

Abstract
We previously reported a five-generation family manifesting an autosomal dominant disorder of facial myokymia and dystonic/choreic movements (FDFM). The dyskinetic episodes are initially paroxysmal but may become constant. With increasing age they may lessen or even disappear. The previous study excluded nine candidate genes chosen for their association with myokymia or chorea and two regions containing single or clustered ion channel genes. We now report identification by whole genome linkage analysis of a broad region on chromosome 3p21-3q21 that segregates with the disease in all ten affected members in three generations who participated in the study. GENEHUNTER-MODSCORE Version 2.0.1 provided a maximum multipoint LOD score of 3.099. No other disorders primarily characterized by myokymia, dystonia, or chorea are known to map to this region. Identification of additional families with FDFM may narrow the critical region and facilitate the choice of candidate genes for further analysis.
Keywords: chorea, movement disorder, episodic disorder
Introduction
We previously described a five-generation family of German ethnic background with both choreiform or dystonic dyskinesia and predominantly facial myokymia (FDFM; OMIM 606703) transmitted in an autosomal dominant pattern (Fernandez et al., 2001). Myokymia is the fine continuous involuntary contraction of muscles that may be visible as worm-like rippling and can be detected by electromyogram. It can affect any muscle group; myokymia of the periorbital muscles may resemble blepharospasm. Chorea is characterized by rapid, jerky, movements that may look dance-like and purposeful but are, in fact, involuntary. Dystonia reflects increased muscle tone that results from involuntary muscle contractions, forcing parts of the body into abnormal, sometimes painful, postures. Dyskinesia is a more general term simply referring to abnormal involuntary movements.
In the family described here, onset of FDFM was in early childhood or adolescence with paroxysmal choreiform or dystonic movements possibly triggered by stressful situations. With age the episodes became more pronounced, prolonged, and frequent, and by age 30 were nearly constant. Later in life some individuals experienced improvement in disease severity. Although intelligence, strength, and dexterity were normal, and lifespan did not appear to be shortened, the disorder was quite socially disabling. Electroencephalographic studies (EEG) were normal in several affected individuals, but one young child had frequent unprovoked staring spells preceded by an aura, with no memory of the event, but no postictal behaviors. An EEG demonstrated interictal epileptiform discharges over the central regions of both cerebral hemispheres. Another affected relative reportedly had a childhood-onset seizure disorder, but documentation was not available. With the exception of acetazolamide in several of the affected individuals, interventions including gabapentin, valproic acid, carbamazepine, amitriptyline and chlordiazepoxide were singularly ineffective.
By targeted genotyping we excluded 11 regions containing genes associated with chorea and myokymia (Fernandez et al., 2001): 1) the Huntington disease gene on chromosome 4p; 2) the paroxysmal dystonic choreoathetosis gene at 2q34; 3) the dentatorubral-pallidoluysian atrophy gene at 12p13; 4) the choreoathetosis/spasticity disease locus on 1p that lies in a region containing a cluster of potassium (K+) channel genes; 5) the episodic ataxia type 1 (EA1) locus on 12p that contains its etiologic gene, KCNA1, and two other voltage-gated K+ channel genes, KCNA5 and KCNA6; 6) the chorea-acanthocytosis locus on 9q21; 7) the Huntington-like syndrome gene on 20p; 8) the paroxysmal kinesigenic dyskinesia locus on 16p11.2-q11.2; 9) the benign hereditary chorea locus on 14q; 10) the SCA type 5 locus on chromosome 11; and 11) the chromosome 19 region that contains several ion channels and the CACNA1A gene, a brain-specific P/Q-type calcium channel gene associated with ataxia and hemiplegic migraine. In addition, by clinical testing in one an affected person in the family, no mutations were found in the caveolin 3 gene (CAV3) at 3p25 that is sometimes associated with myokymia. We now provide an eight-year interval follow-up with description of a newly symptomatic family member, and results of a genome wide scan for the causative gene.
Methods
Family
Under protocols approved by the Institutional Review Board of the University of Washington, subjects were examined and blood samples were obtained from 10 affected members, including a set of identical twins, three unaffected members, and three spouses from three generations of this family (fig. 1). The pedigree position numbers of some individuals differ from those shown previously (Fernandez et al., 2001), but the relative order has been maintained.
Figure 1.

Five-generation pedigree demonstrating apparent autosomal dominant transmission of familial dyskinesia and facial myokymia. Affected individuals are denoted by gray symbols. Haplotypes for nine markers on chromosome 3 are shown for all participating subjects; chromosome segments containing the putative disease-causing allele are shown in black; an ambiguous segment is shown in gray. Arrows identify recombinations in two affected persons that define the minimal linkage region.
