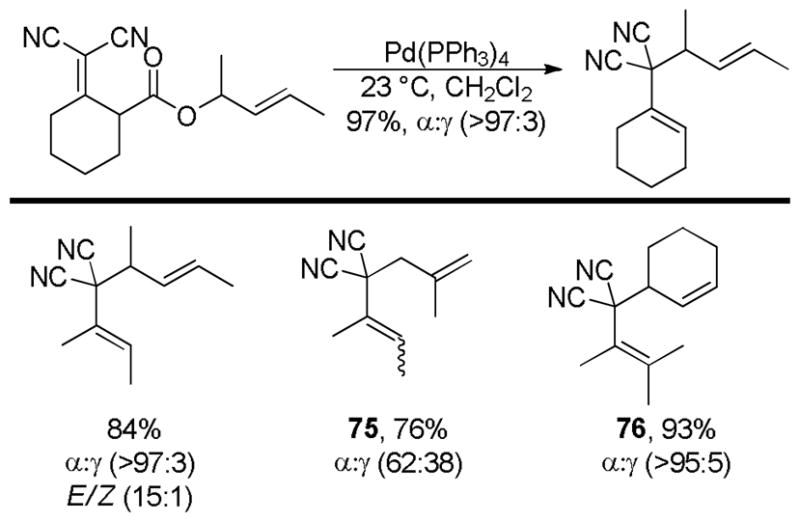
DcA of Vinylogous Malononitriles
The authors also found that the α-allylated kinetic product could undergo a net Pd(0)-mediated Cope rearrangement (Scheme 43).94 This presumably occurs by Pd(0) reionizing the kinetic product (77) to form the sufficiently stable anion which ultimately gives rise to the conjugated product (78) via attack of the Pd-π-allyl from the γ-terminus.95 Notably, the Cope rearrangement of 77 to product 78 did not occur at 70 °C without the Pd(0) catalyst. The same product could be obtained starting directly from the ester (79) by heating the reactant at 70 °C in the presence of catalyst. Thus, the authors could access either the α- or γ-allylated products depending on the temperature of the reaction mixture. In addition they showed that reaction could be made modestly enantioselective if the Trost ligand L-I was used (eq 40).
Scheme 43.
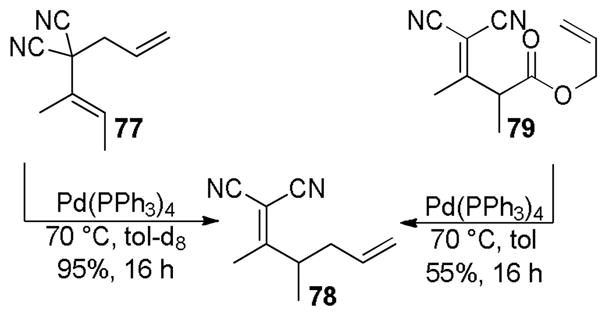
Pd(0)-catalyzed Allylation/Cope Rearrangement
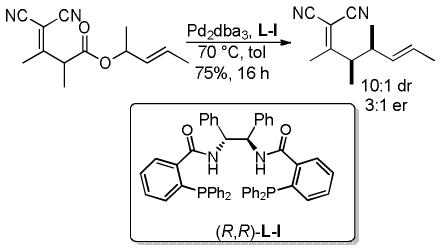 |
(40) |
6.7 α-Sulfonyl Anions
6.7.1 RacemicDecarboxylative Allylation
In all the DcAs developed, successful reaction is reliant on substrate stabilization of the anion that is formed upon decarboxylation. However, a methodology that would allow the DcA of unstabilized hydrocarbon nucleophiles would be valuable. Weaver and Tunge posited that sulfones might allow such a transformation in a two step process (Scheme 44).86 Given the electron withdrawing nature of the sulfone, it was envisioned that the α-sulfonyl carboxylate should be activated towards decarboxylation, generating an α-sulfonyl anion that could undergo C–C bond formation to give homoallylic sulfones. Reductive removal of sulfones is a well-established process, thus allowing the formal allylation of an unactivated hydrocarbon.96 Furthermore, the α-sulfonyl ester reactant is activated for derivatization which facilitates synthesis of more complex sulfones.
Scheme 44.
α-Sulfonyl Esters: Surrogates of Hydrocarbon DcA
The most common method for alkylation of a secondary sulfone, requires cryogenic temperatures, alkyl lithium base, and frequently utilizes stoichiometric, toxic HMPA.97 Thus, Weaver and Tunge believed that a DcA methodology that occurred under neutral conditions and produced only CO2 as the byproduct might be superior to existing methodology for the synthesis of tertiary sulfones. Towards this end, a fully substituted α-sulfonyl ester was synthesized and exposed to Pd(PPh3)4 under a variety of reaction conditions (eq 41). Temperatures that were lower than 95 °C led to sluggish reaction, and other solvents led to increased amounts of protonated product (81), rather than the desired allylated product (80). A screening of ligands revealed that bidentate ligands were superior to Pd(PPh3)4 and the reaction using rac-BINAP as the ligand suppressed the formation of 81, giving the highest yields of homoallylic sulfone 80.
 |
(41) |
Investigation of the reaction scope showed that α,α-dialkyl substituted substrates worked well at 95 °C, using rac-BINAP as the ligand (Chart 19). Substitution was tolerated in the 2-position of the allyl fragment and often led to slightly increased yields. Allyls with β-hydrogens were compatible reaction partners with α-aryl sulfones, but primarily underwent elimination with α,α-dialkyl sulfones. While an α-fluoro substituent slowed the DcA, an α-chloro substrate accelerated the reaction and led to increased yields. An α-phenyl substituent had a more dramatic accelerating effect, allowing use of the simpler Pd(PPh3)4 catalyst at reduced loadings of 1–2 mol %. In addition, the reaction temperature could be reduced to room temperature and the reaction time was significantly shortened (Cond. B, Chart 19). Lastly, it is noteworthy that an alkyl sulfone (specifically a benzyl sulfone) provided similar yields to the phenyl sulfones.
Chart 19.

DcA of α-Sulfonyl Allyl Esters
As with other DcA reactions, the DcA of sulfones is highly regiospecific. For example, upon decarboxylation the benzyl sulfonyl ester 82 led to the homoallylic sulfone that arises from coupling at the carbon that bore CO2 (Scheme 45). No product arose from isomerization to the more thermodynamically stable benzylic α-sulfonyl anion (B). Again, this highlights the ability to generate anions via decarboxylation that are not readily accessible via deprotonation.
Scheme 45.
Regiospecificity of the Anion
Tunge and Weaver further showed that the DcA of sulfones occurs via an outer-sphere mechanism with attack of a sulfonyl anion on the π-allyl ligand (Scheme 46).98 Moreover, they showed that, in contrast to most other decarboxylations, palladium does not catalyze the decarboxylation. Fittingly, the qualitative rates of DcA of sulfones correlated with the expected stability of the intermediate α-sulfonyl anion (Scheme 47). This suggested that decarboxylation is rate-limiting. Thus, substrates that formed α-sulfonyl anions that were benzylically stabilized (D) reacted rapidly, while an anion (A) that could not achieve the ideal conformation of the α-sulfonyl anion required heating at 200 °C in a microwave reactor to convert the reactant to product in just 40% yield.86
Scheme 46.

Outer-sphere DcA
Scheme 47.
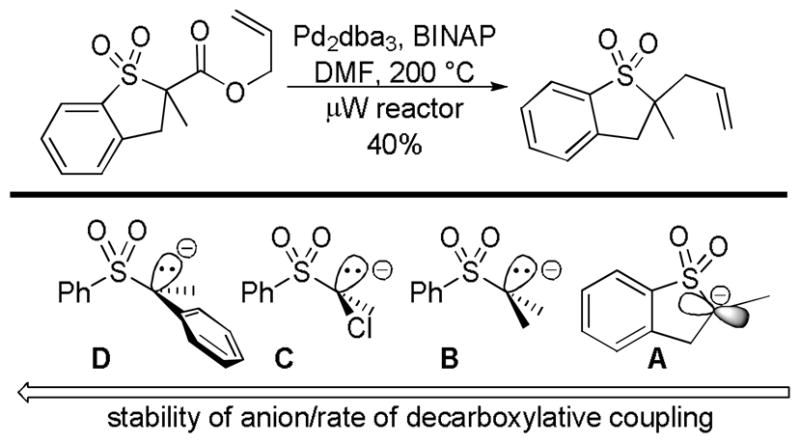
Qualitative Correlation between Stability and Rate
Finally, having established that the sulfone moiety could competently activate the substrate for DcA, the reductive removal of the sulfone was performed. The sulfones were readily cleaved by exposing the substrates to Mg0 turnings in warm methanol (Table 20). As might be expected, α-chlorine atoms were also removed under these conditions, allowing access to either tertiary or secondary coupling products.
Table 20.
Two Step DcA/Reduction
 | |||
|---|---|---|---|
| entry | substrate | product | yield (%) |
| 1 |
|
|
74 |
| 2 |
|
|
83 |
| 3 |
|
|
69 (10:1 l:b) |
| 4 |
|
|
79 (1:1 dr) |
6.7.2 Enantiospecific Decarboxylative Allylation
Having established the racemic reaction, Tunge et al. developed an asymmetric DcA of sulfonyl anions.98 Early work by Corey100 and Cram101 showed that chiral non racemic α-sulfonyl acetic acid derivatives underwent base-catalyzed stereospecific decarboxylative protonation. Indeed, chiral non-racemic α-phenyl propanoic allyl esters underwent DcA to form the corresponding highly enantioenriched homoallylic sulfones (Chart 20). The conservation of enantioenrichement [cee = (100 × % ee product)/% ee starting material] was uniformly high and allylation occurred with retention of stereochemistry. An examination of the substrate scope shows that the yields of the DcA are high and the cee’s are excellent regardless of the substrate. It is noteworthy, that an increase in the concentration led to higher yields than those obtained in their first publication (Cond. B, Chart 20).86
Chart 20.

Stereospecific DcA α-Sulfonyl Esters
Since the DcA of sulfones has been shown to proceed via α-sulfonyl anions (Scheme 46), the ability to achieve stereospecificity at 100 °C is remarkable. Gais reported that a lithio phenyl sulfonyl anion racemized rapidly even at −100 °C.102 However, in order to achieve highly stereospecific DcA, the C–C bond formation must occur faster than racemization. This is possible due to the formation of electrophile and nucleophile in situ in close proximity, where the lifetime of the α-sulfonyl anion is not long enough to allow significant racemization. Tunge et al performed DFT calculations in order to gain insight into the energies associated with the possible modes of racemization of the anion. Specifically, the DFT calculations showed that the barrier to inversion of the α-sulfonyl anion is < 2 kcal/mol (Scheme 48). Thus, the observed stereofidelity is not a result of slow inversion of the anion. Rather, the stereospecificity was attributed to allylation of the most stable conformation of the α-sulfonyl anion (A) via path A and slow rotation about the S–C bond of the α-sulfonyl anion (9.9 kcal/mol barrier, Scheme 48).100–101
Scheme 48.

Orgin of Enantiospecificity
7. sp2-Hybridized Carbon Nucleophiles
7.1 α-Allylation of Acrylonitriles
In Saegusa’s seminal paper describing DcA reactions of β-keto esters he also disclosed the DcA of an α,β-unsaturated α-cyano ester (eq 42).15 While metal-catalyzed decarboxylations of vinyl carboxylates typically require high temperatures,103 the DcA of the α,β-unsaturated α-cyano allyl ester occured at just 50 °C. Decarboxylations of α-cyano acrylic acids do, however, occur under mild conditions under nucleophilic catalysis via conjugate addition intermediates.104 Thus, triphenyl phosphine may play a role in this reaction.
 |
(42) |
7.2 Silyl Enol Ethers
Tsuji reported that silyl enol ethers derived from allyl β-ketoesters would undergo DcA to form allylated silyl enol ethers.105 The reaction likely proceeds via allylation of the silyl enol ether to form A, followed by reformation of the silyl enol ether by decarboxylation (Scheme 49). Snider used this strategy in a 1992 synthesis of velloziolone (Scheme 49).106 In the synthesis, Snider took full advantage of Tsuji’s allylation, which preserves the silyl enol ether nucleophile, by using it in a one-pot transformation to the desired α-iodo ketone.
Scheme 49.
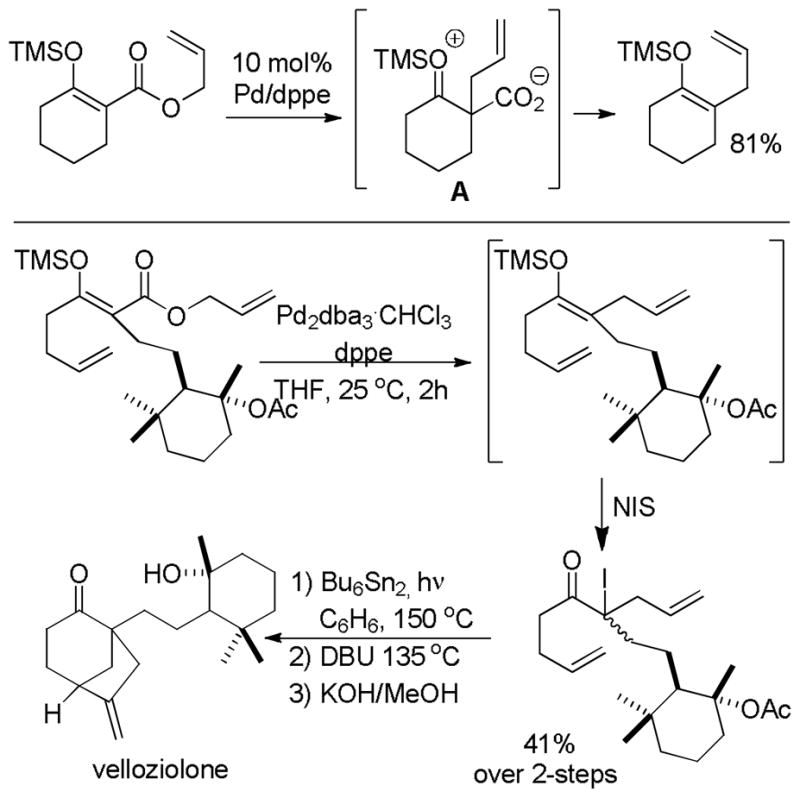
DcA of Silyl Enol Ethers: Synthesis of Velloziolone
7.3 α-Allylation of Coumarins
In search of applications of DcA reactions to biologically relevant substrates, Tunge and Jana found that allyl esters of 3-carboxylcoumarins readily underwent DcA at 25–50 °C in the presence of Pd(PPh3)4 (eq 43).82 Approximately 10% of 6-nitrocoumarin was also formed via the apparent protonation of a vinyl anion equivalent.
 |
(43) |
The mild conditions for this DcA are quite remarkable and puzzling considering that copper-catalyzed decarboxylative metallation of 3-carboxy coumarins takes place at 248 °C in refluxing quinoline.107 Investigation of the substrate scope showed that coumarins with electron withdrawing and donating groups generally worked well (Chart 21), although an amine substituent led to reduced yields (84). Interestingly, a methyl substitutent on the 2-position of the allyl led to a substantial increase in the desired product (85 vs. 83) and substitution at a terminus showed that the reaction takes place with the normal linear selectivity (86, 87). Coupling of an allyl group that has β-hydrogens was also successful (86); however the yield was somewhat reduced due to competing elimination. As expected, the DcA took place preferentially over oxidative addition into aryl bromide bonds, allowing tandem decarboxylative coupling/cross-coupling reactions, albeit, in modest yield (eq 44).
Chart 21.

DcA of 3-Carboxylcoumarins
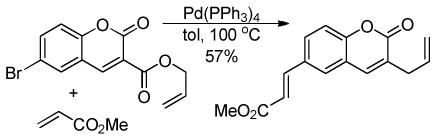 |
(44) |
A variety of substrates related to coumarins also underwent DcA reactions. The ability to form allylated pyrone in 62% yield showed that the benzenoid motif is not necessary. Good yields of DcAs of thiocoumarin and chromones were also realized, suggesting that other heteroaromatic substrates may also be competent partners. It is noteworthy that an open-chain analog of coumarin gave no conversion to product (88). This indicates either that having a phenolic ester is important, or that resonance with the phenolic oxygen of the coumarin is important; the methoxy substitutent in 88 is expected to have less resonance contribution because the acrylate is likely rotated out of conjugation.
8. sp-Hybridized Carbon Nucleophiles
8.1 DcA of Acetylides
In 1980, Saegusa included a single example of DcA of a propiolic allyl ester in his seminal publication that demonstrated the ability to generate homoallylic ketones via decarboxylative allylation.15 Saegusa reported a 21% GC yield and suggested that the reaction does not work well because the substrate possessed only a poor electron withdrawing group (eq 45).
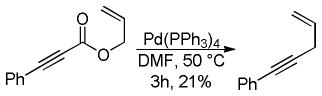 |
(45) |
Skipped enyne products are typically prepared via Stille cross-couplings of alkynyl stannanes. Tunge and Rayabarapu recognized that development of a useful decarboxylative coupling of allyl propiolates would obviate the need for the preparation and use of toxic tin reagents, effectively replacing transmetallation with decarboxylative metalation.108a Thus, they initiated their study with the phenyl cinnamyl propiolate and found that a catalytic amount of Pd(PPh3)4 would facilitate the decarboxylative allylation in good yield in 2 h (Chart 23). These conditions proved to be quite general (Chart 23). The reaction was somewhat tolerant of β-hydrogens; cyclohexenyl and 1,3-dimethyl allyl esters provided products in moderate to good yield. The electronics of the aryl ring do not appear to play a significant role in the outcome of the reaction as oxygen and halogen substituents were both compatible with the DcA. Concerning the acetylide, the reaction was not limited to phenyl propiolates, as the TMS-, cyclohexenyl-, and alkyl-substituted acetylide nucleophiles all underwent coupling in good yield. However, no reaction was observed for terminal alkynes. Moreover the authors showed that when the alkyne substituent was small (i.e. CH3), a bis-allylated alkyne byproduct was formed (89).
Chart 23.

DcA of Allyl Propiolic Esters
Tunge also demonstrated that the intermolecular reaction between the propiolic acid and an allyl acetate could be achieved in similar yields, obviating the need for the two components to be coupled prior to DcA. However, it did require a stoichiometric amount of base to prevent the Pd-catalyzed decarboxylative protonation of the propiolic acid (eq 46).
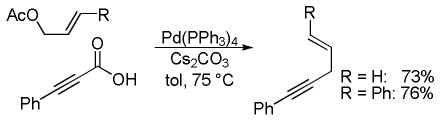 |
(46) |
The rates of the DcA of propiolates were dependent on the allyl substitution and followed the order of monosubstituted aromatic> disubstituted> terminally unsubstituted. This unusual rate dependence suggested that Pd-π-allyl formation was not rate determining, but it did loosely correlate with the expected rate of σ-allyl formation. Additionally, Rayabarapu and Tunge demonstrated that the carbon-carbon bond formation took place via an inner-sphere process, such that the overall reaction led to inversion of an allyl stereocenter (eq 47). Additional control studies demonstrated that a Pd(II) species is intimately involved in the decarboxylation step. Taken together, the mechanism for decarboxylative allylation of alkynes was proposed to involve decarboxylative metalation of the alkyne and reductive elimination of a σ-allyl palladium acetylide (Scheme 50).
Scheme 50.
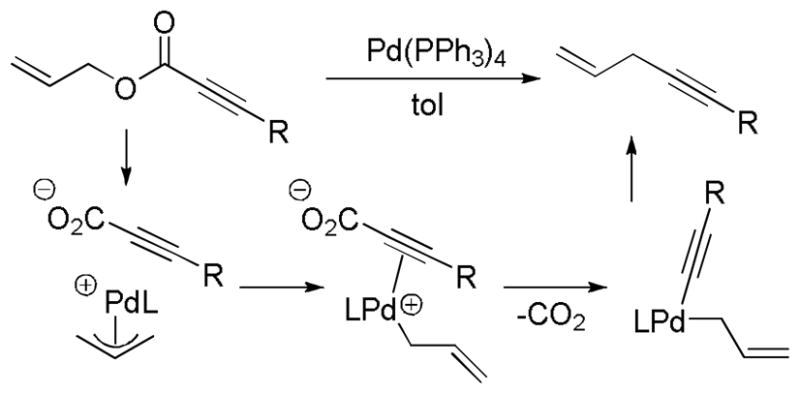
DcA of Allyl Propiolates
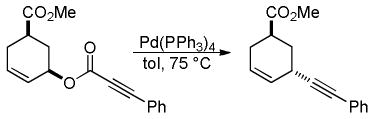 |
(47) |
8.2 Decarboxylative Coupling of Allenes with Acetylides
In 2008, Chung and co-workers developed the DcA of allenyl propiolates in which the carbon-carbon bond formation took place at the exclusively at the internal position of the allene to produce useful conjugated dieneynes (Table 21).109 After some screening, very similar conditions to those reported by Rayabarapu and Tunge108a were found to be useful in effecting the transformation. The reaction was quite general and tolerated rather large allenyl substituents (entry 1). The E:Z selectivity appears to be sterically controlled such that there is little distinction between Me vs. Et (entry 3), but good selectivites are observed when the substituents are sterically different (Ph vs. Me, entry 4). In addition to aryl propiolates, the DcA worked well for silyl and alkenyl propiolates (entries 5 and 7). Several other substrates were tested and found to be unfit for Pd-catalyzed DcA (Chart 24).
Table 21.
DcA of Allenyl Alkynoates
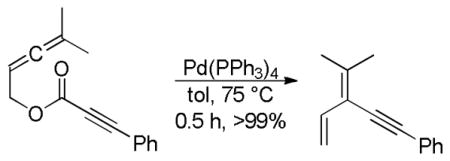 | |||||
|---|---|---|---|---|---|
| entry | product | temp. (°C) | time (h) | yield % | E:Z |
| 1 |
|
100 70 |
1 3 |
38 68 |
-- -- |
| 2 |
|
100 | 4 | 97 | -- |
| 3 |
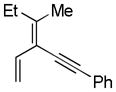
|
100 | 1 | 77 | 1.2:1 |
| 4 |
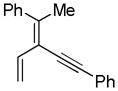
|
70 | 3 | 62 | 19:1 |
| 5 |
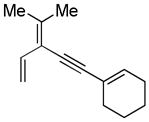
|
100 | 0.5 | 80 | -- |
| 6 |
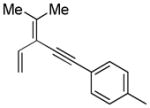
|
100 | 0.5 | 87 | -- |
| 7 |
|
100 | 0.25 | 86 | -- |
Chart 24.

Substrates Unfit for DcA
9 DcA of Heteroatoms
The application of transition metal-catalyzed decarboxylative coupling of allyl alcohol derivatives and heteroatoms has resulted in the development of several new reactions that are complimentary to existing methods, often offering enantioselective variants, altered regioselectivity, or greener alternatives to conventional methods. Specifically, methods to form C–N, C–Se, and C–O bonds via decarboxylation have been developed. The methods are versatile and allow facile access to allylic amines,110 vinyl azetidines,111 vinyl piperidines,111 vinyl hydroquinolines,112 as well as enantioenriched allylic selenides113 and selenocarbonates.113
9.1 Allylic Amination
9.1.1 Reaction Development
It is known that allyl carbamates undergo regio- and stereospecific thermal rearrangements under extreme conditions (eq 48). It was envisioned that a nucleophilic metal might facilitate these reactions under mild conditions in a manner similar to that reported for β-ketoesters.110 Indeed, palladium(0) and ruthenium(II) catalysts were found to efficiently catalyze the decarboxylative allylic amination of allyl carbamates (eqs 49 and 50).
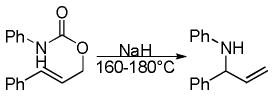 |
(48) |
 |
(49) (50) |
Initial catalyst screening revealed that [Cp*RuCl]4 was an effective catalyst, facilitating the decarboxylative allylic amination albeit with low regioselectivity. Addition of N,N,N′,N′-tetramethylethylenediamine (TMEDA) as a ligand was found to improve the regioselectivity, but this catalyst mixture promoted equilibration of the branched allylic amine to the more thermodynamically stable linear allylic amine.
While the equilibration of the branched and linear products is interesting and allows access to both products, more expedient access to the linear products was sought. Given that Pd(0) catalysts typically favor formation of the linear allylated products,6 it was expected that a Pd(0) catalyst might selectively yield the linear allylic amine. Indeed it was found that Pd(PPh3)4 gave the greatest conversion and highest regioselectivity in CH2Cl2 at 40°C.
An exploration of the scope of the decarboxylative amination showed that the reaction tolerated substitution at the allyl ester fragment (Chart 25), including allyl electrophiles with β–hydrogens that are capable of undergoing elimination—except in the case of aniline-derived carbamates which underwent elimination exclusively. The reaction appeared to be sensitive to the pKa of the corresponding nucleophilic nitrogen species. Carbamates derived from dialkyl amines worked well in all cases, while those derived from aniline and pyrrole, resulted in decreased yields or no product formation at all. Heteroaromatic amines such as pyrazoles, imidazoles, and triazoles were all compatible partners for DcA, however, attempts to form cyclohexenyl pyrrole resulted in elimination (Chart 26).
Chart 25.

Decarboxylative Allylic Amination
Chart 26.

DcA of Heteroaromatic Amines
The authors performed an experiment with an imidazole with a regiochemical indicator (eq 51). They reported that the DcA of carbamate 90 produced two isomeric products 91 and 92 in a 1:1.1 ratio. This experiment suggested that, in the case of heteroaromatic imidazoles, decarboxylation was occurring prior to allylation.
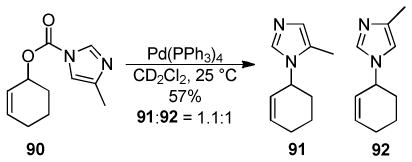 |
(51) |
To explain the observation of elimination with anilines and pyrroles, but not with more basic amines like piperidine, the authors proposed two competing mechanisms. In the first pathway, mechanism A, N,N-dialkyl carbamates undergo allylation of the N-atom prior to decarboxylation, generating a zwitterionic species, which facilitates decarboxylation affording the neutral allylic amine product (Scheme 51). Alternatively, mechanism B involves decarboxylation prior to allylation, generating an anionic nitrogen which can then undergo allylation. Thus, the authors argued that the reaction mechanism is controlled by pKa of the corresponding amine. In the case of dialkyl amines, decarboxylation is unlikely to take place to generate an extremely basic amide. However, decarboxylation of less basic aniline carbamates could take place to generate the corresponding amides. Thus, more basic amides are formed from aniline-derived carbamates, which explained the preference for elimination with those substrates.
Scheme 51.

Two Potential Mechanisms
To summarize, the difference in reactivity is best explained by pKa. Carbamates derived from amines with pKa roughly between 21–32 were shown to favor elimination. In contrast, the reduced basicity of pyrazole, imidazole, and triazole anions allowed the reactions to proceed smoothly (Scheme 52).
Scheme 52.

Correlation of Mechanism to pKa of Corresponding Amine
9.1.2 Synthetic Applications
The DcA of amines has proven to be useful in a number of synthetic efforts. For example, Bates and Dewey have applied the DcA of allyl carbamates in the formal synthesis of swainsonine.114 The paper highlighted the superiority of the DcA over the more traditional base-mediated allylation with allyl bromide (Scheme 53).
Scheme 53.

DcA en Route to Swainsonine
Hoveyda et al. opted to utilize a DcA protocol to achieve a more efficient synthesis of quebrachamine (eq 53) than would be possible using a starndard Calverley protocol (eq 52). In essence, DcA allowed the authors to avoid a two-step deprotection-allylation sequence and form the desired allylic amine in high yield. In doing so, the authors beautifully illustrated that carbamates can not only be used as activating or protecting groups, but they can also deliver useful functionality. In this case, the olefin of the allylic amine was utilized in an enantioselective RCM to establish the absolute stereochemistry of quebrachamine.
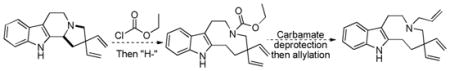 |
(52) |
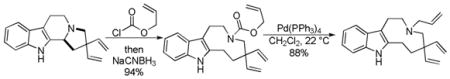 |
(53) |
9.1.3 Intramolecular Decarboxylative Cyclization
Having successfully demonstrated the decarboxylative allylation of acyclic allylic carbamates,110 Tunge investigated the use of cyclic carbamates as precursors to nitrogen-containing heterocycles. More specifically they showed that 1,3-amino alcohols could be activated such that they gave rise to vinyl azetidines and piperidines via intramolecular attack of amide anions on the palladium π-allyl ligands. For example, the trans-vinyl azetidine was formed rapidly from the trans carbamate in a 16:1 dr (Scheme 54). Interestingly, decarboxylative ring contraction of the cis carbamate also afforded the trans azetidine with the same diastereoselectivity. This result indicated that π-allyl palladium isomerization, via π-σ-π epimerization, occurred faster than C–N bond formation.
Scheme 54.
Decarboxylative Ring Contraction
Investigation of the scope of the azetidine synthesis showed that the reaction is highly diastereoselective (Chart 27). Terminal substitution was tolerated while the electronics of the R2 substituent seem relatively unimportant.
Chart 27.

Scope of the Azetidine Formation
Wang and Tunge also investigated the mechanism of the decarboxylative ring contraction. Use of terminally substituted substrate 93, which cannot epimerize via π-σ-π allyl isomerization, allowed the authors to determine that the C–N bond formation took place via outer-sphere attack on the π-allyl ligand to give product with overall retention of stereochemistry, via a double inversion mechanism (Scheme 55). That determination suggested the intermediacy of zwitterionic π-allyl palladium sulfonamides like B.
Scheme 55.

DcA: an Outer-sphere Process
The results also suggested that the preferential formation of the 4-membered azetidine over the 6-membered tetrahydropyridine could be explained by reaction through the thermodynamically favorable syn-Pd-π-allyl intermediate (Scheme 56). Allylic strain in the anti-B conformer led to slower production of the tetrahydropyridine product.
Scheme 56.
Preferential Formation of 4-Membered Ring
To test this hypothesis, a substrate, 94, which would cause the nucleophilic substituent to adopt the anti conformer was subjected to the reaction conditions (eq 54), and did indeed afford the tetrahydropyridine as the kinetic product in high yield. Presumably, this occurs because the Pd-π-allyl (B) adopts a conformer which places the larger phenyl substitutent in the syn position to reduce allylic strain. When a substrate with a smaller methyl substituent (95) was used, the diene was the primary product (eq 55). As a consequence, tertiary C–N bonds cannot be formed via this methodology.
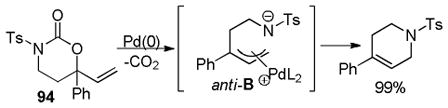 |
(54) |
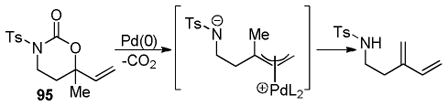 |
(55) |
In addition, it was found that, when vinyl azetidines are formed kinetically, they undergo palladium-catalyzed isomerization to the tetrahydropyridine if given sufficient time (eq 56). Thus, one can access either the vinyl azetidine or tetrahydropyridine products by simply adjusting the duration of the reaction.
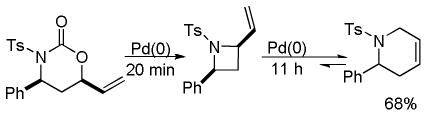 |
(56) |
9.1.4 Enantiospecific Vinyl Aziridine Formation
Yamamoto demonstrated that vinyl oxazolidinones also undergo decarboxylative ring contraction to form vinyl aziridines (eq 57).115 The activated vinyl aziridines are epimerizable in the presence of Pd(0) and Yamamoto took advantage of this to convert mixtures of 1,2-amino alcohol diastereomers to highly diastereomerically enriched, enantiopure aziridines (eq 57). Here the stereochemistry of amino acids can be used to control the stereochemistry in the thermodynamically preferred cis-vinyl aziridines. The enantioenriched vinyl aziridines produced by this method were further functionalized to afford (E)-alkene dipeptide isosteres.
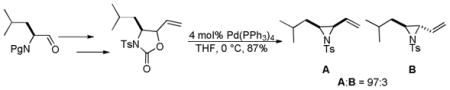 |
(57) |
9.1.5 Enantioselective Allylic Amidation
In 2007, Singh and Han reported an asymmetric Ir(I)-catalyzed decarboxylative amidation of Cbz-protected allylic carbamates (Chart 28).116 The DcA provides access to highly enantioenriched allylic carbamates that can be readily deprotected to form allylic amines. Notably, the reaction is quite tolerant of allyl electrophiles that possess β-hydrogens. Furthermore, a reaction between two different allylic imide reactants resulted in complete crossover, suggesting that the reaction proceeded via π-allyl iridium intermediates rather than through a [3,3]-rearrangement. In this report, the authors generally utilized an equivalent of DBU as well as proton sponge to promote the reaction. Given the need for the base and the tolerance of β-hydrogens on the allyl moiety it is probable that C–N bond formation occurs prior to decarboxylation. In support of this assertion, the authors reported that the lithium-amide of benzyl carbamate did not undergo successful reaction with an Ir-π-cinnamyl complex; a mechanism that involved decarboxylation prior to allylation would need to proceed via similar intermediates. Thus, the data support a mechanism involving enantioselective allylation followed by decarboxylation, where reaction of the catalyst with one of the enantiotopic faces of the olefin is probably the enantiodescriminating step.
Chart 28.

Enantioselective Decarboxylative Allylic Amidation
9.1.6 Enantiospecific Allylic Amidation
Soon after their first report, Singh and Han reported a complementary enantiospecific decarboxylative amidation.117 In this report optically enriched branched Cbz-protected carbamates were converted with high enantiospecificity to their corresponding allylic carbamates. The enantiospecific methodology allowed Han to address a shortcoming in their enantioselective decarboxylative amidation which was limited to substrates with sterically small allyl substituents. The authors reported that the reaction of terminally unsubstituted allyl carbamates reacted more smoothly, with the stereochemistry of the carbinol carbon being effectively transferred to the product (Chart 29). Thus, epimerization viaπ–σ–π isomerization was slow on the reaction timescale. Interestingly, the authors used a chiral ligand for the reaction, however, the absolute stereochemistry of the ligand had no effect on the reaction. Thus, there was no evidence for a matched/mismatched catalyst-substrate pair. Ultimately, the chiral phosphoramidite ligand was utilized primarily because catalysts generated from standard achiral ligands, like P(OPh)3, were ineffective.
Chart 29.

Enantiospecific Decarboxylative Allylic Amidation
9.2 Allylic Etherification
9.2.1 Alkyl Allyl Ethers
Similar to the formation of allylic amines (vida supra), allylic ethers have been generated by DcA of allyl carbonates. Most of these reactions have focused on allyl aryl carbonates but a few examples of DcAs of allyl alkyl carbonates have been reported. Specifically, in 1981, Guibe reported a single example of DcA of a primary alcohol generated from the carbonate generated from glycerol (eq 58).118 Similarly, Tsuji reported an 88% yield of the O-allylation product when screening for production of another desired product (eq 59).119 One potential reason for relatively sparse examples of DcA to generate alkyl allyl ethers is because the methods have not yet been shown to afford advantages compared to more straightforward, traditional allylations.
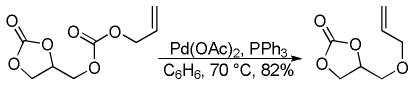 |
(58) |
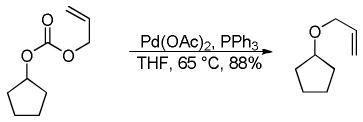 |
(59) |
9.2.2 Allyl Aryl Ethers
DcA has been used more widely to make aryl allyl ethers. In an early report, Rama screened Ni, Pd, and Rh catalysts for their ability to facilitate the asymmetric DcA of crotyl phenyl carbonates. Interestingly, each of these metals produced the branched product as the major product. In addition to the catalyst screening, the authors also reported the first enantioselective decarboxylative etherification, however the ee’s were poor at best (23% ee, eq 60).120
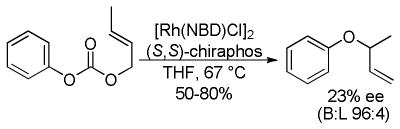 |
(60) |
A few years later, Larock, made significant strides in the development of the DcA of allyl aryl carbonates.121 His DcA of allyl aryl carbonates has several noteworthy aspects: first the reaction is typically moderately regioselective for the branched product, unless a conjugated linear product (91) is possible (Chart 30). Typically when olefin geometry is an issue, the E-olefin was formed preferentially. It is noteworthy that bulky ortho substituents on the phenol were also well tolerated. Finally, the reaction occurs with overall retention of configuration, presumably via a double inversion mechanism.58
Chart 30.

Pd-Catalyzed DcA of Aryl Allyl Carbonates
In 2008, Lacour et al. applied their ruthenium catalyst system to the asymmetric DcA of aryl allyl carbonates (Chart 31).122 In general, good ee’s and conversions were observed unless the aryl component had a nitro group. Interestingly, the authors showed that the ee deteriorates after the reaction has reached completion, concomitant with an increase in the amount of the thermodynamic linear product. A similar result was seen by Waetzig and Tunge in their allylic amination.110 Furthermore, subjecting two products to a crossover experiment led to complete crossover, providing evidence for reversibility of the C–O bond formation. Thus, the branched ethers are the kinetic products and achieving their synthesis with high enantioselectivity requires that the reaction be stopped before competing reionization/racemization becomes problematic.
Chart 31.

Ru-Catalyzed Asymmetric DcA of Allyl Aryl Carbonates
More recently, Trivedi and Tunge demonstrated that an anionic iron catalyst is competent at facilitating the DcA of allyl aryl carbonates and that the reaction takes place in high yields (Chart 32).123 They demonstrated that the reaction is regioselective, giving the same regioisomeric product regardless of whether the linear or branched carbonate reactant was used. This selectivity lies in contrast to related iron-catalyzed allylations that proceed with regiospecificity.124 Instead, the selectivities are similar to those reported by Larock using a Pd-catalyst, though the yields are somewhat higher with the iron catalyst.121 That said, the major advantage of this methodology is the use of the iron catalyst compared to the more commonly utilized precious metal catalysts, which are far more scarce and costly. Lastly, while the O-allylation products were typically formed, the authors demonstrated the synthesis of C-allylated phenols via a tandem DcA/Claisen rearrangement (eq 61).
Chart 32.

Fe-Catalyzed DcA of Allyl Aryl Carbonates
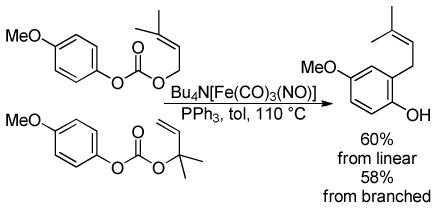 |
(61) |
9.3 Allylic Selenation
Tunge and Waetzig showed that allyl selenides are available via decarboxylative coupling of allyl selenocarbonates.113 They reasoned that the ability of the allylic selenide derivatives to readily undergo stereospecific [2,3] sigmatropic rearrangements to afford allylic amines, chlorides, and alcohols would make the selenide a versatile synthon (Scheme 57).
Scheme 57.
Enantioenriched Allyl Selenides are Useful Intermidiates
9.3.1 Racemic Reaction
Traditional methods of generating nucleophilic selenium include super-stoichiometric amounts of SmI2 or selenium-tin reagents.125 In hopes of developing a decarboxylative alternative, Tunge and Waetzig demonstrated that palladium can facilitate decarboxylative coupling of easily accessible allylic selenocarbonates.113 The racemic reaction was facilitated by Pd(PPh3)4 catalyst, affording allylic selenides in moderate to excellent yields (Chart 33). Once again, the stereochemistry of the reaction suggests that the decarboxylative allylation occurs via an outer-sphere mechanism, giving products with retention of stereochemistry.
Chart 33.