Genetic analyses
Genomic DNA was extracted from leukocytes or Epstein-Barr virus transformed B lymphocyte lines by standard methods. A whole genome linkage analysis at an average 10 cM resolution was performed using ABI PRISM ® Linkage Mapping screening set Version 2.5. Using an MJ Research PTC-240 or PTC-100 Thermal Cycler, genomic DNA (40 ng) was PCR amplified in 10 μl volumes containing 0.4 U FastStart Taq DNA Polymerase (Roche Diagnostics, Mannheim, Germany), and final concentrations of 1.5 mM MgCl2, 200 mM of each dNTP, and 0.3-0.5 μM each of the forward (fluorescence-labeled) and reverse primers. The PCR began with a heating step of 95°C for 4 min, followed by 30 cycles at 94°C for 45 sec, 57°C for 45 sec, 72°C for 60 sec, and a final extension step at 72°C for 7 min. Products were size-separated on an ABI PRISM ® 3130 XL Genetic Analyzer (Applied Biosystems). Power and multipoint linkage analyses for the genome scan were performed with SLINK (Ott 1989; Weeks et al., 1990) and GENEHUNTER Version 1.2 (Kruglyak and others 1996), respectively. For program constraints, unaffected at-risk individuals were not included in the latter analysis, and although both twins were genotyped, only one was included. Haplotypes were constructed manually for all regions that provided positive LOD scores. For regions that could not be eliminated by this procedure, and to narrow the critical region of linkage, additional markers were identified from the Marshfield genetic map, obtained from Bioneer (Alameda, CA) and genotyped as previously described (Raskind et al., 2005). Fine mapping analyses, which also included the unaffected at-risk individuals, were done with GENEHUNTER-MODSCORE Version 2.0.1 (Dietter et al., 2007) and program-generated haplotypes were also corroborated by hand.
Results
Family Description
We previously described in detail the clinical characteristics of disease in 5 of 18 family members affected with FDFM (Fernandez et al., 2001). An additional member of the youngest generation subsequently developed symptoms of this disease at age 5 (individual V-3 in fig. 1), including mild persistent involuntary twitches of facial muscles and outstretched fingers and hands. He and his two affected sisters have shown modest improvement on acetazolimide. A video of affected family members has been archived with the Movement Disorders Society (Bird 2002).
Linkage Analysis
A simulation study performed under the assumptions of 0.0001 disease allele frequency, 90% penetrance, and four alleles of equal frequency suggested that samples from 14 family members, including one member of the identical twin pair but not individual V-3 whose sample was obtained after the study was begun, could provide a maximum LOD score of 2.39 at a recombination rate (θ) = 0.0. The estimated maximum LOD score rose to 3.099 when individual V-3 was included.
Using the same penetrance estimate and also without the sample from individual V-3, a whole genome scan across all 22 autosomes at a 10 cM level was performed. Multipoint analysis identified five regions of interest with LOD scores above 0.0, including chromosomes 2q14.1-2q22.3, 3p21.1-3q21.3, 4q28.3, 6q23.3-6q27, and 18q11.2-18qter (data not shown). The region on chromosome 6 was eliminated by haplotype analysis. Genotypes of individual V-3, whose DNA was obtained after completion of the genome scan, allowed exclusion of the chromosome 18 locus. Additional markers in the regions of interest on chromosomes 2, 3, and 4, were genotyped (table I). Maximum multipoint LOD scores of .43 and 0.11 were obtained for chromosomes 2 and 4, respectively. Furthermore, results from manual haplotyping were inconsistent with linkage to either of these chromosomal regions. The finer scale mapping on chromosome 3 provided a maximum LOD score of 3.099 (fig. 2). Recombinant events in affected individuals V-3 and III-2 defined the proximal and distal boundaries of the minimal linkage region at D3S1582 and D3S3606, respectively (fig. 1). Although a recombination in individual IV-10 might narrow the region further, the possibility of incomplete penetrance of FDFM in this at-risk person must be considered.
Table I.