Decarboxylative Allylic Selenation
9.3.2 Kinetic Resolution
After establishing the racemic reaction, a screening of chiral nonracemic ligands was performed, however the results were initially puzzling. The reaction proceeded to ~50% conversion in 2 h and then ~55–80% in 24 h. Moreover, efforts to increase the conversion by heating or extending the reaction time had a negative effect on the enantioselectivity of the product. One explanation of such observations could be a kinetic resolution of the starting selenocarbonate. Indeed, when palladium ligated by the naphthyl Trost ligand-L-VII was utilized, the conversion stopped at 53%. Highly enantioenriched starting material, as well as enantioenriched allylic selenide were recovered from the resulting mixture (eq 62, 63).
 |
(62) |
 |
(63) |
Cyclohexenyl selenocarbonate proved to be a good substrate for kinetic resolution with a selectivity factor of 31.9 (eq 62) and addition of the ester further increased the selectivity factor to 80.1 (eq 63). The observed kinetic resolution is likely a consequence of rate determining ionization, which allows the chiral ligand to distinguish between the enantiomers of the substrate. Ionization was proposed to be followed by fast decarboxylation and fast bond formation.
Given the high selectivity factor, enantioenriched allylic selenides were isolated in good yields, allowing access to enantioenriched allylic amines, chlorides, and alcohols. To demonstrate this transfer of stereochemistry the selenide was converted to an allylic aniline and allyl chloride with a high degree of stereotransfer (eqs 64,65).
 |
(64) |
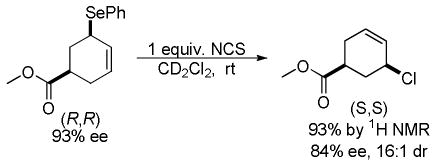 |
(65) |
10 Interceptive Decarboxylative Allylations (IDcA)
10.1 IDcA of Cyclic and Acyclic β-Ketoesters
As noted above, allyl esters can be thought of as masked nucleophiles and electrophiles which are both contained in a single moiety. Treatment of these compounds with an appropriate transition metal catalyst effectively reveals both reactive intermediates.126 If DcA reactions are performed in the presence of a sufficiently reactive electrophile, these intermediates can be intercepted before their combination to form DcA products (eq 66). In this section of the review, we aim to detail the scope of interceptive DcA (IDcA) reactions that have been developed over the past two decades.
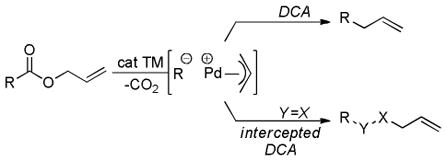 |
(66) |
10.1.1 Intramolecular IDcA
In 1989, Tsuji reported that an enolate intermediate, formed by decarboxylation, could be intercepted by a tethered Michael acceptor (Scheme 58).127 A brief investigation of potential reaction conditions found that the monodentate phosphine ligand PPh3 was more effective than a bidentate phosphine ligand dppe [1,2-bis(diphenylphosphino)ethane] at promoting the IDcA reaction. In addition to ligand choice, the source of the palladium catalyst was also important; reactions performed with Pd(PPh3)4 rather than Pd(OAc)2/(PPh3)2 facilitated product formation much more efficiently. The reaction was suggested to proceed via π-allyl palladium enolate (A) formation followed by Michael addition to generate a new π-allyl palladium enolate intermediate (B). The newly formed palladium enolate can then react with the electrophilic Pd π-allyl complex to generate the observed allylated ketone product (Scheme 58). As illustrated in Scheme 58, this methodology provides a potentially useful route for the generation of quaternary spirocyclic carbon centers.
Scheme 58.

Decarboxylative Michael Addition and Allylation
10.1.2 Intermolecular IDcA
Nearly a decade after Tsuji’s seminal report, Yamamoto reported the intermolecular β-acetonation-α-allylation of activated olefins with allyl acetoacetate.128 Like the previous intramolecular reaction of Tsuji, the intermolecular IDcA proceeded in the presence of a catalytic amount of Pd(PPh3)4 in THF (Chart 34). As depicted in Chart 34, Yamamoto reported that the reaction allowed additions to both arylidene malononitriles and arylidene-α-cyano esters. Products derived from the latter acceptors contain two adjacent stereocenters which were formed with minimal diastereoselectivity. In addition to the acrylonitrile derivatives utilized by Yamamoto, Tunge later showed that Meldrum’s acid adducts of aldehydes were sufficiently electrophilic to undergo palladium-catalyzed IDcA, and further demonstrated the compatibility of these reactions with substituted allyl groups.129 Thus, Meldrum’s acid adducts of aldehydes are competent electrophiles for IDcA, while simple benzylidene malonates [RCH=C(CO2Et)2] are not.128 Yamamoto attributed the lack of reactivity of RCH=C(CO2Et)2 to “steric inhibition of resonance” where imperfect coplanarity of the two ester groups reduces the electrophilicity of the alkene. Indeed, the more planar benzylidene malononitriles and Meldrum’s acid adducts are many orders of magnitude more electrophilic than benzylidene malonates.130
Chart 34.

Decarboxylative β-Acetonation α-Allylation
Yamamoto proposed a mechanism for the IDcA in which decarboxylation preceeds Michael addition and allylation. While this proposed mechanism is reasonable, the mechanistic discussion in section 4.2 of this review suggests an alternative mechanism in which formation of a stabilized enolate, Michael addition and allylation occur prior to decarboxylation (Scheme 59).111 Decarboxylation of the β-ketoacid would then lead to the observed products.
Scheme 59.

Proposed Mechanism for IDcA
10.1.3 Ruthenium-catalyzed IDcA
In 2005, Tunge and co-workers reported the ruthenium-catalyzed decarboxylative insertion of electrophiles into allyl β-ketoesters.129 Exposure of allyl β-ketoesters to 2.5 mol % [Cp*RuCl]4 and 10 mol % bipyridine (bpy) in the presence of sufficiently electrophilic Michael acceptors led to good yields of IDcA products (Scheme 60). While a benzylidene malononitrile with a p-acetyl substitutent underwent smooth IDcA (96), the reaction was not successful with a more electron-rich p-hydroxy substituted Michael acceptor (97); instead only the standard DcA product was formed. This lack of reactivity highlights the fact that, for successful IDcA, the intercepting electrophile must be more electrophilic than the ruthenium-π-allyl complex (estimated E ~ −10).130 An α-cyano coumarin was also sufficiently electrophilic to intercept the DcA reaction to produce 98, although that reaction proceeded in lower yield compared to other olefinic coupling partners (Scheme 60). Addition to the cyclic olefin did, however, proceed with good diastereoselectivity. Another interesting olefinic coupling partner used was the benzylidene Meldrum’s acid adduct. Tunge reported that decarboxylative addition of allyl β-ketoesters to such alkenes produced products that could be hydrolyzed to form γ,δ-unsaturated acids like 99. This is significant since these products are not directly obtainable from additions to mono-activated olefins like cinnamic acid.
Scheme 60.

Ruthenium-Catalyzed Decarboxylative Insertion of Electrophiles
10.1.4 Regioselective IDcA
Tunge and Wang also demonstrated the first regioselective IDcA of benzylidene malononitriles to give the branched allylation product (eq 67). A large breadth of work investigating metal-catalyzed allylic alkylations suggested that the regioselectivity of allylation could be controlled by appropriate selection of transition metal catalyst.32,36,131 This was confirmed by treatment of a β-ketoester with benzylidenemalononitrile and either [Cp*RuCl]4/bpy or Pd(PPh3)4. While the ruthenium catalyst delivered only the branched Michael addition-allylation product,131b a palladium catalyst selectively formed linear allylation product (eq 67).
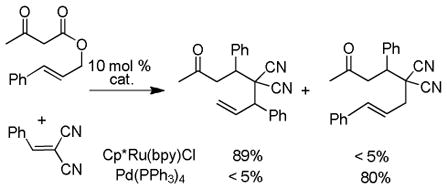 |
(67) |
10.1.5 Asymmetric IDcA
In 2010, Stoltz et al. reported the asymmetric decarboxylative addition of cyclic β-ketoesters to activated olefins (Scheme 61).132 Building on the earlier observations of Yamamoto128b and Tunge,129 Stoltz developed an asymmetric IDcA reaction that proceeded well in the presence of electron deficient arylidene malononitriles (Table 22, entries 1–4) and benzylidene Meldrum’s acid derivatives (Table 22, 100). Cyclic β-ketoesters and arylidene malononitriles smoothly underwent the IDcA in the presence of the PHOX-ligated palladium catalyst, allowing generation of adjacent quaternary and tertiary carbon stereocenters with high enantioselectivity and diastereoselectivity. Similar to their proposed mechanism for DcA of β-ketoesters, Stoltz proposes that an η1-allyl, O-bound palladium enolate intermediate I, is intercepted in an enantioselective fashion with prochiral electrophiles (Scheme 61, path B).132
Scheme 61.

Asymmetric IDcA of Cyclic β-Ketoesters
Table 22.
Asymmetric Interceptive DcA of Cyclic β-Ketoesters
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | R′ | yield | dr (A:B) | A | B |
| 1 | Me |
|
97% | 1:>20 | — |
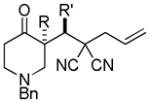 89% ee |
| 2 | Et | 99% | 1:>20 |
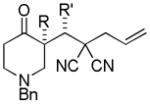 71% ee |
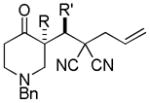 97% ee |
|
| 3 | Me |
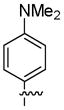
|
54% | 1:>20 | — |
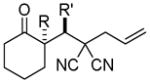 97% ee |
| 4 | Et |
|
92% | 1:>2.3 |
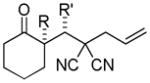 89% ee |
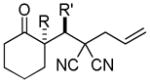 96% ee |
|
| ||||||
 100, 66% 1:1.2 d.r. 43% ee (major) |
 101, 76% |
|||||
Table 22 highlights a number of substrates and olefin coupling partners for which both excellent diastereoselectivity and enantioselectivity were reported. The asymmetric transformation is tolerant of small alkyl groups in the α-position as well as tertiary amines in the backbone of the cyclic β-ketoesters, however the yield is diminished with electron-donor substituents on the arylidene malononitrile (entry 3). This reduced yield may reflect less effective trapping of the enolate with the less electrophilic Michael acceptor. The benzylidene Meldrum’s acid adduct was also a suitable reaction partner (100), however the reaction suffers from low diastereo- and enantioselectivity. Lastly, use of alkylidene malononitriles resulted in deprotonation of the malononitrile followed by allylation to provide 101 (Table 22).133
The proposed catalytic cycle for asymmetric IDcA involves ionization of the allyl β-ketoester with the Pd(PHOX) catalyst followed by loss of CO2 (Scheme 62). This process generates the η1-allyl palladium enolate intermediate A, a proposed key intermediate in the normal DcA reaction. Conjugate addition delivers intermediate B. The ensuing stabilized malononitrile anion attacks the π-allyl palladium complex delivering product and regenerates the Pd(0) catalyst. Interestingly, compared to the products of normal DcA reactions, the IDcA products have the opposite absolute configuration at the α-carbon (Scheme 61). However, the stereochemistry of addition of the electrophile is the same as that observed for protonation of the enolate intermediates.71 Thus, it appears that the intermediate palladium enolates react with external electrophiles and palladium allyl electrophiles with the opposite sense of stereoinduction.
Scheme 62.
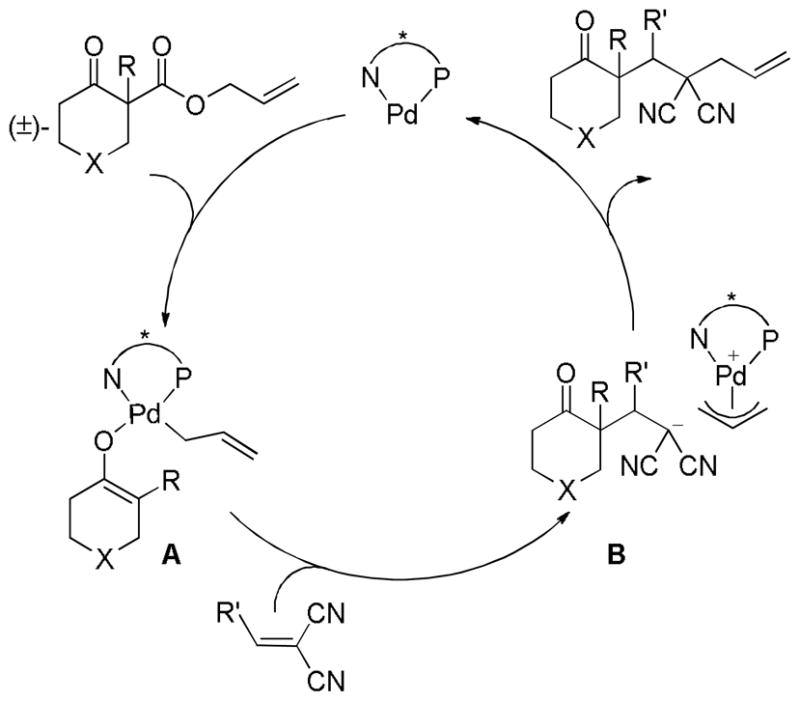
Proposed Cycle for Asymmetric IDcA
10.2 IDcA of γ-Methylidene-δ-Valerolactones
Recenlty, Hayashi and Shintani have extended the palladium-catalyzed decarboxylation of allyl ester moieties to malonate-derived valerolactones. In doing so, they developed an elegant method for the palladium-catalyzed decarboxylative formation of 1,4-dipoles A from γ-methylidene-δ-valerolactones (Scheme 63).134 Subjecting the γ-methylidene-δ-valerolactones to a catalytic amount of palladium resulted in the ionization of the allyl moiety followed by loss of CO2 to form the zwitterionic species A. Since the intramolecular attack of the enolate on the π-allyl ligand of A to form a 4-membered ring is disfavored, Hayashi proposed that one could intercept such intermediates even with relatively weak electrophiles to form B.134b,134d,134g,h At this point, regioselective attack on the π-allyl palladium moiety generates 2 potential products. Attack at the terminal carbon of the palladium π-allyl leads to an exo-methylene cyclohexane product (path a, Scheme 63). On the other hand, attack at the central carbon of the π-allyl palladium results in a spiro-[2.4]heptane product (path b, Scheme 63).135
Scheme 63.

Proposed Mechanism for Decarboxylative [4+2] Cycloadditions
10.2.1 [4+2] Cycloadditions with α,β-Unsaturated Electrophiles
Taking full advantage of these mechanistic manifolds, Hayashi and Shintani developed the palladium-catalyzed decarboxylative [4+2] cycloaddition of γ-methylidene-δ-valerolactones 102 with electron deficient α,β-unsaturated ketones, esters and nitriles (eq 68).134b Cycloadducts 103 were obtained when valerolactones were treated with Michael acceptors in the presence of 5 mol % CpPd(η3- C3H5) and bulky phosphine ligands [(t-Bu)2P(o-PhC6H4), P(o-Tol)3]. Interestingly, when smaller monodentate and bidentate phosphines (PPh3, BINAP, dppf) or phosphite ligands [P(OMe)3, P(OiPr)3] were employed, the reaction did not result in exo-methylene cyclohexanes, 103. Instead, the major products obtained were the spiro[2.4]heptanes 104 (eq 68).
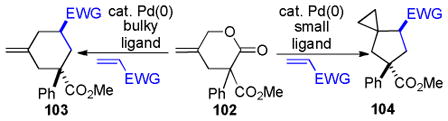 |
(68) |
Based on the observed selectivity, Hayashi investigated the scope of substituents that are tolerated on the γ-methylidene-δ-valerolactones and activated olefin coupling partners (Chart 35). The spiro[2.4]heptane formation with methyl acrylate is tolerant of a variety of α-aromatic substituted valerolactones and the respective products were obtained in good yield and moderate diastereoselectivities. Employment of tert-butyl acrylate as the acceptor led to substantially higher diastereoselectivities as compared to methyl and ethyl acrylates. In addition, α,β-unsaturated cyclic ketones and esters were both competent olefins for cycloaddition, providing the tricyclic products 105 and 106 in good yield, albeit with minimal diastereoselectivities (Chart 35).
Chart 35.

Regioselective Decarboxylative Formation of Spiro[2.4]heptanes
10.2.2 [4+2] Cycloadditions with Isocyanates
Shintani and Hayashi extended their IDcA method to the palladium-catalyzed asymmetric decarboxylative [4+2] lactamization of γ-methylidene-δ-valerolactones with isocyanates.134d The reaction is proposed to proceed through a similar 1,4-zwitterionic intermediate (A, Scheme 63) which is efficiently trapped with aryl isocyanates to afford enantioenriched lactams (Scheme 64). Once again, ligand effects could be utilized to access either the spiro lactam 107 or the 6-membered lactam 108.
Scheme 64.

Decarboxylative Cycloadditions with Isocyanates
Hayashi reported that treatment of racemic lactone reactant with a palladium catalyst in the presence of alkyl isocyanates and chiral phosphoramidite ligand L-XI at 40 °C delivered the spiro-γ-lactam 109 in good yield, however with relatively low enantioselectivity (55% ee, Scheme 64). Interestingly, the seemingly minor modification of adding o-methoxy groups to the ligand (phosphoramidite L-XI→L-XII, Scheme 64) combined with changing from an alkyl isocyanate to an aryl isocyanate resulted in the formation of the highly enantioenriched δ-lactam 110 (93% ee).
The interesting reversal of the regiochemistry, and its dependence on subtle changes to the ligand, was further probed via an investigation of the effect of the electronic nature of the ligand on the regioselectivity of product formation (Table 23).136 The regioselectivity of the lactamization is not affected by the aryl α-substituent of the lactone. Instead, the observed regioselectivity is dependent on the electronic effects imparted by the ligand on the metal center. Employment of electron rich aryl-substituted phosphines favors nucleophilic attack at the terminal carbon of the π-allyl palladium complex, affording the 6-membered lactams exclusively (Table 23). On the contrary, reactions using electron deficient aryl-substituted phosphines favored nucleophilic attack at the central carbon of the π-allyl palladium complex which resulted in formation of the spiro[2.4]lactams (Table 23). These results suggest that minor alteration of ligand electronics greatly influence the electrophilicity of the central and terminal carbons of the π-allylmetal complex, which in turn has a significant influence on the regiochemistry of nucleophilic substitution. To explain this, the authors proposed that the reactive orbitals on the allyl moiety (n and π* orbitals) are energetically similar and easily attenuated via choice of phosphine ligand.137 In addition, stronger nucleophiles, like those derived from alkyl isocyanates, were suggested to have better orbital overlap with the central carbon leading to spiro[2.4]lactams (Scheme 63).137b
Table 23.
Regioselectivity of Decarboxylative Lactamizations
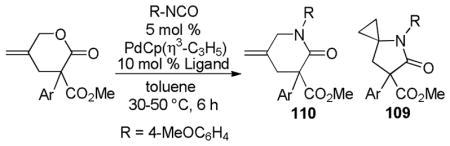 | ||||
|---|---|---|---|---|
| entry | Ar | ligand | 110 : 109 | % yielda |
| 1 | 4-MeOC6H4 | P(4-MeOC6H4)3 | >99:1 | 80 |
| 2 | 4-MeOC6H4 | P(4-CF3C6H4)3 | 4:96 | 64 |
| 3 | 4-MeC6H4 | P(4-MeOC6H4)3 | >99:1 | 86 |
| 4 | 4-MeC6H4 | P(4-CF3C6H4)3 | 5:95 | 73 |
| 5 | 3-MeC6H4 | P(4-MeOC6H4)3 | >99:1 | 85 |
| 6 | 3-MeC6H4 | P(4-CF3C6H4)3 | 5:95 | 62 |
| 7 | ferrocenyl | P(4-MeOC6H4)3 | 97:3 | 76 |
| 8 | ferrocenyl | P(4-CF3C6H4)3 | 6:94 | 71 |
isolated yield
Based on the discovery that electron rich ligands could give rise to δ-lactams with enantioselectivity (Scheme 64), Hayashi and Shintani investigated the electronic effects of the aryl isocyanates on the outcomes of their IDcA. These studies revealed that electron-rich aryl substituted isocyanates were smoothly incorporated into the decarboxylated lactones in good yield and with high enantioselectivity (Chart 36), while an electron poor aromatic isocyanate resulted in reduced yields and lower enantioselectivity. A screen of the α-substituents on the lactone revealed that electron-rich and electron-poor aromatic groups as well as heteroaromatic substituents are tolerated well under the reaction conditions. Interestingly, α-alkyl substituents were tolerated as well, however these substrates provided products in slightly lower yields and diminished enantioselectivities (e.g. 111, Chart 36). Nonetheless, the decarboxylative functionalization of α-alkyl malonates usually requires more forcing conditions to effect decarboxylation (see section 2.1.2).18 Thus, the observed decarboxylation of an α-alkyl malonate at 30 °C may provide insight regarding ways to lower the barriers for decarboxylation.136
Chart 36.

Asymmetric Generation of Lactams
To further investigate the reactivity of lactones with α-alkyl substituents, Hayashi and Shintani performed a series of experiments with deuterium labeled lactones (Scheme 65). When deuterium labeled α-benzyl lactone 112-d2 was subjected to 5 mol % Pd(PPh3)4 in toluene at 30 °C, 30% of the deuterated lactam 113-d2 was obtained. The observation of nearly equal deuterium incorporation in the ring and exo-methylene positions of 113 is consistent with the formation of a symmetrical π-allyl intermediate. Importantly, analysis of recovered lactone 112-d2 also showed 40% deuterium incorporation into the exo-methylene carbon. The deuterium incorporation into the exo-methylene carbon of the lactone suggests that ionization of the allyl carboxylate is a more facile process than decarboxylation to reveal the 1,4 zwitterionic intermediate (Scheme 63). This could be attributed to the difficulty of decarboxylation of the α-alkyl malonate derivative. In order to test this hypothesis, deuterium labeled α-phenyl lactone 114-d2 was subjected to the identical reaction conditions. After 3 minutes, analysis of the product lactam 115-d2 revealed equal amounts of deuterium incorporation in the exo-methylene as well as into the δ-position. However, analysis of recovered lactone 114-d2 revealed that there was only trace amounts of deuterium incorporation into the exo-methylene. This observation suggests that palladium-catalyzed ionization of the α-aryl-δ-valerolactones is succeeded by rapid decarboxylation.
Scheme 65.

Isotopic labeling experiments
In order to further investigate the mechanism of the decarboxylative isocyanate insertion, Hayashi and Shintani reported that subjecting enantioenriched (+)- or (−)-114 (Scheme 66) to the palladium catalyst resulted in the identical enantiomeric lactam (S)-115. Additionally, when racemic lactone (+/−)-114 is used, the ee of the lactone 114 remains at less than 15% throughout the reaction. The combination of these two observations suggest that the lactone substrate is not kinetically resolved under the reaction conditions.
Scheme 66.

Origin of Enantioselectivity: Formation of δ-Lactams
As detailed in scheme 66, reaction of the proposed zwitterionic intermediate with aryl isocyanate is the stereochemistry-determining step. As Hayashi explains, ligand coordination to the metal center blocks the si face of the enolate, which facilitates C–C bond formation with the aryl isocyanate from the re face, generating an intermediate which is poised to form the product via intramolecular nucleophilic attack.
10.2.3 [4+2] Cycloadditions with Phosphinate Protected Imines
In a similar manner to the palladium-catalyzed decarboxylative formation of δ-lactams from δ-lactones, Hayashi and Shintani reported accessing piperidine-3-carboxylic acids diastereoselectively via [4+2] decarboxylative cycloaddition of γ-methylidene-δ-valerolactones with phosphinate ester protected imines (eq 69).134g Use of the diphenylphosphinoferrocene (dppf) in toluene provided the highest yields with optimal diastereoselectivity. The reaction is proposed to proceed similarly to the reaction with isocyanates. However, in contrast to the reaction with isocyanates, the reaction with imines results in the formation of vicinal stereocenters.
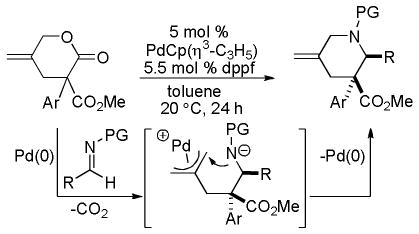 |
(69) |
The overall yield of the reaction was not substantially influenced by the α-aryl substituent of the lactone; electron rich, electron poor, and heteroaromatic substituents and alkyl substituents were all tolerated under the reaction conditions. Likewise, electronically differing aromatic imine substituents were also well suited for the cycloaddition reaction (Chart 37). However, a conjugated styryl substituted imine was not an ideal coupling partner, resulting in a significantly lower yield and low diastereoselectivity (116, Chart 37). This result is fitting with a sterically driven diastereoselection where the size of the imine substituent has an important influence on the diastereoselectivity.
Chart 37.

Piperidine-3-Carboxylic Acids via Decarboxylation of γ-Methylidene-δ-Valerolactones
10.2.4 [4+3] Cycloadditions with Nitrones
Utilizing the same pro-zwitterionic γ-methylidene-δ-valerolactones, Hayashi and Shintani developed a method for the stereoselective synthesis 1,2-oxazepines via a palladium-catalyzed [4+3] cycloaddtion with nitrones (eq 70).134a,134c Exposure of a lactone to a catalytic amount of palladium and aryl substituted nitrone delivered the highly functionalized oxazepines 117. The reaction was suggested to proceed in a step-wise fashion through intermediate A (eq 70).
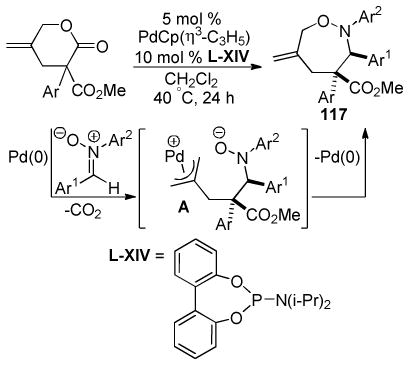 |
(70) |
An investigation of the scope of the diastereoselective [4+3] cycloaddition revealed that electron deficient, heteroaromatic, and α-naphthyl valerolactones were all tolerated well (Chart 38). Similarly, various aryl substituted nitrones provided the product oxazepines in good to excellent yield with moderate to good diastereoselectivity. Similar to their previous results, the authors reported an enantioselective variant of the [4+3] cycloadditions utilizing chiral phosphoramidite ligand L-XI (Chart 38).
Chart 38.

Decarboxylative Synthesis of Functionalized 1,2-Oxazepines
10.2.5 [4+3] Cycloadditions with Azomethine Imines
In addition to nitrones, Hayashi and co-workers were able to extend their palladium-catalyzed decarboxylative [4+3] cycloadditions to azomethine imine coupling partners (eq 71). These reactions provided the exo-methylene bicyclic heterocycles 118 in good yield with moderate diastereoselectivity.
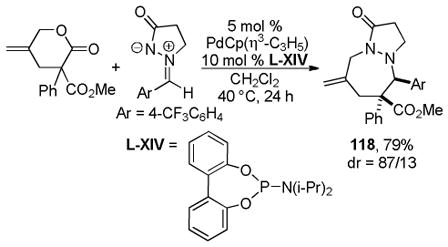 |
(71) |
10.2.6 Heptanyl Carbocycles and Heterocyles via IDcA
In keeping with the strategy of synthesizing 7-membered rings by IDcA reactions with 1,3-dipoles, Hayashi and Shintani reported the synthesis of heptanyl carbocycles and heterocycles from the palladium-catalyzed cycloaddition of γ-methylidene-δ-valerolactones with 1,1-dicyanocyclopropane and N-tosyl aziridines (Scheme 67).138
Scheme 67.

Decarboxylative Cycloaddition with Dicyanocyclopropane and N-tosyl Aziridines
As depicted in scheme 67, exposure of γ-methylidene-δ-valerolactones to palladium catalyst and 1,1-dicyanocyclopropane resulted in the heptanyl carbocycle (±)-119 as long as the ester contains an α-aryl substiuent; similar treatment with N-tosyl aziridines provided the analogous 7-membered cyclic amines (±)-120. However, as discussed above, α-alkyl substituted lactones do not readily decarboxylate which resulted in the formation of 9-membered lactones like 121 (Scheme 67). As before, an asymmetric variant of the reaction using a chiral phosphoramidite ligand L-XVI provided enantioenriched carbo- and heterocyclic esters 119 and 120 in good yield and enantioenrichment. A proposed catalytic cycle and mechanistic rationale for the products obtained is detailed in scheme 68.
Scheme 68.

Proposed Catalytic Cycle for Cycloheptane Formation
10.2.7 [4+1] Cycloadditions with Isocyanides
As detailed above, Hayashi has developed a variety of palladium-catalyzed decarboxylative [4+2] and [4+3] cycloadditions. A more recent report involved the palladium-catalyzed [4+1] cycloaddition between isocyanides and decarboxylated γ-methylidene-δ-valerolactones to deliver cyclopentenyl imines (eq 72).134e Treatment of lactone with a catalytic amount of Pd(PPh3)4 facilitated ionization and decarboxylation followed by insertion of the isocyanide to deliver A. Under the reaction conditions A undergoes olefin isomerization to form the observed cyclopentenylimine 122.
 |
(72) |
The results of an investigation of the electronic effects of the aryl substituents on both the valerolactones and the isocyanide reagents are highlighted in Chart 39. The data suggest that, of the various N-substituted isocyanides that were tested, both aryl and alkyl substituted isocyanides gave products in moderate to good yield. However, an electron-rich, p-substituted aryl isocyanides (R = p-MeOC6H4) required a change in catalyst from Pd(PPh3)4 to the more active CpPd(η3-C3H5)/dppf catalyst. The authors also developed an asymmetric palladium-catalyzed decarboxylative [4+1] cycloaddition using CpPd(η3-C3H5) and (R)-DTMB-Segphos ligand (123, Chart 39), but have not elaborated on the scope of this transformation.
Chart 39.

Cyclopentenimines via Decarboxylation of γ-Methylidene-δ-Valerolactones
10.2.8 [4+2] Cycloadditions with Isatins and other Activated Ketones
Another interesting application of the γ-methylidene-δ-valerolactones was the synthesis of spirooxindole derivatives via decarboxylative [4+2] cycloaddition with isatins and other activated ketones (eq 73).134f This report represents an interesting transformation, since ketones are normally not competent partners for Pd-catalyzed IDcA reactions. Hayashi and co-workers reported that treatment of γ-methylidene-δ-valerolactones with palladium catalyst in the presence of phosphoramidite ligand L-XVII and various isatins produced spirooxindole products in excellent yields and diastereoselectivities (eq 73).
 |
(73) |
Investigating the scope of the decarboxylative cycloaddition revealed that simple benzyl protected isatins are converted to the spiro-oxindole product in high yield and excellent diastereoselectivity (entry 1, Table 24). Moreover, the reaction also proceeded smoothly with both electron rich as well as electron deficient isatins (entries 2–4). Last, Hayashi reported successful decarboxylative [4+2] cycloadditions in the presence of other activated ketones (entries 5 and 6).
Table 24.
Decarboxylative cycloadditions with activated ketones
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | ketone | product | % yield(dr) | entry | ketone | product | % yield(dr) |
| 1 |
|
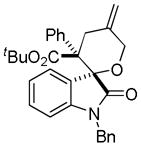
|
93(95/5) | 4 |
|
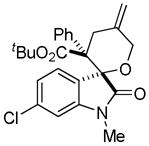
|
94(95/5) |
| 2 |
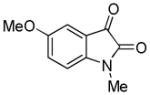
|
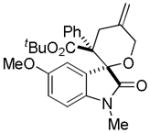
|
93(98/2) | 5 |
|
|
94(98/2) |
| 3 |
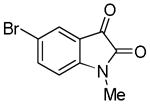
|
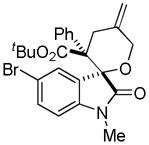
|
92(95/5) | 6 |
|
|
98 - |
Hayashi was also able to develop an asymmetric variant of the decarboxylative [4+2] cycloaddition with isatins utilizing the chiral phosphoramidite ligand L-XII, allowing for the formation of adjacent quaternary and tertiary stereocenters (eq 74). Treatment of α-3-thienyl γ-methylidene-δ-valerolactones with 5 mol % of palladium catalyst and 10 mol % L-XII in the presence of isatin at 0 °C in THF delivered the enantioenriched spirooxindole in good enantiomeric excess.
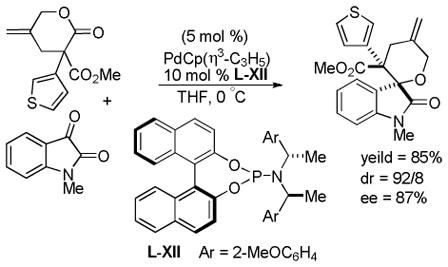 |
(74) |
10.3 Diphenylglycinate Esters
10.3.1 IDcA of Amino Acid Derivatives
In 2009, Chruma et al. reported that treatment of amino acid derived allyl diphenylglycinate imines 124 with catalytic palladium and benzylidenemalononitrile led to successful IDcA (Scheme 69).126 It was proposed that transition metal-catalyzed ionization of the allyl ester 124 and loss of CO2 generates the 2-azaallyl anion A, which is intercepted by the benzylidene malononitrile to form B. The resultant anion then reacts with the π-allyl palladium intermediate delivering allylated imine 125 as a (1:1) mixture of diastereomers. Interestingly, an investigation of the solvent effects on the diastereoselectivity of the IDcA with diphenylglycinate imines revealed that the reactions proceeded smoothly even in mixtures of toluene/H2O and MeCN/H2O. This observation is quite interesting in light of the fact that related DcA reactions are often quite sensitive to protic conditions.76,86,92
Scheme 69.

IDcA of Amino Acid Esters
Chruma also investigated the scope of each component of the IDcA reaction of allyl diphenylglycinate esters (Chart 40). Notably, arylidene malononitriles bearing electron-rich or electron poor aromatics and heteroaromatics are smoothly converted to products under the above conditions with yields ranging from 77–99% (Chart 40). Aliphatic isobutylidene malononitriles proved to be competent olefins for interception of the DcA (126), however, neopentylidene malononitriles were not favorable coupling partners (127), instead the normal DcA product 127′ resulted. A brief investigation of the effect of the substituent on the diphenylglycinate moiety (R1) showed that, with electron deficient aromatics and esters, the reaction proceeded smoothly; however, an electron rich (128) aromatic substituent led to competing formation of the regioisomeric allylation product 128′. As previously noted,129 benzylidene Meldrum’s acid adducts are sufficiently reactive to engage in many IDcA reactions. Thus, Chruma and co-workers investigated the scope of additions to aldehyde adducts of Meldrum’s acid. Indeed, both electron rich and electron deficient aromatic β-substituted arylidene Meldrum’s olefins were competent for IDcA reactions (i.e. 129), however the reactions occurred with lower yields than with their arylidene malononitrile counterparts. Remarkably, Chruma also reported the IDcA using an electron deficient aldehyde to produce 130 as a mixture of diastereomers. To our knowledge, this is the only successful intermolecular IDcA reaction with an aldehyde electrophile.89
Chart 40.

IDcA of Allyl Diphenylglycinates
Chruma proposed that the intercepted DcA reaction was initiated by oxidative addition of the allyl carboxylate leading to A. Loss of CO2 from this intermediate generates the N-bound Pd(II) intermediate B. At this point the reaction can proceed in one of three potential directions: protonation, allylation, or addition to the Michael acceptor. Based on the lack of protonated product for reactions performed in the presence of H2O, Chruma speculated that the α-amino anion equivalent remains tightly coordinated to the metal center and introduction of the activated arylidene moiety produces intermediate C (Scheme 70). Reductive elimination, or Tsuji-Trost allylation completes the construction of the IDcA product.
Scheme 70.

Proposed Catalytic Cycle for IDcA
10.3.2 Tandem IDcA Heck-Coupling
In addition to extending the scope of IDcA reactions to allyl diphenylglycinate esters, Chruma and co-workers were able to demonstrate the synthetic utility of the products by extending the method to a stepwise, one-pot IDcA-Heck cyclization (eq 75).
 |
(75) |
10.4 Allylic Alkynoate Esters
In 2005, Tunge reported that the palladium-catalyzed DcA of allylic alkynoates proceeded through palladium allyl acetylide complexes A (Scheme 71).108a Byproduct 130, observed when R′=Me, suggested that intermediates A could be intercepted by alkynes. Li and Liang recognized that use of more reactive alkynes, specifically benzynes derived from ortho-silyl triflates, would lead to a more general interceptive DcA to give products 131.108b
Scheme 71.

Interception of palladium acetylides
10.4.1 IDcA with Benzyne
En route to determining the optimal reaction conditions, Li and Liang performed catalyst, fluoride source, solvent, and temperature screens and found that Pd(PPh3)4, CsF, and MeCN at 60 °C provided optimal conversion to product.108b The scope of the IDcA of allylic alkynoates is detailed in Chart 41. Investigation of the alkynyl substituents of the propiolate esters revealed that both heteroaryl and electron rich aryl propiolates provided excellent yields. Alkyl propiolates also proved to be competent substrates under the reaction conditions, however products were formed in lower yields (Chart 41). A reaction with an electron deficient aryl propiolate (R=4-FC6H4) also formed product in low yield. A crotyl ester (R1= CH3) was also smoothly converted to product, albeit with relatively low linear to branched selectivity (l:b = 3:1). Longer chain aliphatic substituents were also tolerated and only the linear products were observed (132, Chart 41). Li and Liang also investigated the scope of the substituents on the aryne moietiey, showing that alkyl substituted benzyne and naphthyne compounds effectively intercepted the DcA; however, electron deficient aryne moieties, greatly reduced the product yield (133, Chart 41).
Chart 41.

IDcA of Allylic Alkynoates with Benzynes
Terminal cinnamyl propiolic esters were also investigated as reactive partners for the IDcA under the above reaction conditions. It was reported that a terminal propiolate and 2-(trimethylsilyl)phenyl trifluoromethanesulfonate underwent decarboxylative coupling, however the reactants formed an unexpected product 134 (eq 76). Compound 134 could potentially be rationalized by a Sonogashira-like coupling from an intermediate IDcA product.
 |
(76) |
Several mechanisms were proposed to rationalize the formation of IDcA products (Scheme 70). For simplicity, the substituents on both allyl and aryne moieties have been ignored. Beginning with allylic alkynoate, exposure to the Pd(0) catalyst could generate π-allyl palladium propiolate II, or rapid decarboxylation could deliver π-allyl palladium alkynyl intermediate I. This palladium allyl acetylide complex can react via two related paths. Path A involves migratory insertion of the Pd-acetylide onto the aryne functionality which results in aryl palladium-π-allyl intermediate III, subsequent reductive elimination results in a new sp2-sp3 C–C bond. Path B involves migratory insertion of the π-allyl palladium moiety onto the aryne functionality resulting in aryne-palladium-alkyne intermediate IV. The ensuing reductive coupling generates a new sp-sp2 C–C bond. A plausible alternative reactive manifold is path C which entails migratory insertion of aryne into the π-allyl palladium bond resulting in aryl-palladium-propiolate intermediate V. Loss of CO2 would lead to intermediate IV, which proceeds to product through reductive elimination (Scheme 72).
Scheme 72.