Whole genome screen markers and additional markers in regions of interest.
| Markers | UniSTS# (Additional Markers) |
KcM1 |
|---|---|---|
| D2S347 | 131.51 | |
| D2S2271 | 48424 | 133.65 |
| D2S112 | 141.62 | |
| D2S151 | 152.04 | |
| D2S2241 | 24241 | 156.92 |
| D3S1289 | 71.41 | |
| D3S1582 | 26051 | 72.21 |
| D3S1300 | 80.32 | |
| D3S1285 | 91.18 | |
| D3S1566 | 97.75 | |
| D3S3681 | 109.22 | |
| D3S1271 | 117.76 | |
| D3S1278 | 129.73 | |
| D3S1267 | 139.12 | |
| D3S3606 | 726 | 143.94 |
| D3S1292 | 146.60 | |
| D4S402 | 124.45 | |
| D4S1615 | 34042 | 128.31 |
| D4S1575 | 132.05 | |
| D4S1565 | 71100 | 143.84 |
| D4S424 | 144.56 |
Location on the Marshfield map (Map)
Figure 2.
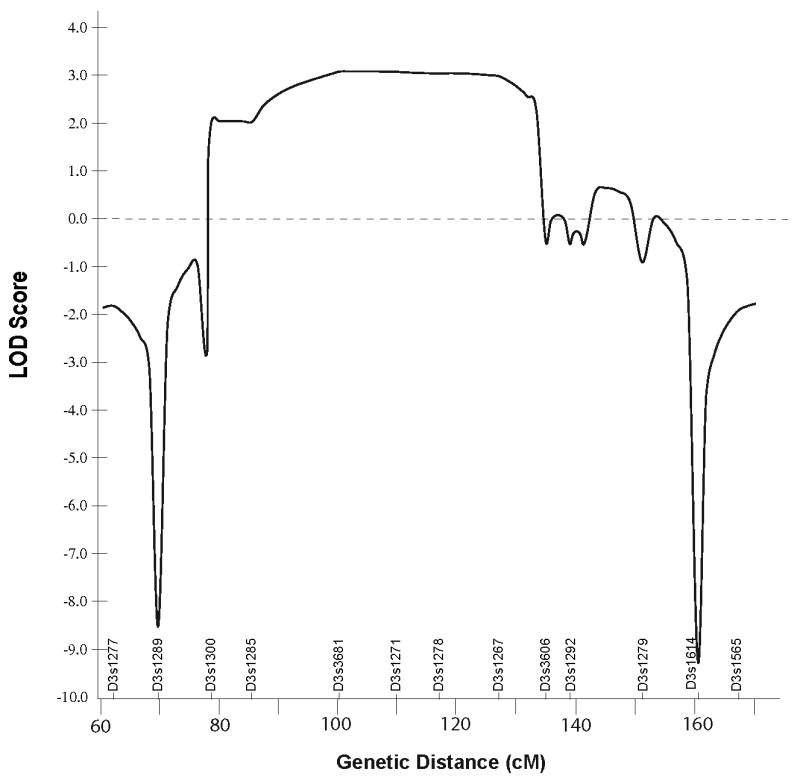
Multipoint LOD plot of results for 25 markers on chromosome 3 that demonstrates a broad region with positive scores.
Discussion
This report provides additional information about the natural history of FDFM. The age of onset ranges from early childhood to late adolescence. We describe a new affected person (V-3) whose symptoms began at age 5 and who had modest benefit from acetazolamide, as did other relatives including his affected sisters.
The availability of this additional subject in the family, coupled with advances in genomics and statistical genetics analytic programs, permitted a successful genome-wide linkage study for FDFM. The 71.73 cM region of linkage corresponds to approximately 74Mb and contains 245 annotated genes, of which 47 are associated with disorders (NCBI Entrez Gene, Build 36.3). In addition, the region contains numerous pseudogenes, hypothetical proteins and open reading frames. If the recombinant event in individual IV-10, who is unaffected at age 60, is considered, the critical region could be reduced to 52.76 cM. Disorders with similar features to FDFM may help prioritize candidate genes.
Myokymia is also a prominent feature of the autosomal dominant disorders episodic ataxia type 1 (EA1; OMIM 160120) (Browne et al., 1994; Jen et al., 2007) and myokymia with neonatal epilepsy (BFNC/myokymia; OMIM 606437) (Cooper and Jan 2003; Dedek et al., 2001). It has been documented occasionally in a variety of other disorders, including several spinocerebellar ataxias. EA1 is caused by mutations in the KCNA1 gene on chromosome 12p13. Like FDFM, the symptoms tend to improve after early adulthood (Brunt and van Weerden 1990). Mutations in KCNA1 can also cause myokymia and spasticity in the absence of ataxia (Chen et al., 2007). BFNC/myokymia is caused by a missense mutation within the putative voltage sensor of the potassium channel KCNQ2 on chromosome 20q13.3 (Jen et al., 2007).