Proposed Catalytic Cycles for IDcA of Allylic Alkynoates
10.5 AllylCarbonate Esters
10.5.1 Decarboxylative Carbonylation of AcyclicCarbonates
It is well know that allylic carbonates are efficient allylating reagents vis-a-vis the Tsuji-Trost reaction.139 In the same vein, Tsuji reported another mode of interceptive DcA involving the decarboxylative carbonylation of acyclic allyl carbonates with carbon monoxide to form β,γ-unsaturated esters.140 Specifically, allyl ethyl carbonate was converted to a mixture of the β,γ-unsaturated ester and ethyl allyl ether in the presence of a catalytic Pd(0) source under an atmosphere of carbon monoxide (eq 77). The regiochemistry of the reaction was investigated with the branched isomeric 3-hept-1-enyl carbonate, which formed the linear substituted-β,γ-unsaturated ester, albeit in low yield (eq 78). Similarly, a diallyl carbonate was allowed to react with the palladium catalyst and CO. While decarboxylative carbonylation to form β,γ-unsaturated allyl esters was facile (eq 79), even prolonged exposure to the reaction condition did not result in further IDcA of the allyl acetate derivative to form the anhydride product.
 |
(77) |
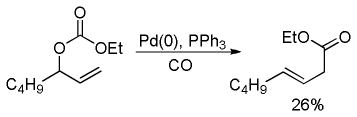 |
(78) |
 |
(79) |
Optimized reaction conditions for the decarboxylative carbonylation were established after a solvent, temperature, CO pressure, and ligand screen. Traditional solvents such as toluene, benzene, THF, and acetonitrile were not ideal for the decarboxylative carbonylation reaction. For this reason, the reactions were performed neat. The ideal reaction temperature was found to be 50 °C, and 5–10 atm was the most effective CO pressure. While the entirety of reactions with allyl carbonates detailed in the schemes above were performed with monodentate phosphine ligands, Pd(OAc)2 and bidentate bis-diphenylphosphinoethane (dppe) also formed an active catalyst for the decarboxylative carbonylation of ethyl allyl carbonate (eq 80). In addition to obtaining the target β,γ-unsaturated ethyl ester 135, Tsuji also reported generating the mono- and diallylated β,γ-unsaturated ethyl esters 136 and 137.
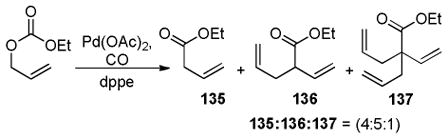 |
(80) |
The mechanism proposed by Tsuji for the decarboxylative carbonylation of ethyl allyl carbonates is detailed in Scheme 73. Ionization of allyl ethyl carbonate with a catalytic amount of Pd(0) generates π-allyl-palladium-carbonate A which decarboxylates to form the π-allyl-palladium-alkoxide B. Migratory insertion of CO generates C and subsequent reductive elimination, or acyl substitution,141 affords the γ,δ-unsaturated ester product and regenerates the palladium catalyst.
Scheme 73.

Proposed Mechanism for Decarboxylative Carbonylation
10.5.2 Decarboxylative Carbonylation of Cyclic Carbonates
In 1987, Bando and Tamaru reported the extension of carbonylative DcA reactions to a cyclic carbonate leading to 2-vinyl-γ-butyrolactones 138 (eq 82).141b As the reaction conditions were optimized, Tamaru found that the product selectivity was solvent dependent. A mixture of dioxane/THF proved to be the ideal solvent system, achieving product yields up to 89%. The authors further noted that reactions in polar protic solvents did not result in the formation of the butyrolactone 138, rather a mixture of isomerized butyrolactone 139 and a β,γ-unsaturated ester 140 was obtained (eq 81). An investigation of the scope of the reaction revealed that non-substituted vinyl carbonates (R1=R2=R3=H) were converted to product, albeit with reduced yield (141, Chart 42). A carbonate with a tert-butyl substituent (R1=tBu) was subjected to the above reaction conditions and converted to the disubstituted-γ-butyrolactone (142), however the reaction proceeded with no diastereoselectivity. Lastly, it is notable that 3-methyl vinyl carbonate was converted to the α-quaternary butyrolactone 143 in relatively good yield (Chart 42); typically carbonylation of sterically hindered carbon centers is difficult.
Chart 42.

γ-Butyrolactones via Decarboxylative Carbonylation
 |
(81) (82) |
The stereoselectivity of the decarboxylative carbonylation reaction was revealed via a series of reactions with diastereomerically enriched substrates. For example, a nearly 1:1 mixture of cis:trans carbonate was stereoselectively converted to the α-vinyl-β-benzyl-γ-butyrolactone as a single diastereomer (eq 83). The stereoconvergence can be attributed to the facile π–σ–π interconversion of the intermediate π-allyl complex.1d On the other hand, a cis-carbonate, which cannot epimerize through π–σ–π interconversion, underwent stereospecific conversion to the trans-γ-butyrolactone (eq 84). Under the same conditions, the trans-carbonate 144 yielded nearly equal amounts of both the cis-and trans-γ-butyrolactones 145 and 146 (Scheme 74). Interestingly, 146 was obtained with a cis-configuration about the olefin. To explain the observed diastereoselectivities, Tamaru and Bando proposed that the palladium-catalyzed ionization of the allylic carbonate proceeded with inversion of stereochemistry (A, Scheme 74).142 Subsequent CO2 loss and CO insertion is acheived with retention of stereochemistry, thus rationalizing the stereospecific formation of the cis-145. However, the high energy cis-intermediate A can isomerize to the trans-palladacycle C via the intermediacy of an η1-allyl palladium complex. As such, rotation about the Pd–C bond in B, followed by CO insertion, explains the observed formation of the cis-olefinic product 141 (Scheme 74).
Scheme 74.

Rationale for Observed Stereochemistry for the Decarboxylative Carbonylation
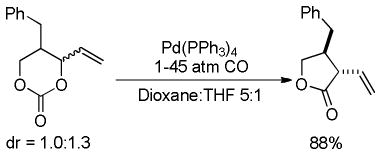 |
(83) |
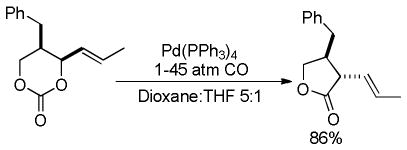 |
(84) |
In a similar manifold, Tamaru and Bando employed cyclic vinyl carbonates along with isocyanates to access cyclic vinyl carbamates 147 (Scheme 75).141a,143 In this case, the intermediate π-allyl-palladium alkoxide A is intercepted with isocyanates (instead of CO) to form amides B, subsequent attack of the carbamate on the π-allyl-palladium moiety results in cyclic carbamate 147.
Scheme 75.

Decarboxylative Isocyanation
A probe into the scope of the reaction showed that both electron rich and electron poor aryl isocyanates underwent carbamate formation smoothly, and the carbonate-carbamate transformation tolerated substitution at all carbons with the exception of C-3 (Chart 43). Investigating the scope of isocyanates further revealed that reactions with the N-alkyl isocyanate, benzyl isocyanate, did not result in carbamate formation. Fitting with the proposed formation of an intermediate carbamate anion, the highest yields were obtained with N-substituents that could stabilize the intermediate carbamate anion. Thus, tosyl isocyanate was chosen as the isocyanate of choice for investigating the diastereoselectivity of the reaction (Chart 43).141a,143
Chart 43.

Palladium-Catalyzed Decarboxylative Carbamate Formations
Investigating the effect of the carbonate substituent at R2 revealed that methyl and phenyl groups were compatible, however the diastereoselectivity for formation of the trans product was much better for the phenyl substituent (148, 149, Chart 43). Carbonates with both aromatic as well as bulky alkyl groups at R1 provided products in high yield (150, 151). For these cases, it is noteworthy that the reaction conditions determined the major diastereomer that was obtained. At lower temperature and reduced reaction times the cis-diastereomers were favored; however at a higher temperature and longer reaction time the trans-diastereomers were favored. These results suggest a kinetic preference for formation of the cis-product which can isomerize to the more stable trans isomer via ionization and π–σ–π equilibration upon prolonged reaction. As expected, reactions beginning with diastereomerically enriched carbonates that cannot undergo epimerization via π–σ–π equilibration stereospecifically form either cis- or trans-152 from the cis- and trans-carbonates respectively (Chart 43).
Bando and Tamaru’s rationale for the observed diastereoselectivity for products 148 and 149 is depicted in scheme 76. Palladium-catalyzed ionization of either cis- or trans-carbonate proceeds with inversion at C-3, subsequent decarboxylation and addition to the isocyanate results in intermediates A and D. Equilibration of A and D via η1-allyl palladium intermediates is much faster than cyclization to form the carbamates. Thus, the diastereoselectivity is controlled by the rates of ring closure. The transition state to form the cis-product is destabilized by a developing 1,3-diaxial interaction which favors the formation of the trans-product.
Scheme 76.

Rationale for Observed Diastereoselecitivites
10.5.3 Decarboxylative Michael Addition/Allylation
Yamamoto further expanded the concept of inserting activated olefins into the intermediates of palladium-catalyzed decarboxylation of allyl carbonates.128a,133 Specifically, treatment of acyclic allylic ethyl carbonates with a palladium catalyst in the presence of electron deficient olefins delivered alkoxyallylation products (eq 85). Similar to his IDcA reactions using β-ketoesters,128b Yamamoto, investigated how the electrophilicity of benzylidine and alkylidine malononitriles affected the palladium-catalyzed decarboxylative alkoxyallylation of electron deficient olefins. Arylidene malononitriles with electron donating and electron withdrawing substituents underwent IDcA with allyl ethyl carbonates in high yield (entries 3 and 4, Table 25). A bulky alkylidene malononitrile that does not have acidic hydrogens was also sufficiently reactive to undergo IDcA in high yield (entry 6). Allylic carbonates with substituents on the allyl moiety were also subjected to the reaction conditions in the presence of benzylidene malononitriles and provided the linear allylation product selectively (entry 7, Table 25).
Table 25.
Palladium-Catalyzed Alkoxyallylation of Alkylidene Malononitriles
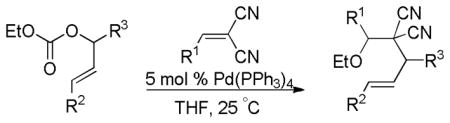 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | R3 | yield %a(l:b) |
| 1 | Ph | H | H | 92 |
| 2 | 4-MeC6H4 | H | H | 81 |
| 3 | 4-MeOC6H4 | H | H | 92 |
| 4 | 4-MeO2CC6H4 | H | H | 85 |
| 5 | 1-Napthyl | H | H | 90 |
| 6 | tBu | H | H | 99 |
| 7 | Ph | Me | H | 92 (78:22) |
isolated via column chromatography
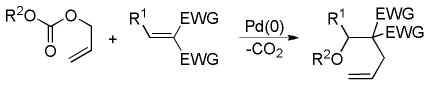 |
(85) |
The proposed catalytic cycle for the decarboxylative alkoxyallylation reaction begins with ionization of the allyl ester moiety and loss of CO2 (Scheme 77). The resulting ethoxide then reacts with the electrophilic arylidene malononitrile, forming a new C–O bond to deliver ion pair intermediate B. Subsequent regioselective attack of the stabilized nucleophile on the cationic π-allyl palladium moiety generates alkoxyallylated product and regenerates the Pd(0) catalyst.
Scheme 77.

Proposed Mechanism for Decarboxylative Alkoxyallylation
10.6 Allyl Carbamate Esters
10.6.1 Decarboxylative Carbonylation AcyclicCarbamates
In 1984, Tsuji reported the decarboxylative carbonylation of allylic carbonates to form β,γ-unsaturated esters.140b In an analogous fashion, Yamamoto and Miyazawa reported the decarboxylative carbonylation of acyclic allylic carbamates (eq 86).144
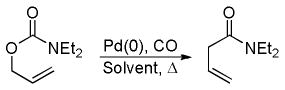 |
(86) |
The reaction entailed treatment of allyl diethyl amido carbamates with a catalytic source of Pd(0) under an atmosphere of CO to afford β,γ-unsaturated diethylamides (eq 86). The specific reaction conditions used to convert the carbamates into amides were 5 mol% Pd2(dba)3·CHCl3, 20 mol % PPh3, under 80 atm of CO, at 100 °C. As before, the reactions were performed under solvent-free conditions. Under these conditions, methallyl and cinnamyl carbamates were converted to β,γ-unsaturated diethylamides 153 and 154 in moderate to good yields (Chart 44). Likewise, aliphatic allyl carbamates were converted to the corresponding amides in moderate yields; however, the control of the olefin geometry was not high (155, 156). An acryloyl carbamate was likewise converted to the substituted β,γ-unsaturated amide 157 with high regio- and diastereoselectivity; however, the reaction suffered from a low yield due to significant competing elimination.
Chart 44.
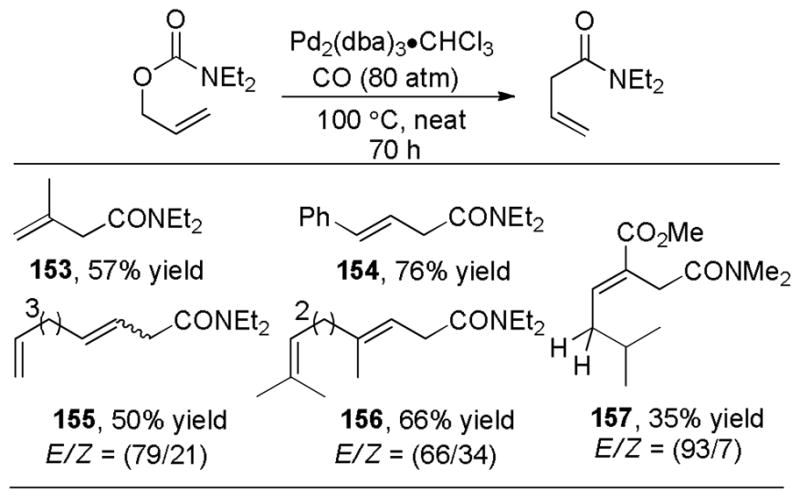
Decarboxylative Carbonylation of Allyl Carbamates
10.6.2 Decarboxylative Carbonylation of Cyclic Carbamates
Similar to the aforementioned decarboxylative carbonylation of allylic carbonates, Tamaru reported the decarboxylative carbonylation of allylic carbamates in 1992 (Scheme 78).145 The reaction involved smooth conversion of cyclic allyl carbamates to γ-lactams 158 in the presence of catalytic palladium under an atmosphere of CO in dioxane solvent. The authors further showed that ionization of a 1:1 mixture of diastereomeric carbamates generated the product as a single diastereomer via facile epimerization of intermediate A.
Scheme 78.

Decarboxylative Carbonylation of Cyclic Vinyl Carbamates
Tamaru and co-workers further investigated the effect of substituents on the tertiary amide of the cyclic carbamates on the IDcA reaction. As detailed in scheme 79, when the substituent on the tertiary amide is benzyl, the reaction was not successful (159c→162). The lack of reactivity was attributed to the inability of allyl carbamates with electron donor substituents on nitrogen to decarboxylate.146 Moreover, when reactions were performed in ethanol with electron withdrawing Ts or COPh N-substituents, cyclic lactams were not observed (e.g. 159a, 159b). Instead, Tamaru suggested that intermediate A (R= Ts, COPh) is protonated in the polar protic solvent, allowing for the formation of the acyclic amino ester products 160 and 161 (Scheme 79).
Scheme 79.

Decarboxylative Carbonylation of Cyclic Vinyl Carbamates
10.6.3 Decarboxylative Carbonylation of Oxazolidinones
In 2000, Knight extended the decarboxylative carbonylation method to the asymmetric synthesis of 3,6-dihydro-1H-pyridin-2-ones 163 from non-racemic 5-vinyloxazolidin-2-ones (eq 87).147 Similar to the report by Tamaru, if the nitrogen of the oxazolidinone was Boc-protected (R= Boc), the authors obtained acyclic the β,γ-unsaturated amino ester 164 (eq 88). However, when the amide nitrogen was not protected (R=H), the reaction resulted in formation of dihydropyridinone 163 (eq 87). Optimization of the lactam formation showed that decarboxylative carbonylation proceeded in good yield with 5 mol % Pd(OAc)2(PPh3)2 under 65 atm of CO pressure, in EtOH at 70 °C, although extended reaction times (120 h) were required. With these reaction conditions in hand, Knight investigated the scope of the reaction and showed that substrates with aliphatic substituents at R1 were converted in good yield to the respective dihydropyridinones (Chart 45). In contrast, a substrate with substitution at R3 (R3 = Me) did not afford any product and only the starting material was recovered from the reaction solution. This lack of reactivity could be attributed to the difficulty of carbonylation of a sterically hindered tertiary carbon center.
Chart 45.

Dihydropyridinones via Decarboxylation of Vinyloxazolidinones
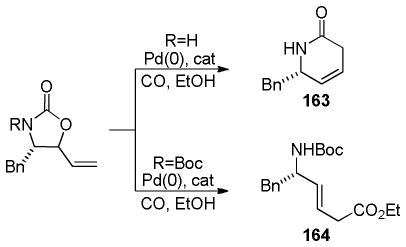 |
(87) (88) |
A mechanistic rationale for the formation of the dihydropyridones involves initial ionization of the ester moiety to generate a syn-π-allyl palladium carbamate intermediate A (Scheme 80). To investigate the π-allyl palladium formation, Knight found that treatment of a mixture of vinyl oxazolidinone diastereomers (R1=i-Pr, R2=R3=H) with Pd(PPh3)4 resulted in recovery of only the trans-vinyl oxazolidinone. Based on this observation, Knight postulated that ring opening of the vinyl carbamates is reversible. This result also suggested that decarboxylation to form primary amine intermediate B is not facile. A sluggish decarboxylation allows for the π-allyl palladium intermediate to isomerize to the anti-A configuration that is more favorable for formation of the cyclic lactam.148 Subsequent decarboxylation and carbonylation followed by cyclization delivered dihydropyridinone. It should be noted that the maintanence of the enantioenriched center indicates that reversible β-hydride elimination from any π-allyl intermediates is likely not occuring. Knight also extended the palladium-catalyzed decarboxylative carbonylation method to the diastereospecific,148 diastereoselective,149 and enantiospecific150 synthesis of 3,6-dihydro-1H-pyridin-2-ones.
Scheme 80.

Proposed Catalytic Cycle for Decarboxylative Carbonylation
10.6.4 Cycloadditionsof Divinyl Oxazolidinones
In addition to their cycloadditions with CO, Knight showed that vinyl oxazolidinones undergo a palladium-catalyzed [3+2] cycloaddition with activated olefins to afford highly substituted pyrrolidines (Chart 46).151 The decarboxylative cycloaddition occurs with benzylidene malononitriles (165), benzylidene Meldrum’s acid adducts (166), and a cyano chromone (167) to provide substituted pyrrolidines in good yield.151
Chart 46.
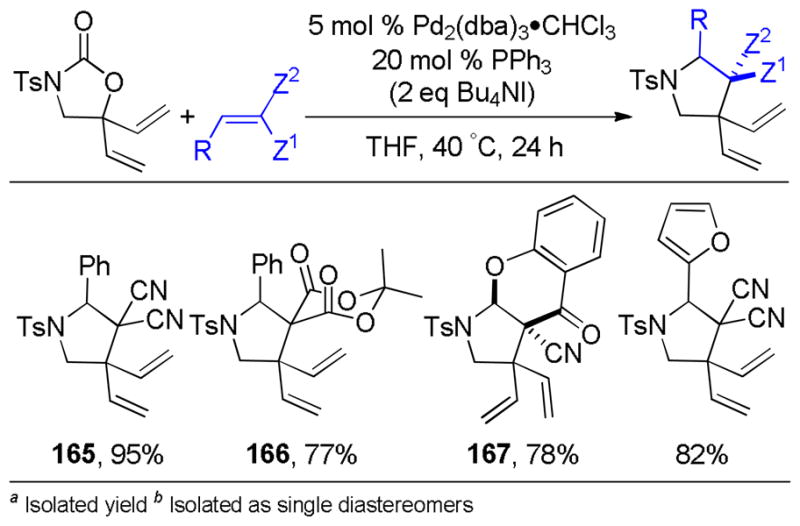
Decarboxylative [3+2] Cycloaddition with Activated Olefins
10.6.5 Diastereoselective Cycloadditions of Vinyl Oxazinones
Building on investigations of palladium-catalyzed reactions of allylic carbamates by Tsuji,146 Tamura,145 Cook,152 and Tunge,110–111 Tunge and Wang developed a similar palladium-catalyzed IDcA of vinyl oxazinones with Michael acceptors to form highly substituted 4-vinylpiperidines (Scheme 81). Specifically, treatment of rac-168 with one equivalent of benzylidene malononitrile, and 5 mol% Pd(PPh3)4 in CH2Cl2 produced vinyl piperidine 169 as a single diastereomer (Scheme 79). The reaction was believed to proceed via the ionized and decarboxylated intermediate tosyl amide A. This ambiphilic intermediate can undergo addition to benzylidene malononitrile, reversibly generating the zwitterionic intermediate B. Both cis- and trans-168 were converted to the same product, showing that epimerization through π-σ-π allyl interconversion is more rapid than cyclization.153
Scheme 81.

Diastereoselective Cycloadditions of Vinyl Oxazinones
An investigation of the scope of the IDcA demonstrated that the reaction tolerated substitution at the carbinol carbon (R2=Me), providing products in high yield (entries 5–7, Table 26). Substitution at the 5-position of the carbonate (R1) and at the vinyl carbon (R3) generated lower yields; however, the products were generated with good diastereoselectivity (entries 1,2). Carbamates lacking any substitution underwent cycloaddition with low diastereoselectivity, favoring the trans diastereomer (entries 3, 4, Table 26). Likewise, for substrates in which R2=methyl the reactions proceeded in excellent yield, but exhibited minimal trans diastereoselectivity (entries 5 and 6). This trans selectivity is amplified when a larger Ph substituent was used in the R2 position (entry 7, Table 26). To explain the observed selectivities, the authors speculated that reversible addition of the tosyl amide to the benzylidene malononitrile would allow the cyclization to take place through the lowest energy conformer. The observed diastereoselectivities were then rationalized using a model that placed the large substituents in the equatorial positions about a six-membered cyclization transition state (A or B, Chart 47). Thus, if either R1 or R2 was large, high diastereoselectivities resulted.
Table 26.
Yields and Diastereoselectivies of IDcA
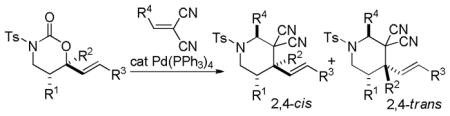 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | R4 | yield (dr)a,b |
| 1 | Ph | H | Ph | p-AcOC6H4 | 53 (>19:1) |
| 2 | Ph | H | Ph | Ph | 54 (10:1) |
| 3 | H | H | H | p-AcOC6H4 | 99 (1:2.8) |
| 4 | H | H | H | Ph | 85 (1:3) |
| 5 | H | CH3 | H | p-AcOC6H4 | 92 (1:2) |
| 6 | H | CH3 | H | Ph | 99 (1:2) |
| 7 | H | Ph | H | Ph | 87 (1:>19) |
Yield and syn/anti ratio of isolated product.
>19:1 indicates that the minor diastereomer was not detected by 1H NMR spectroscopy.
Chart 47.
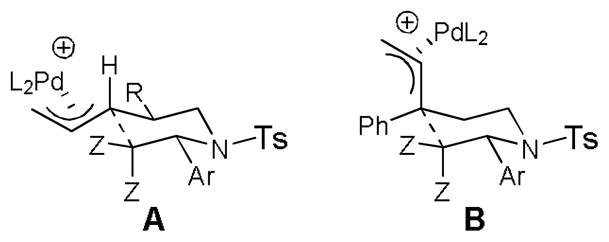
Rationale for Observed Diastereoselectivity
10.6.6 Asymmetric Cycloaddition of Benzoxazinanone
In 2008, Tunge and Wang reported a similar palladium-catalyzed decarboxylation of benzoxazinanone 170 followed by cycloaddition with arylidene malononitriles to generate highly substituted dihydroquinolines 171 (eq 89).112 In analogy to the intermediates derived from simple vinyl oxazinanones, vinyl benzoxazinones are proposed to form zwitterionic intermediates A. These intermediates are equivalent to aza-ortho-xylylenes that are coordinated to, and polarized by palladium (eq 89).
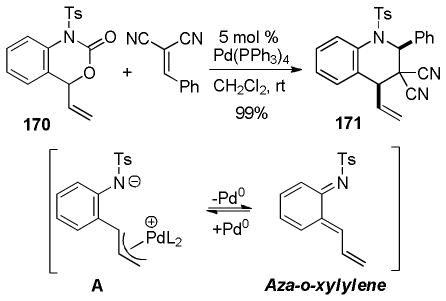 |
(89) |
Ligand studies directed at developing an asymmetric cycloaddition revealed that the anthracenyl Trost ligand (L-III) provided both high enantioselecitivity and diastereoselectivity (Chart 48). The reaction conditions chosen to probe the scope of the decarboxylative aza-o-xylylene formation and subsequent cycloaddition with arylidene malononitriles entailed: 5 mol % Pd2(dba)3 and 10 mol % L-III in CH2Cl2 at room temperature for 4–6 hours. Electron rich and electron poor vinyl benzoxazinanones were screened for cycloaddition, enantioselection, and diastereoselection as well as compatibility with various arylidene malononitriles. In general, the cycloadditions proceeded smoothly under the reaction conditions delivering products with good yield, high enantioselectivities, and diastereoselectivities (Chart 48). As detailed above, more electron-rich benzylidene malononitriles are not as well suited for the cycloaddition, thus electron deficient arylidene malononitriles proved to be ideal reactive partners for cycloaddition. In addition, vinyl benzoxazinones that lacked a substituent para- to the nitrogen provided products in substantially lower yield.
Chart 48.

Diastereo- and Enantioselective Decarboxylative Cycloaddition
Tunge and Wang proposed a stepwise cycloaddition mechanism that is based on their previous determination that such reactions proceed through zwitterionic intermediates like A (Scheme 82).111 Treatment of 170 with the active catalyst generates a palladium polarized aza-o-xylylene intermediate A (Scheme 82). Subsequent conjugate addition to the benzylidene malononitrile resulted in two potential equilibrating intermediates B or C. The stereochemical influence of the chiral ligand suggests that the cyclization of one of these intermediates is favored. Thus, Tunge speculated that the reaction proceeded via an initial reversible conjugate addition followed by a stereochemical defining cyclization.
Scheme 82.

Stereochemical Rationale
10.6.7 IDcA of Acyclic Carbamates
Previously, Tunge reported that cyanocoumarins were competent partners for the ruthenium-catalyzed decarboxylative insertion of electrophiles into β-ketoesters.129 Further investigation proved that the 3-cyanocoumarins were indeed sufficiently reactive toward the palladium-catalyzed IDcA with a cyclic allyl carbamate to generate the tricyclic product 172 (Scheme 83). Moreover, syn-addition to the 3-cyanocoumarin set three contiguous stereocenters with high diastereoselectivity. Similar studies were published in the same year by Yamamoto, in which acyclic N-phenyl carbamates 173 underwent intercepted decarboxylative allylation with 3-cyanocoumarins in the presence of catalytic palladium to generate α-allyl, β-amino dihydrocoumarins 174 (Scheme 83).154 Yamamoto also extended the scope of the reaction to 3-substitued cyano- and formylchromones (Chart 49). The reactions of 3-cyanochromenones with N-phenyl carbamates were tolerant of methyl-, bromo-, and chloro- substituents on the phenyl ring, providing products in good to excellent yields. Moreover, the products were formed as single diastereomers resulting from trans addition to the olefin. Examples utilizing the 3-formylchromones suggested that conversion to product was more favorable with electron withdrawing substituents on the phenyl ring.
Scheme 83.

Interception with 3-Cyanocoumarin
Chart 49.

α-Allyl-β-amino Chromones
Another competently reactive olefin for interceptive DcA was 2-cyanocyclohexenone (eq 90). Interestingly, 2-cyanocyclopentenone was not converted to product 177 under the above reaction conditions (eq 91). Other unreactive olefins include simple acrylonitriles (e.g. PhHC=CHCN) as well as other activated olefins [PhHC=C(CN)CO2H, PhHC=C(CN)SO2Ph, and H2C=C(CN)2]. Further investigation of the scope of carbamates that add to cyanocyclohexenone showed electron rich aryl, or alkyl, N-substitutents provided the best results (eq 90, R = p-MeC6H4, p-OMeC6H4, C6H11, CH3). Notably, the trans stereoselectivity for addition to 2-cyanocyclohexeneone is the same as that observed for additions to the chromones. Yamamoto suggested that the observed stereoselectivity is explained by the disfavored 1,3 diaxial interactions of the large π-allyl-palladium (Chart 50). It was further suggested that equilibration via hetero bis-π-allylpalladium complex B, facilitates the formation of the more energetically favorable equatorial π-allypalladium species (C), leading to the observed anti-stereoselectivities.155 That model of stereoselection assumes an inner sphere allylation mechanism,155 however, related stabilized nucleophiles typically react via an outer-sphere mechanism.1d In such a case, the stereochemistry can be attributed to allylation anti to the bulky carbamate.
Chart 50.
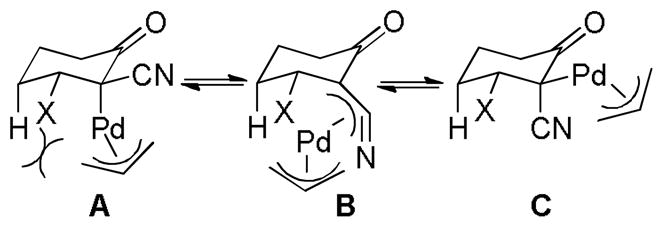
Yamamoto’s Proposed Stereochemical Rationale
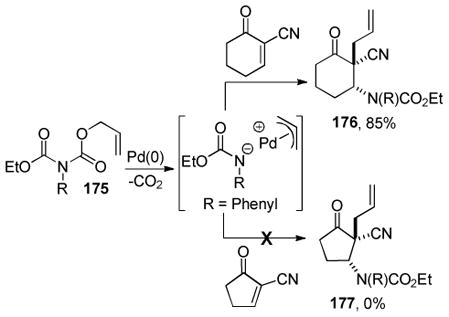 |
(90) (91) |
Lastly, Yamamoto showed that treatment of simple allyl amides with Pd(0) in the presence of benzylidenemalononitrile does not result in intercepted DcA product (eq 92). This result highlights the importance of generating the carbamate nucleophile via decarboxylation.
 |
(92) |
10.7 IDcA of Propargyl Carbonates
In 1996 Bruneau and Dixneuf reported a palladium-catalyzed decarboxylative dihydrofuran synthesis that represents an interesting type of IDcA reaction (eq 93).156 Specifically, treatment of cyclic propargyl carbonates with acrylates and a catalytic amount of palladium generated alkenyl-2,5-dihydrofuran upon loss of CO2 (eq 93). An investigation into the scope of the reaction revealed that aliphatic alkynyl substituents are reactive coupling partners with ethyl acrylate to produce dihydrofurans (Chart 51). The same was observed for reactions using acrylamides.
Chart 51.

Palladium-Catalyzed Decarboxylative Synthesis of Dihydrofurans
 |
(93) |
Dixneuf and Bruneau proposed the following mechanism to explain this interesting transformation (Scheme 84). Exposure of the cyclic propargyl carbonate to potassium bromide and the palladium catalyst results in nucleophilic ring opening and decarboxylation to provide decarboxylated allenylpalladium intermediate A (Scheme 84). Heck-type coupling of intermediate A with the activated olefin forms intermediate C and a palladium-hydride. Subsequent hydridopalladation of the allenyl moiety results in π-allyl palladium intermediate D, which can undergo nucleophilic attack from the alkoxide moiety to afford the observed dihydrofuran product (Scheme 84).
Scheme 84.

Catalytic Cyclic for IDcA of Propargyl Carbonates
11 Decarboxylative Benzylation
11.1 Introduction to Decarboxylative Benzylations
Much of the chemistry discussed thus far has relied on the ability of metals to undergo oxidative addition with allyl acetate derivatives to form π-allyl metal complexes and then form a nucleophile via decarboxylation (vide supra). Analogous decarboxylative benzylation reactions have recently started to appear in the literature. To close this review, we will discuss these advancements.
As decarboxylative allylation had its beginnings with the Tsuji-Trost reaction of malonates, decarboxylative benzylation eminated from related reactions with stabilized nucleophiles (Scheme 85). Starting in the early 1990’s, various benzyl acetates and carbonates were shown to couple with nucleophiles in the presence of palladium catalysts (Scheme 85, A to C). Since that time, the breadth of nucleophilic coupling partners has steadily increased and this work has been recently reviewed.157 For example, malonate-type nucleophiles,158 boronic acids,159 and more recently, direct arylation methods,160 have been utilized to make new benzyl C–C bonds though palladium-benzyl intermediates. With respect to heteroatomic nucleophiles, catalytic benzylations of phenols,161 amines,158d and sulfonyl nucleophiles162 have all been disclosed. Although this review focuses on the decarboxylative benzylation of relatively unstabilized nucleophiles (Scheme 85, B to C), some relevant information can be gained by briefly discussing aspects of the previous catalytic benzylations of stabilized nucleophiles.
Scheme 85.

Comparison of π-Benzyl Substitution Reactions
The earliest examples of palladium-catalyzed benzylation reactions were limited to naphthyl or quinolyl acetates, since simple benzyl acetates provided poor conversion.158b The decreased rate of π-benzyl formation from benzyl acetates compared to aromatics with extended π-systems has been ascribed to the disruption of aromaticity upon π-benzyl formation.158b In the case of naphthyl, or other benzyl derivates where aromaticty isn’t completely disrupted, the rate of π-benzyl formation is greater than that observed for simple benzyl derivates (Scheme 86). Nonetheless, certain palladium catalysts have been shown to promote π-benzyl formation of even simple benzyl esters.158d It should also be noted that, while it is often assumed that palladium π-benzyl intermediates are involved in these reactions, π-benzyl complexes may prefer to form the corresponding σ-benzyl isomers, and the σ-benzyl isomers may be the actual reactive intermediates.163
Scheme 86.

Relative Rates of Palladium π-Allyl/Benzyl Formation
As stated, benzyl substitution reactions where a pro-nucleophile and a benzyl acetate or carbonate are coupled using a palladium catalyst has been reviewed in detail.157b Conversely, decarboxylative benzylations are still in their infancy; decarboxylative benzylations of phenols,161 diphenylglycinate imines,164 acetylides,165 and ketones165 have only recently been reported.
11.2 Decarboxylative Benzylation of Various Nucleophiles
11.2.1 Decarboxylative Benzyl Ether Synthesis
The first general method for decarboxylative benzylation was reported by Kuwano and coworkers who showed that aryl benzyl carbonates would undergo decarboxylative coupling to form benzyl phenyl ethers (Chart 52).161 Importantly, such benzyl ethers are frequently used as protecting groups in organic synthesis. In contrast to common methods for benzyl ether synthesis which require basic or acidic conditions and often utilize stoichiometric coupling reagents,166 Kuwano’s palladium-catalyzed decarboxylative benzylation of alcohols allows benzyl ethers to be synthesized under neutral conditions with no stoichiometric additives.
Chart 52.

Decarboxylative Benzylation of Phenols
Kuwano investigated a number of palladium catalysts and reaction conditions for decarboxylative benzylation, eventually achieving quantitative formation of benzyl phenyl ether utilizing 1 mol % of CpPd(allyl) precatalyst with DPEphos as a ligand at 60 °C. Although alkyl benzyl carbonates could not be coupled, various benzyl phenyl carbonate derivatives provided high yields of product under the optimal reaction conditions (Chart 52).
With regard to the phenolic coupling component, electron rich, electron poor, and sterically encumbered substrates all provided high yields (Chart 52); however, in order to achieve a high yield for benzylation of a nitrophenol, longer reaction times (22 hours) and a high catalyst loading (5 mol % Pd) were required. Presumably, this was due to the decreased nucleophilicity of the in-situ generated 4-nitrophenoxide. This decarboxylative strategy was also applicable for benzylation with diarylmethanes as well as p-methoxybenzyl groups, both of which are important alcohol protecting groups in organic synthesis.
11.2.2 Decarboxylative Benzylation of Diphenylglycinate Imines
Building off their previous work on the decarboxylative allylation of allyl diphenylglycinate imines,89 Chruma and coworkers reported the palladium-catalyzed decarboxylative benzylation of diphenylgycinate imines in early 2010 (Scheme 87).167 In this approach, the palladium catalyst promotes the formation of the π-benzyl complex and, upon decarboxylation, generates the 2-aza-allyl anion coupling partner. As observed in the related allylation, the α-imino anion undergoes benzylation at the least hindered carbon to generate the desired benzylated adduct 178.
Scheme 87.

Decarboxylative Benzylation of aza-Allyl Anions
Under the optimal reaction conditions, yields varied from good to poor with some degree of predictability. Electron-donating substituents that stabilize the π-benzyl complex allowed for favorable ionization and provided higher yields of product (178, Chart 53). Conversely, when the benzyl substituent was substituted with a para-nitro substituent, the yield was drastically lowered (180). In addition, electron-withdrawing groups on the imine component, which are expected to accelerate decarboxylation, also facilitate the reaction. The importance of a stabilized azaallyl anion can be seen in the comparison of 179 and 181, as the benzylation of the p-cyano substituted anion led to 81% yield of the desired product while the analogous unsubstituted product was formed in a modest yield of 44%. Ultimately, the combination of electron rich benzyl electrophiles with electron deficient amino acid derivatives provided the highest yields of products (178, Chart 53). In general, deviation from the optimal electronics of the coupling partners led to less favorable π-benzyl formation or decarboxylation and decreased the yields of the desired products. That said, many examples, including couplings of heteroarenes (184, 185), were shown to lead to desired product formation with an average yield of 53%. In addition to the desired benzylation products, the mass balance was stated to emanate from decarboxylative protonation of the azaallyl anion and acetoxylation of the π-benzyl intermediate (from the Pd(OAc)2 precatalyst).
Chart 53.

Select Examples of Benzylated α-Imino Anions
11.2.3 Decarboxylative Benzylation of Acetylides
As stated earlier, π-benzyl formation occurs more readily with extended aromatic systems than with simple benzyl esters. Tunge and coworkers have demonstrated that various polyaromatic benzyl propiolates and benzyl β-keto esters undergo high yielding decarboxylative coupling in the presence of a palladium catalyst (Chart 54).165
Chart 54.

Decarboxylative Coupling of Benzyl Propioliate and β-Keto Esters
Specifically, the decarboxylative benzylation of propiolic esters was shown to be catalyzed by Pd(PPh3)4 at 110° C in toluene (Chart 55); the related allylation chemistry takes place at 75 °C.108 The higher temperature required for benzylation likely reflects a change in the rate-limiting step from decarboxylation (DcA) to ionization (for benzylation). With regard to the benzyl component, various isomers of naphthalene, quinoline and indole were all viable coupling partners. Phenyl and alkyl propiolates likewise led to high yielding decarboxylative coupling with the aforementioned benzyl derivatives. While the terminal propiolate yielded no decarboxylative benzylation, TMS-protected alkynes provided high yields of the protected benzyl acetylenes (Chart 55). Importantly, the neutral conditions of the reaction allow the synthesis of base-sensitive benzylic alkynes that are prone to isomerize to allenes when synthesized by typical methods that utilize base or stoichiometric organometallic reagents.165
Chart 55.

Decarboxylative Coupling of Benzyl Propioliates
While Tunge’s method provides access to a variety of benzylated alkynes under neutral conditions, the reaction is still limited to couplings of benzyl groups with extended conjugation. Li reported an intermolecular palladium-catalyzed decarboxylative coupling of propiolic acids with benzyl halides, that circumvents this problem (Chart 56).168 Because benzyl halides undergo more facile ionization than benzyl acetates, Li was able to couple electron-deficient benzyl moieties that were not viable partners for Tunge’s decarboxylative benzylation. In Li’s approach, the active palladium/XPhos(L-XVI) catalyst facilitates the oxidative addition into the benzyl halide (Scheme 88). This palladium(II) species then facilitates decarboxylation followed by reductive eliminaton to generate the desired product.
Chart 56.

Palladium-Catalyzed Benzylation of Propiolic Acids
Scheme 88.