Epilepsy is a less frequent feature of FDFM. The episodic ataxias EA2, EA3, EA5, and EA6 are not characterized by myokymia but include epilepsy as a symptom with varying frequency, and they, along with FDFM, may respond to acetazolamide (Jen et al., 2007). EA2 (OMIM 108500) and EA5 (OMIM 601949) are caused by mutations in genes that code for components of calcium channels, the CACNA1A gene on chromosome 19p13 (Strupp et al., 2007) and the CACNB4 gene on chromosome 2q22-q23 (Escayg et al., 2000), respectively. EA6 (OMIM 60011) is caused by mutation of SLC1A3, a glutamate transporter gene on chromosome 5p13 (Jen et al., 2005). The genes for EA3 (OMIM 606554) and EA7 (OMIM 611907) (Kerber et al., 2007) that map to chromosomes 1q42 and 19q13, respectively, have not yet been identified. Two other disorders deserve mention as they share both the episodic nature of FDFM and choreiform dyskinesia. Paroxysmal nonkinesigenic choreoathetosis (OMIM 118800) is caused by mutations in the N-terminal alpha helix of the protein of the myofibrillogenesis regulator-1 gene (MR1) (Rainier et al., 2004). Paroxysmal kinesigenic choreoathetosis (OMIM 128200) maps to chromosome 16p11.2-q12.1, but the gene has not yet been identified.
FDFM also shares some clinical features with nonparoxysmal disorders. Myokymia is observed in spinocerebellar ataxia (SCA) type 14 (Stevanin et al., 2004) and rarely in SCA2 (Cancel et al., 1997). Facial fasculations but no true myokymia are common in SCA3/MJD (Berciano et al., 2006), SCA5 (11q13). Interestingly, although EA2 is allelic to SCA6, patients with SCA6 do not exhibit myokymia. Genes in or related to those in the “ataxiome” (Lim et al., 2006) that map to the critical region might be considered candidates. The families of genes responsible for the inherited choreas and dystonias may also be informative for FDFM.
High priority candidates within the critical region include the calcium channel gene CACNA2D3 (Hanke et al., 2001) and the KCTD6 gene that catalyzes transmembrane transfer of potassium ions through voltage-gated channels (Van Bogaert et al., 2007). The Ca2+ dependent secretion activator gene CADPS is also of potential interest, although it is not reported to express in muscle (Sadakata et al., 2006). Work on these genes will be pursued. However, considering the number of genes contained in the large region of interest, the most efficacious steps toward identification of the FDFM gene would be to ascertain additional families whose disease links to the same region.
Acknowledgments
The authors wish to thank the members of the family whose cooperation with, and interest in, the research is essential. The research was supported in part by funds from the Department of Veterans Affairs (WHR, MM, JW, HL, and TDB) and NIH 2 T32 DC000033 (BP).
Literature Cited
- Berciano J, Infante J, García A, de Pablos C, Amer G, Polo J, Volpini V, Combarros O. Stiff man-like syndrome and generalized myokymia in spinocerebellar ataxia type 3. Mov Disord. 2006;21(7):1031–1035. doi: 10.1002/mds.20865. [DOI] [PubMed] [Google Scholar]
- Bird TD. Familial dyskinesia and facial myokymia. Mov Disord. 2002;17:747. [Google Scholar]
- Browne D, Gancher S, Nutt J, Brunt E, Smith E, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Brunt E, van Weerden T. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain. 1990;113(Pt 5):1361–1382. doi: 10.1093/brain/113.5.1361. [DOI] [PubMed] [Google Scholar]
- Cancel G, Durr A, Didierjean O, Imbert G, Burk K, Lezin A, Belal S, Benomar A, Abada-Bendib M, Vial C, Guimaraes J, Chneiweiss H, Stevanin G, Yvert G, Abbas N, Saudou F, Lebre AS, Yahyaoui M, Hentati F, Vernant JC, Klockgether T, Mandel JL, Agid Y, Brice A. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet. 1997;6(5):709–715. doi: 10.1093/hmg/6.5.709. [DOI] [PubMed] [Google Scholar]
- Chen H, von Hehn C, Kaczmarek LK, Ment LR, Pober BR, Hisama FM. Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia. Neurogenetics. 2007;8(2):131–135. doi: 10.1007/s10048-006-0071-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Jan LY. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60(4):496–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch T, Steinlein O. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98(21):12272–12277. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietter J, Mattheisen M, Furst R, Ruschendorf F, Wienker TF, Strauch K. Linkage analysis using sex-specific recombination fractions with GENEHUNTER-MODSCORE. Bioinformatics. 2007;23(1):64–70. doi: 10.1093/bioinformatics/btl539. [DOI] [PubMed] [Google Scholar]
- Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, Johnston J, Baloh R, Sander T, Meisler MH. Coding and noncoding variation of the human calcium-channel beta(4)-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531–1539. doi: 10.1086/302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Raskind W, Wolff J, Matsushita M, Yuen E, Graf W, Lipe H, Bird T. Familial dyskinesia and facial myokymia (FDFM): a novel movement disorder. Ann Neurol. 2001;49(4):486–492. [PubMed] [Google Scholar]
- Hanke S, Bugert P, Chudek J, Kovacs G. Cloning a calcium channel alpha2delta-3 subunit gene from a putative tumor suppressor gene region at chromosome 3p21.1 in conventional renal cell carcinoma. Gene. 2001;264(1):69–75. doi: 10.1016/s0378-1119(00)00600-4. [DOI] [PubMed] [Google Scholar]
- Jen J, Graves T, Hess E, Hanna M, Griggs R, Baloh R investigators C. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130(10):2484–2493. doi: 10.1093/brain/awm126. [DOI] [PubMed] [Google Scholar]
- Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65(4):529–534. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol. 2007;64(5):749–52. doi: 10.1001/archneur.64.5.749. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J Neurosci Res. 1999;57:280–289. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. American Journal of Human Genetics. 1996;58(6):1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Xu Y, Huang Y, Ahn AH, Auburger GW, Pandolfo M, Kwiecinski H, Grimes DA, Lang AE, Nielsen JE, Averyanov Y, Servidei S, Friedman A, Van Bogaert P, Abramowicz MJ, Bruno MK, Sorensen BF, Tang L, Fu YH, Ptacek LJ. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet. 2004;13(24):3161–3170. doi: 10.1093/hmg/ddh330. [DOI] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel A, Szabó G, Rual J, Fisk C, Li N, Smolyar A, Hill D, Barabási AL, Vidal M, Zoghbi HY. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;127(7):1335–1347. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- Marshfield Map. http://research.marshfieldclinic.org/genetics/GeneticResearch/data/Maps/
- Ott J. Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainier S, Thomas D, Tokarz D, Ming L, Bui M, Plein E, Zhao X, Lemons R, Albin R, Delaney C, Alvarado D, Fink JK. Myofibrillogenesis regulator 1 gene mutations cause paroxysmal dystonic choreoathetosis. Arch Neurol. 2004;61(7):1025–1029. doi: 10.1001/archneur.61.7.1025. [DOI] [PubMed] [Google Scholar]
- Raskind WH, Igo RP, Chapman NH, Berninger VW, Thomson JB, Matsushita M, Brkanac Z, Holzman T, Brown M, Wijsman EM. A genome scan in multigenerational families with dyslexia: identification of a novel locus on chromosome 2q that contributes to phonological decoding efficiency. Molecular Psychiatry. 2005;10(7):699–711. doi: 10.1038/sj.mp.4001657. [DOI] [PubMed] [Google Scholar]
- Sadakata T, Itakura M, Kozaki S, Sekine Y, Takahashi M, Furuichi T. Differential distributions of the Ca2+ -dependent activator protein for secretion family proteins (CAPS2 and CAPS1) in the mouse brain. J Comp Neurol. 2006;495(6):735–753. doi: 10.1002/cne.20947. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Hahn V, Lohmann E, Bouslam N, Gouttard M, Soumphonphakdy C, Welter ML, Ollagnon-Roman E, Lemainque A, Ruberg M, Brice A, Durr A. Mutation in the catalytic domain of protein kinase C gamma and extension of the phenotype associated with spinocerebellar ataxia type 14. Arch Neurol. 2004;61(8):1242–1248. doi: 10.1001/archneur.61.8.1242. [DOI] [PubMed] [Google Scholar]
- Strupp M, Zwergal A, Brandt T. Episodic ataxia type 2. Neurotherapeutics. 2007;4(2):267–273. doi: 10.1016/j.nurt.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Van Bogaert P, Azizieh R, Désir J, Aeby A, De Meirleir L, Laes J, Christiaens F, Abramowicz M. Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann Neurol. 2007;61(6):579–586. doi: 10.1002/ana.21121. [DOI] [PubMed] [Google Scholar]
- Weeks D, Ott J, Lathrop G. SLINK: a general simulation program for linkage analysis. Am J Hum Genet. 1990;47:A204. abstr. [Google Scholar]