Palladium-Catalyzed Benzylation of Propiolic Acids
With regard to the propiolic acid derivative, the reaction was accepting of various aryl and alkyl substituents, while an unsubstituted propiolic acid failed to give the desired product (Chart 56). Many benzyl bromides and chlorides were viable coupling partners as long as the benzyl halide was not too electron deficient (186, Chart 56).
11.2.4 Decarboxylative Benzylation of Enolates
In analogy to the seminal decarboxylative allylations described by Tsuji and Saegusa in 1980,14–15 decarboxylative benzylations of β-keto esters were recently reported by the Tunge group (Chart 57).165 Utilizing Pd(PPh3)4 as a catalyst, many α-benzyl ketones could be accessed in good yield. However, a Pd/PBu3 catalyst system generally provided superior results for coupling of α,α-disubstituted esters. Under the appropriate reaction conditions, benzyl ketones with quaternary, tertiary, and secondary α-carbon centers could be synthesized in good to high yields (Chart 57). Interestingly decarboxylative benzylation leading to 187 took place at room temperature. While a variety of benzyl derivatives with extended conjugation underwent decarboxylative benzylation, a simple benzyl electrophile required an electron-donating substituent to couple in moderate yield (188).
Chart 57.

Decarboxylative Coupling of Benzyl β-Keto Esters
Notably, the benzylation was regiospecific for C–C bond formation at the site of decarboxylation (Chart 58). For example, decarboxylative benzylation of substrates with other acidic sites lead to site-specific benzylation at the position that bore CO2. Thus, unsymmetrical 1,4-diketones could be readily synthesized by benzylation. Once again, this demonstrates how the regiospecificity of decarboxylative coupling can be taken advantage of to synthesize materials that would be difficult to access using standard base-mediated enolate chemistry.
Chart 58.

Regiospecific Decarboxylative Benzylation
In conclusion, metal catalyzed decarboxylative benzylation is a viable approach to electrophilic benzylation reactions that are difficult to accomplish otherwise. Nucleophilic coupling partners disclosed thus far include phenols,161 diphenylglycinate imines,164 acetylides,165 and ketones.165 With these strong initial publications, it can be expected that this field of chemistry will flourish over the upcoming years.
12. Conclusions
Decarboxylative allylations have received significant attention by many research groups. While decarboxylative coupling reactions offer a “greener” alternative to standard allylation and benzylation reactions, significant progress still has to be made to realize this promise. A major limitation of current methods is that anion stabilizing groups are often required to achieve decarboxylative metalation. The development of decarboxylative allylations of simple alkyl or alkenyl nucleophiles would significantly expand the scope of products that can be accessed via DcA. In addition, the development of asymmetric allylations has been mostly limited to enolate allylation and cycloadditions. Thus, the discovery of methods for asymmetric decarboxylative allylation of a wide variety of nucleophiles is critical for continued advancement of this field. Moreover, developing interceptive decarboxylations of less activated pro-nucleophiles would allow the synthesis of many relevant chemical building blocks. Nonetheless, if researchers can continue to advance the state of the art of decarboxylative couplings, as has been seen in the last 6–7 years, DcA reactions are likely to become widely used by synthetic chemists. Ultimately, continued innovation will likely require a better mechanistic understanding of the diverse decarboxylative couplings presented herein.
Chart 22.
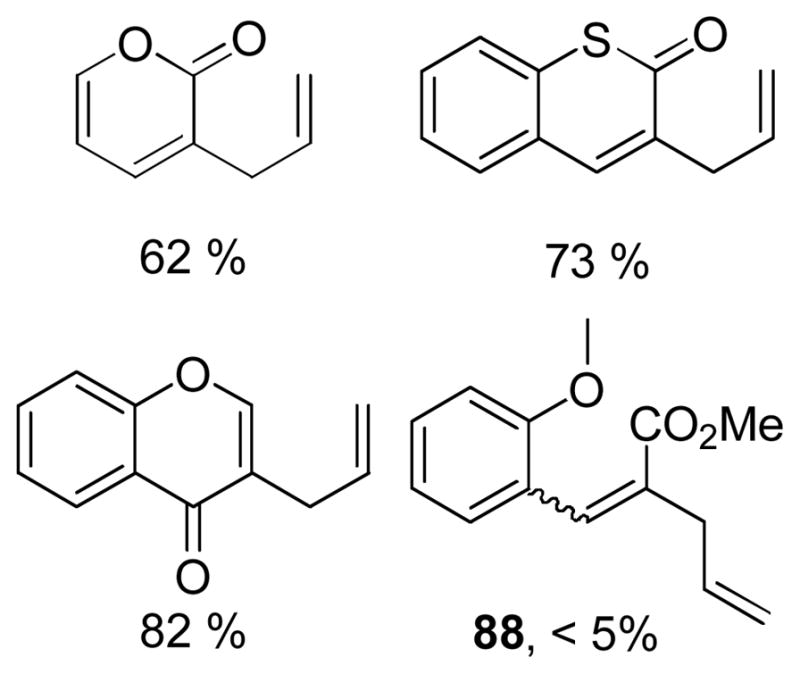
DcA of Heteroaromatics
Acknowledgments
We thank the National Institute of General Medical Sciences (1R01GM079644) and the National Science Foundation (CHE-054808) for funding of various aspects of research described in this review.
References
- 1.(a) Torborg C, Beller M. Adv Synth Catal. 2009;351:3027. [Google Scholar]; (b) McGlacken GP, Fairlamb IJS. Eur J Org Chem. 2009;2009:4011. [Google Scholar]; (c) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 2.(a) Negishi E-i, Wang G, Rao H, Xu Z. J Org Chem. 2010;75:3151. doi: 10.1021/jo1003218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Terao J, Kambe N. Acc Chem Res. 2008;41:1545. doi: 10.1021/ar800138a. [DOI] [PubMed] [Google Scholar]; (c) Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Marion N, Nolan SP. Acc Chem Res. 2008;41:1440. doi: 10.1021/ar800020y. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kantchev EAB, O’Brien CJ, Organ MG. Angew Chem Int Ed. 2007;46:2768. doi: 10.1002/anie.200601663. [DOI] [PubMed] [Google Scholar]; (g) Phan NTS, Sluys MVD, Jones CW. Adv Synth Catal. 2006;348:609. [Google Scholar]; (h) Corbet JP, Mignani G. Chem Rev. 2006;106:2651. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]; (i) Trost BM. J Org Chem. 2004;69:5813. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; (j) Echavarren AM, Cárdenas DJ. In: Metal-Catalyzed Cross-Coupling Reactions. 2. Meijere A, Diederich F, editors. Wiley; 2004. [Google Scholar]; (K) Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (l) Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; (c) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Acc Chem Res. 1995;28:154. [Google Scholar]
- 4.(a) Austeri M, Buron F, Constant S, Lacour J, Linder D, Muller J, Tortoioli S. Pure Appl Chem. 2008;80:967. [Google Scholar]; (b) Mohr JT, Stoltz BM. Chem Asian J. 2007;2:1476. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mohr JT, Ebner DC, Stoltz BM. Org Biomol Chem. 2007;5:3571. doi: 10.1039/b711159m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) You SL, Dai LX. Angew Chem Int Ed. 2006;45:5246. doi: 10.1002/anie.200601889. [DOI] [PubMed] [Google Scholar]; (e) Tunge JA, Burger EC. Eur J Org Chem. 2005:1715. [Google Scholar]; (f) Tsuji J. Proc Jpn Acad, B. 2004;80:349. [Google Scholar]
- 5.Tsuji J, Takahashi H, Morikawa M. Tetrahedron Lett. 1965;6:4387. [Google Scholar]
- 6.Tsuji J. In: Handbook of Organopalladium Chemistry in Organic Synthesis. Negishi E-i, Meijere A., editors. Vol. 2. Wiley; 2002. p. 1669. [Google Scholar]
- 7.(a) Trost BM, Self CR. J Org Chem. 1984;49:468. [Google Scholar]; (b) Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2591. [Google Scholar]
- 8.Negishi E, Matsushita H, Chatterjee S, John RA. J Org Chem. 1982;47:3188. [Google Scholar]
- 9.Braun M, Meier T. Angew Chem, Int Ed. 2006;45:6952. doi: 10.1002/anie.200602169. [DOI] [PubMed] [Google Scholar]
- 10.Trost BM, Schroeder GM. J Am Chem Soc. 1999;121:6759. [Google Scholar]
- 11.Graening T, Hartwig JF. J Am Chem Soc. 2005;127:17192. doi: 10.1021/ja0566275. [DOI] [PubMed] [Google Scholar]
- 12.Nesmeyanov AN, Lutsenko IF, Ananchenko SN. Org Khim. 1950;132:136. [Google Scholar]
- 13.Rund JV, Plane RA. J Am Chem Soc. 1964;86:367. [Google Scholar]
- 14.Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199. [Google Scholar]
- 15.Tsuda T, Chujo Y, Nishi S, Tawara K, Saegusa T. J Am Chem Soc. 1980;102:6381. [Google Scholar]
- 16.Chattopadhyay K, Jana R, Day VW, Douglas JT, Tunge JA. Org Lett. 2010;12:3042. doi: 10.1021/ol101042x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuji J, Yamada T, Minami I, Yuhara M, Nisar M, Shimizu I. J Org Chem. 1987;52:2988. [Google Scholar]
- 18.Imao D, Itoi A, Yamazaki A, Shirakura M, Ohtoshi R, Ogata K, Ohmori Y, Ohta T, Ito Y. J Org Chem. 2007;72:1652. doi: 10.1021/jo0621569. [DOI] [PubMed] [Google Scholar]
- 19.Tardibono LP, Patzner J, Cesario C, Miller MJ. Org Lett. 2009;11:4076. doi: 10.1021/ol901518g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu I, Ishii H. Tetrahedron. 1991;50:487. [Google Scholar]
- 21.Sinha SC, Dutta S, Sun J. Tetrahedron Lett. 2000;41:8243. [Google Scholar]
- 22.Shimizu I, Ishii H, Tasaka A. Chem Lett. 1989;18:1127. [Google Scholar]
- 23.(a) Szabó KJ. Chem Eur J. 2004;10:5268. doi: 10.1002/chem.200400261. [DOI] [PubMed] [Google Scholar]; (b) Cardenas DJ, Echavarren AM. New J Chem. 2004;28:338. [Google Scholar]; (c) Goliaszewski A, Schwartz J. Tetrahedron. 1985;41:5779. [Google Scholar]
- 24.Waetzig SR, Rayabharapu DK, Weaver JD, Tunge JA. Angew Chem, Int Ed. 2006;45:4977. doi: 10.1002/anie.200600721. [DOI] [PubMed] [Google Scholar]
- 25.Shintani R, Tsuji T, Park S, Hayashi T. Chem Commun. 2010;46:1697. doi: 10.1039/b924416f. [DOI] [PubMed] [Google Scholar]
- 26.Tsuda T, Okada M, Nishi S, Saegusa T. J Org Chem. 1986;51:421. [Google Scholar]
- 27.(a) Trost BM, Verhoeven TR. J Am Chem Soc. 1980;102:4730. doi: 10.1021/ja00418a063. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Verhoeven TR. J Org Chem. 1976;41:3215. [Google Scholar]
- 28.Tsuda T, Tokai M, Ishida T, Saegusa T. J Org Chem. 1986;51:5216. [Google Scholar]
- 29.Tsuji J, Minami I, Shimizu I. Tetrahedron Lett. 1983;24:1793. [Google Scholar]
- 30.Tsuji J, Minami I, Shimizu I. Chem Lett. 1984:1721. [Google Scholar]
- 31.(a) Belda O, Moberg C. Acc Chem Res. 2003;37:159. doi: 10.1021/ar030239v. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Lautens M. J Am Chem Soc. 1987;109:1469. [Google Scholar]; (c) Trost BM, Lautens M. J Am Chem Soc. 1982;104:5543. [Google Scholar]
- 32.Burger EC, Tunge JA. Org Lett. 2004;6:2603. doi: 10.1021/ol049097a. [DOI] [PubMed] [Google Scholar]
- 33.Koelle U. Chem Rev. 1998;98:1313. doi: 10.1021/cr960363a. [DOI] [PubMed] [Google Scholar]
- 34.Trost BM, Fraisse PL, Ball ZT. Angew Chem, Int Ed. 2002;41:1059. doi: 10.1002/1521-3773(20020315)41:6<1059::aid-anie1059>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Constant S, Tortoioli S, Mueller J, Linder D, Buron F, Lacour J. Angew Chem, Int Ed. 2007;46:8979. doi: 10.1002/anie.200704019. [DOI] [PubMed] [Google Scholar]
- 36.Burger EC, Tunge JA. Org Lett. 2004;6:4113. doi: 10.1021/ol048149t. [DOI] [PubMed] [Google Scholar]
- 37.(a) Trost BM. Acc Chem Res. 2002;35:695. doi: 10.1021/ar010068z. [DOI] [PubMed] [Google Scholar]; (b) Fuji K, Kinoshita N, Tanaka K. Chem Commun. 1999:1895. [Google Scholar]
- 38.Yan B, Spilling CD. J Org Chem. 2008;73:5385. doi: 10.1021/jo8004028. [DOI] [PubMed] [Google Scholar]
- 39.Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 40.(a) Trost BM, Xu J. J Am Chem Soc. 2005;127:2846. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 42.(a) McDougal NT, Virgil SC, Stoltz BM. Synlett. 2010:1712. doi: 10.1055/s-0030-1258094. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schulz SR, Blechert S. Angew Chem Int Ed. 2007;46:3966. doi: 10.1002/anie.200604553. [DOI] [PubMed] [Google Scholar]
- 43.Trost BM, Bream RN, Xu J. Angew Chem Int Ed. 2006;45:3109. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 44.Levine SR, Krout MR, Stoltz BM. Org Lett. 2008;11:289. doi: 10.1021/ol802409h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura M, Hajra A, Endo K, Nakamura E. Angew Chem Int Ed. 2005;44:7248. doi: 10.1002/anie.200502703. [DOI] [PubMed] [Google Scholar]
- 46.Burger EC, Barron BR, Tunge JA. Synlett. 2006:2824. [Google Scholar]
- 47.(a) Steiner DD, Mase N, III, CFB Angew Chem Int Ed. 2005;44:3706. doi: 10.1002/anie.200500571. [DOI] [PubMed] [Google Scholar]; (b) Enders D, Huettl MRM. Synlett. 2005:991. [Google Scholar]; (c) Shibata N, Suzuki E, Takeuchi Y. J Am Chem Soc. 2000;122:10728. [Google Scholar]; (d) Cahard D, Audouard C, Plaquevent JC, Roques N. Org Lett. 2000;2:3699. doi: 10.1021/ol006610l. [DOI] [PubMed] [Google Scholar]; (e) Davis FA, Zhou P, Murphy CK, Sundarababu G, Qi H, Han W, Przeslawski RM, Chen BC, Carroll PJ. J Org Chem. 1998;63:2273. [Google Scholar]
- 48.(a) Belanger E, Houze C, Guimond N, Cantin K, Paquin JF. Chem Commun. 2008:3251. doi: 10.1039/b803097a. [DOI] [PubMed] [Google Scholar]; (b) Kuwano R, Naoki I, Murakami M. Chem Commun. 2005:3951. doi: 10.1039/b505105c. [DOI] [PubMed] [Google Scholar]
- 49.Trost BM, Lehr K, Michaelis DJ, Xu J, Buckl AK. J Am Chem Soc. 2010;132:8915. doi: 10.1021/ja103771w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trost BM, Xu J, Reichle M. J Am Chem Soc. 2007;129:282. doi: 10.1021/ja067342a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2008;130:11852. doi: 10.1021/ja8038954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burger EC, Tunge JA. Chem Commun. 2005:2835. doi: 10.1039/b503568f. [DOI] [PubMed] [Google Scholar]
- 53.Trost BM, Fraisse PL, Ball ZT. Angew Chem Int Ed. 2002;41:1059. doi: 10.1002/1521-3773(20020315)41:6<1059::aid-anie1059>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 54.Constant S, Tortoioli S, Müller J, Lacour J. Angew Chem Int Ed. 2007;46:2082. doi: 10.1002/anie.200604573. [DOI] [PubMed] [Google Scholar]
- 55.He H, Zheng X-J, Li Y, Dai L-X, You S-L. Org Lett. 2007;9:4339. doi: 10.1021/ol7019394. [DOI] [PubMed] [Google Scholar]
- 56.Madrahimov ST, Markovic D, Hartwig JF. J Am Chem Soc. 2009;131:7228. doi: 10.1021/ja902609g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:6840. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Fiaud JC, Aribe-Zouioueche L. Tetrahedron Lett. 1982;23:5279. [Google Scholar]; (b) Bäckvall JE, Nordberg RE, Vågberg J. Tetrahedron Lett. 1983;24:411. [Google Scholar]
- 59.Showen RL. In: Transition States of Biochemical Processes. 1. Gandour RD, RLS, editors. Spinger: Plenum; New York: 1978. [Google Scholar]
- 60.Darensbourg DJ, Holtcamp MW, Khandelwal B, Klausmeyer KK, Reibenspies JH. Inorg Chem. 1995;34:2389. [Google Scholar]
- 61.Hay R, Caughley B. Aust J Chem. 1967;20:1829. [Google Scholar]
- 62.Swain CG, Bader RFW, Esteve RM, Griffin RN. J Am Chem Soc. 1961;83:1951. [Google Scholar]
- 63.(a) Pocker Y, Davison BL, Deits TL. J Am Chem Soc. 1978;100:3564. [Google Scholar]; (b) Sauers CK, Jencks WP, Groh S. J Am Chem Soc. 1975;97:5546. [Google Scholar]
- 64.Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem Eur J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 65.Carcache DA, Cho YS, Hua Z, Tian Y, Li Y-M, Danishefsky SJ. J Am Chem Soc. 2006;128:1016. doi: 10.1021/ja056980a. [DOI] [PubMed] [Google Scholar]
- 66.Herrinton PM, Klotz KL, Hartley WM. J Org Chem. 1993;58:678. [Google Scholar]
- 67.Tanaka T, Okamura N, Bannai K, Hazato A, Sugiura S, Tomimori K, Manabe K, Kurozumi S. Tetrahedron. 1986;42:6747. [Google Scholar]
- 68.Nicolaou KC, Vassilikogiannakis G, Mägerlein W, Kranich R. Angew Chem Int Ed. 2001;40:2482. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 69.Trost BM, Strege PE. J Am Chem Soc. 1975;97:2534. [Google Scholar]
- 70.(a) Fernández F, Gómez M, Jansat S, Muller G, Martin E, Flores-Santos L, García PX, Acosta A, Aghmiz A, Giménez-Pedrós M, Masdeu-Bultó AM, Diéguez M, Claver C, Maestro MÁ. Organometallics. 2005;24:3946. [Google Scholar]; (b) Faller JW, Wilt JC, Parr J. Org Lett. 2004;6:1301. doi: 10.1021/ol0497382. [DOI] [PubMed] [Google Scholar]; (c) Tamura R, Hegedus LS. J Am Chem Soc. 1982;104:3727. [Google Scholar]
- 71.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 72.De Lorbe JE, Lotz MD, Martin SF. Org Lett. 2010;12:1576. doi: 10.1021/ol100273p. [DOI] [PubMed] [Google Scholar]
- 73.Lavallée J-F, Deslongchamps P. Tetrahedron Lett. 1988;29:5117. [Google Scholar]
- 74.(a) Tricotet T, Brueckner R. Tetrahedron Lett. 2006;47:8499. [Google Scholar]; (b) Burns AC, Forsyth CJ. Org Lett. 2008;10:97. doi: 10.1021/ol7024058. [DOI] [PubMed] [Google Scholar]
- 75.Trost BM, Stiles DT. Org Lett. 2007;9:2763. doi: 10.1021/ol070971k. [DOI] [PubMed] [Google Scholar]
- 76.McFadden RM, Stoltz BM. J Am Chem Soc. 2006;128:7738. doi: 10.1021/ja061853f. [DOI] [PubMed] [Google Scholar]
- 77.Enquist JA, Jr, Stoltz BM. Nature. 2008;453:1228. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Negishi E-i, Tan Z, Liang B, Novak T. Proc Natl Acad Sci. 2004;101:5782. doi: 10.1073/pnas.0307514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seto M, Roizen JL, Stoltz BM. Angew Chem, Int Ed. 2008;47:6873. doi: 10.1002/anie.200801424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.White DE, Stewart IC, Seashore-Ludlow BA, Grubbs RH, Stoltz BM. Tetrahedron. 2010;66:4668. doi: 10.1016/j.tet.2010.04.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jana R, Trivedi R, Tunge JA. Org Lett. 2009;11:3434. doi: 10.1021/ol901288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Waetzig SR, Tunge JA. J Am Chem Soc. 2007;129:4138. doi: 10.1021/ja077070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Waetzig SR, Tunge JA. J Am Chem Soc. 2007;129:14860. doi: 10.1021/ja077070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burger EC, Tunge JA. J Am Chem Soc. 2006;128:10002. doi: 10.1021/ja063115x. [DOI] [PubMed] [Google Scholar]
- 86.Weaver JD, Tunge JA. Org Lett. 2008;10:4657. doi: 10.1021/ol801951e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.(a) Friestad GK, Korapala CS, Ding H. J Org Chem. 2006;71:281. doi: 10.1021/jo052037d. [DOI] [PubMed] [Google Scholar]; (b) Hirabayashi R, Ogawa C, Sugiura M, Kobayashi S. J Am Chem Soc. 2001;123:9493. doi: 10.1021/ja011125m. [DOI] [PubMed] [Google Scholar]; (c) Bloch R. Chem Rev. 1998;98:1407. doi: 10.1021/cr940474e. [DOI] [PubMed] [Google Scholar]
- 88.(a) Toney MD. Arch Biochem Biophys. 2005;433:279. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]; (b) Grigg R, Idle J, McMeekin P, Vipond D. J Chem Soc, Chem Commun. 1987;49 [Google Scholar]
- 89.Yeagley AA, Chruma JJ. Org Lett. 2007;9:2879. doi: 10.1021/ol071080f. [DOI] [PubMed] [Google Scholar]
- 90.Fields WH, Khan AK, Sabat M, Chruma JJ. Org Lett. 2008;10:5131. doi: 10.1021/ol801986m. [DOI] [PubMed] [Google Scholar]
- 91.Grenning AJ, Tunge JA. Org Lett. 2010;12:740. doi: 10.1021/ol902828p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Recio A, Tunge JA. Org Lett. 2009;11:5630. doi: 10.1021/ol902065p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Walters MA, Hoem AB, McDonough CS. J Org Chem. 1996;61:55. [Google Scholar]
- 94.Nakamura H, Iwama H, Ito M, Yamamoto Y. J Am Chem Soc. 1999;121:10850. [Google Scholar]
- 95.Trost BM, Bunt RC. J Am Chem Soc. 1998;120:70. [Google Scholar]
- 96.Brown AC, Carpino LA. J Org Chem. 1985;50:1749. [Google Scholar]
- 97.Simpkins NS. Sulfones in Organic Synthesis. In: Baldwin JE, Maynus PD, editors. Tetrahedron Organic Chemistry Series. Vol. 10. Pergamon Press; Oxford: 1993. [Google Scholar]
- 98.Weaver JD, Ka BJ, Morris DK, Thompson W, Tunge JA. J Am Chem Soc. 2010;132:12179. doi: 10.1021/ja104196x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bors DA, Streitwieser A. J Am Chem Soc. 1986;108:1397. [Google Scholar]
- 100.(a) Corey EJ, Koenig H, Lowry TH. Tetrahedron Lett. 1962:515. [Google Scholar]; (b) Corey EJ, Lowry TH. Tetrahedron Lett. 1965:803. [Google Scholar]; (c) Corey EJ, Lowry TH. Tetrahedron Lett. 1965:793. [Google Scholar]
- 101.Cram DJ, Wingrove AS. J Am Chem Soc. 1963;85:1100. [Google Scholar]
- 102.(a) Gais HJ, Hellmann G, Günther H, Lopez F, Lindner HJ, Braun S. Angew Chem, Int Ed. 1989;28:1025. [Google Scholar]; (b) Gais HJ, Hellmann G. J Am Chem Soc. 1992;114:4439. [Google Scholar]
- 103.(a) Oda N, Yoshida Y, Nagai S, Ueda T, Sakakibara J. Chem Pharm Bull. 1987;35:1796. [Google Scholar]; (b) Ross RM, Burnett ML. J Am Chem Soc. 1949;71:3562. [Google Scholar]
- 104.Corey EJ, Fraenkel G. J Am Chem Soc. 1953;75:1168. [Google Scholar]
- 105.Tsuji J, Ohashi Y, Minami I. Tetrahedron Lett. 1987;28:2397. [Google Scholar]
- 106.Snider BB, Buckman BO. J Org Chem. 1992;57:4883. [Google Scholar]
- 107.(a) Posakony J, Hirao M, Stevens S, Simon JA, Bedalov A. J Med Chem. 2004;47:2635. doi: 10.1021/jm030473r. [DOI] [PubMed] [Google Scholar]; (b) Worden LR, Kaufman KD, Weis JA, Schaaf TK. J Org Chem. 1969;34:2311. [Google Scholar]; (c) Adams R, Bockstahler TE. J Am Chem Soc. 1952;74:5346. [Google Scholar]
- 108.(a) Rayabarapu DK, Tunge JA. J Am Chem Soc. 2005;127:13510. doi: 10.1021/ja0542688. [DOI] [PubMed] [Google Scholar]; (b) Pi SF, Tang BX, Li JH, Liu YL, Liang Y. Org Lett. 2009;11:2309. doi: 10.1021/ol900643r. [DOI] [PubMed] [Google Scholar]
- 109.Sim SH, Park H-J, Lee SI, Chung YK. Org Lett. 2008;10:433. doi: 10.1021/ol702577g. [DOI] [PubMed] [Google Scholar]
- 110.Mellegaard-Waetzig SR, Rayabarapu DK, Tunge JA. Synlett. 2005:2759. [Google Scholar]
- 111.Wang C, Tunge JA. Org Lett. 2006;8:3211. doi: 10.1021/ol0610744. [DOI] [PubMed] [Google Scholar]
- 112.Wang C, Tunge JA. J Am Chem Soc. 2008;130:8118. doi: 10.1021/ja801742h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Waetzig SR, Tunge JA. Chem Commun. 2008:3311. doi: 10.1039/b806949b. [DOI] [PubMed] [Google Scholar]
- 114.Bates RW, Dewey MR. Org Lett. 2009;11:3706. doi: 10.1021/ol901094h. [DOI] [PubMed] [Google Scholar]
- 115.Ibuka T, Mimura N, Aoyama H, Akaji M, Ohno H, Miwa Y, Taga T, Nakai K, Tamamura H, Fujii N, Yamamoto Y. J Org Chem. 1997;62:999. doi: 10.1021/jo962094u. [DOI] [PubMed] [Google Scholar]
- 116.Singh OV, Han H. J Am Chem Soc. 2007;129:774. doi: 10.1021/ja067966g. [DOI] [PubMed] [Google Scholar]
- 117.Singh OV, Han H. Org Lett. 2007;9:4801. doi: 10.1021/ol702115h. [DOI] [PubMed] [Google Scholar]
- 118.Guibe F, M’Leux YS. Tetrahedron Lett. 1981;22:3591. [Google Scholar]
- 119.Minami I, Shimizu I, Tsuji J. J Organomet Chem. 1985;296:269. [Google Scholar]
- 120.Consiglio G, Scalone M, Rama F. J Mol Catal. 1989;50:L11. [Google Scholar]
- 121.Larock RC, Lee NH. Tetrahedron Lett. 1991;32:6315. [Google Scholar]
- 122.Austeri M, Linder D, Lacour J. Chem Eur J. 2008;14:5737. doi: 10.1002/chem.200800619. [DOI] [PubMed] [Google Scholar]
- 123.Trivedi R, Tunge JA. Org Lett. 2009;11:5650. doi: 10.1021/ol902291z. [DOI] [PubMed] [Google Scholar]
- 124.(a) Plietker B, Dieskau A. Eur J Org Chem. 2009;2009:775. [Google Scholar]; (b) Plietker B, Dieskau A, Möws K, Jatsch A. Angew Chem Int Ed. 2008;47:198. doi: 10.1002/anie.200703874. [DOI] [PubMed] [Google Scholar]; (c) Plietker B. Angew Chem Int Ed. 2006;45:1469. doi: 10.1002/anie.200503274. [DOI] [PubMed] [Google Scholar]; (d) Plietker B. Angew Chem Int Ed. 2006;45:6053. doi: 10.1002/anie.200602261. [DOI] [PubMed] [Google Scholar]; (e) Zhou B, Xu Y. J Org Chem. 1988;53:4419. [Google Scholar]; (f) Xu Y, Zhou B. J Org Chem. 1987;52:974. [Google Scholar]
- 125.(a) Zielinska-Blajet M, Siedlecka R, Skarzewski J. Tetrahedron Asym. 2007;18:131. [Google Scholar]; (b) Iwaoka M, Tomoda S. Top Curr Chem. 2000;208:55. [Google Scholar]
- 126.Yeagley AA, Lowder MA, Chruma JJ. Org Lett. 2009;11:4022. doi: 10.1021/ol901745x. [DOI] [PubMed] [Google Scholar]
- 127.Nokami J, Watanabe H, Mandai T, Kawada M, Tsuji J. Tetrahedron Lett. 1989;30:4829. [Google Scholar]
- 128.(a) Patil NT, Yamamoto Y. Synlett. 2007:1994. [Google Scholar]; (b) Shim JG, Nakamura H, Yamamoto Y. J Org Chem. 1998;63:8470. doi: 10.1021/jo980818r. [DOI] [PubMed] [Google Scholar]
- 129.Wang C, Tunge JA. Org Lett. 2005;7:2137. doi: 10.1021/ol050466s. [DOI] [PubMed] [Google Scholar]
- 130.(a) Lemek T, Mayr H. J Org Chem. 2003;68:6880. doi: 10.1021/jo0344182. [DOI] [PubMed] [Google Scholar]; (b) Kaumanns O, Mayr H. J Org Chem. 2008;73:2738. doi: 10.1021/jo702590s. [DOI] [PubMed] [Google Scholar]; (c) Kaumanns O, Lucius R, Mayr H. Chem Eur J. 2008;14:9675. doi: 10.1002/chem.200801277. [DOI] [PubMed] [Google Scholar]
- 131.(a) Tunge JA, Burger EC. Eur J Org Chem. 2005:1715. [Google Scholar]; (b) Trost BM, Fraisse PL, Ball ZT. Angew Chem Int Ed. 2002;41:1059. doi: 10.1002/1521-3773(20020315)41:6<1059::aid-anie1059>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 132.Streuff J, White DE, Virgil SC, Stoltz BM. Nature Chem. 2010;2:192. doi: 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.(a) Nakamura H, Sekido M, Ito M, Yamamoto Y. J Am Chem Soc. 1998;120:6838. [Google Scholar]; (b) Sekido M, Aoyagi K, Nakamura H, Kabuto C, Yamamoto Y. J Org Chem. 2001;66:7142. doi: 10.1021/jo0158332. [DOI] [PubMed] [Google Scholar]
- 134.(a) Shintani R, Murakami M, Hayashi T. J Am Chem Soc. 2007;129:12356. doi: 10.1021/ja073997f. [DOI] [PubMed] [Google Scholar]; (b) Shintani R, Park S, Hayashi T. J Am Chem Soc. 2007;129:14866. doi: 10.1021/ja077236o. [DOI] [PubMed] [Google Scholar]; (c) Shintani R, Murakami M, Hayashi T. Pure Appl Chem. 2008;80:1135. [Google Scholar]; (d) Shintani R, Park S, Shirozu F, Murakami M, Hayashi T. J Am Chem Soc. 2008;130:16174. doi: 10.1021/ja807662b. [DOI] [PubMed] [Google Scholar]; (e) Park S, Shintani R, Hayashi T. Chem Lett. 2009;38:204. [Google Scholar]; (f) Shintani R, Hayashi S-y, Murakami M, Takeda M, Hayashi T. Org Lett. 2009;11:3754. doi: 10.1021/ol901348f. [DOI] [PubMed] [Google Scholar]; (g) Shintani R, Murakami M, Hayashi T. Org Lett. 2009;11:457. doi: 10.1021/ol802569q. [DOI] [PubMed] [Google Scholar]; (h) Shintani R, Tsuji T, Park S, Hayashi T. Chem Commun. 2010:1697. doi: 10.1039/b924416f. [DOI] [PubMed] [Google Scholar]
- 135.Hoffmann HMR, Otte AR, Wilde A, Menzer S, Williams DJ. Angew Chem, Int Ed. 1995;34:100. [Google Scholar]
- 136.Shintani R, Tsuji T, Park S, Hayashi T. J Am Chem Soc. 2010;132:7508. doi: 10.1021/ja1023223. [DOI] [PubMed] [Google Scholar]
- 137.(a) Curtis MD, Eisenstein O. Organometallics. 1984;3:887. [Google Scholar]; (b) Carfagna C, Galarini R, Linn K, Lopez JA, Mealli C, Musco A. Organometallics. 1993;12:3019. [Google Scholar]
- 138.Shintani R, Murakami M, Tsuji T, Tanno H, Hayashi T. Org Lett. 2009;11:5642. doi: 10.1021/ol902326s. [DOI] [PubMed] [Google Scholar]
- 139.Tsuji J, Shimizu I, Minami I, Ohashi Y. Tetrahedron Lett. 1982;23:4809. [Google Scholar]
- 140.(a) Tsuji J, Sato K, Okumoto H. Tetrahedron Lett. 1982;23:5189. [Google Scholar]; (b) Tsuji J, Sato K, Okumoto H. J Org Chem. 1984;49:1341. [Google Scholar]
- 141.(a) Bando T, Harayama H, Fukazawa Y, Shiro M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1994;59:1465. [Google Scholar]; (b) Tamaru Y, Bando T, Hojo M, Yoshida Z. Tetrahedron Lett. 1987;28:3497. [Google Scholar]; (c) Jayasree S, Seayad A, Sarkar BR, Chaudhari RV. J Mol Catal A Chem. 2002;181:221. [Google Scholar]; (d) Jang EJ, Lee KH, Lee JS, Kim YG. J Mol Catal A Chem. 1999;138:25. [Google Scholar]; (e) El Ali B, Alper H. J Org Chem. 1993;58:3595. [Google Scholar]
- 142.Tsuji J. Tetrahedron. 1986;42:4361. [Google Scholar]
- 143.Tamaru Y, Bando T, Kawamura Y, Okamura K, Yoshida Z, Shiro M. J Chem Soc, Chem Comm. 1992:1498. [Google Scholar]
- 144.Miyazawa M, Wang SZ, Takeda H, Yamamoto K. Synlett. 1992:323. [Google Scholar]
- 145.Bando T, Tanaka S, Fugami K, Yoshida Z, Tamaru Y. Bull Chem Soc Jpn. 1992;65:97. [Google Scholar]
- 146.Minami I, Ohashi Y, Shimizu I, Tsuji J. Tetrahedron Lett. 1985;26:2449. [Google Scholar]
- 147.Knight JG, Ainge SW, Harm AM, Harwood SJ, Maughan HI, Armour DR, Hollinshead DM, Jaxa-Chamiec AA. J Am Chem Soc. 2000;122:2944. [Google Scholar]
- 148.Knight JG, Tchabanenko K. Tetrahedron. 2002;58:6659. [Google Scholar]
- 149.Anderson TF, Knight JG, Tchabanenko K. Tetrahedron Lett. 2003;44:757. [Google Scholar]
- 150.Knight JG, Lawson IM, Johnson CN. Synthesis. 2006:227. [Google Scholar]
- 151.(a) Knight JG, Tchabanenko K, Stoker PA, Harwood SJ. Tetrahedron Lett. 2005;46:6261. [Google Scholar]; (b) Knight JG, Stoker PA, Tchabanenko K, Harwood SJ, Lawrie KWM. Tetrahedron. 2008;64:3744. [Google Scholar]
- 152.(a) Cook GR, Shanker PS. Tetrahedron Lett. 1998;39:3405. [Google Scholar]; (b) Cook GR, Sun L. Org Lett. 2004;6:2481. doi: 10.1021/ol049087+. [DOI] [PubMed] [Google Scholar]
- 153.Auburn PR, Mackenzie PB, Bosnich B. J Am Chem Soc. 1985;107:2033. [Google Scholar]
- 154.Patil NT, Huo Z, Yamamoto Y. J Org Chem. 2006;71:6991. doi: 10.1021/jo061110c. [DOI] [PubMed] [Google Scholar]
- 155.Kamijo S, Jin T, Yamamoto Y. J Am Chem Soc. 2001;123:9453. doi: 10.1021/ja016355f. [DOI] [PubMed] [Google Scholar]
- 156.Darcel C, Bruneau C, Albert M, Dixneuf PH. Chem Commun. 1996:919. [Google Scholar]
- 157.(a) Liegault B, Renaud JL, Bruneau C. Chem Soc Rev. 2008;37:290. doi: 10.1039/b704255h. [DOI] [PubMed] [Google Scholar]; (b) Kuwano R. Synthesis. 2009:1049. [Google Scholar]
- 158.(a) Legros JY, Fiaud JC. Tetrahedron Lett. 1992;33:2509. [Google Scholar]; (b) Legros JY, Toffano M, Fiaud JC. Tetrahedron. 1995;51:3235. [Google Scholar]; (c) Legros J-Y, Primault Gl, Toffano M, Rivière M-A, Fiaud J-C. Org Lett. 2000;2:433. doi: 10.1021/ol991274y. [DOI] [PubMed] [Google Scholar]; (d) Kuwano R, Kondo Y, Matsuyama Y. J Am Chem Soc. 2003;125:12104. doi: 10.1021/ja037735z. [DOI] [PubMed] [Google Scholar]; (e) Kuwano R, Kondo Y. Org Lett. 2004;6:3545. doi: 10.1021/ol048540e. [DOI] [PubMed] [Google Scholar]
- 159.Kuwano R, Yokogi M. Org Lett. 2005;7:945. doi: 10.1021/ol050078q. [DOI] [PubMed] [Google Scholar]
- 160.Mukai T, Hirano K, Satoh T, Miura M. Org Lett. 2010;12:1360. doi: 10.1021/ol1002576. [DOI] [PubMed] [Google Scholar]
- 161.Kuwano R, Kusano H. Org Lett. 2008;10:1979. doi: 10.1021/ol800548t. [DOI] [PubMed] [Google Scholar]
- 162.Kuwano R, Kondo Y, Shirahama T. Org Lett. 2005;7:2973. doi: 10.1021/ol0509787. [DOI] [PubMed] [Google Scholar]
- 163.(a) Narahashi H, Shimizu I, Yamamoto A. J Organomet Chem. 2008;693:283. [Google Scholar]; (b) Becker Y, Stille JK. J Am Chem Soc. 1978;100:838. [Google Scholar]
- 164.Fields WH, Chruma JJ. Org Lett. 2009;12:316. doi: 10.1021/ol902651j. [DOI] [PubMed] [Google Scholar]
- 165.Torregrosa RRP, Ariyarathna Y, Chattopadhyay K, Tunge JA. J Am Chem Soc. 2010;132:9280–9282. doi: 10.1021/ja1035557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.(a) Poon KWC, Dudley GB. J Org Chem. 2006;71:3923. doi: 10.1021/jo0602773. [DOI] [PubMed] [Google Scholar]; (b) Shintou T, Kikuchi W, Mukaiyama T. Bull Chem Soc Jpn. 2003;76:1645. [Google Scholar]; (c) Iversen T, Bundle DR. J Chem Soc Chem Comm. 1981:1240. [Google Scholar]
- 167.Fields WH, Chruma JJ. Org Lett. 2010;12:316. doi: 10.1021/ol902651j. [DOI] [PubMed] [Google Scholar]
- 168.Zhang W-W, Zhang X-G, Li J-H. J Org Chem. 2010;75:5259. doi: 10.1021/jo1010284. [DOI] [PubMed] [Google Scholar]