

Abstract
Host defense to the apicomplexan parasite Toxoplasma gondii is critically dependent on CD8+ T cells, whose effector functions include the induction of apoptosis in target cells following the secretion of granzyme proteases. Here we demonstrate that T. gondii induces resistance of host cells to apoptosis induced by recombinant granzyme B. Granzyme B induction of caspase-independent cytochrome c release was blocked in T. gondii-infected cells. Prevention of apoptosis could not be attributed to altered expression of the Bcl-2 family of apoptotic regulatory proteins, but was instead associated with reduced granzyme B-mediated, caspase-independent cleavage of procaspase 3 to the p20 form in T. gondii-infected cells, as well as reduced granzyme B-mediated cleavage of the artificial granzyme B substrate, GranToxiLux. The reduction in granzyme B proteolytic function in T. gondii-infected cells could not be attributed to altered granzyme B uptake or reduced trafficking of granzyme B to the cytosol, implying a T. gondii-mediated inhibition of granzyme B activity. Apoptosis and GranToxiLux cleavage were similarly inhibited in T. gondii-infected cells exposed to the natural killer-like cell line YT-1. The endogenous granzyme B inhibitor PI-9 was not up-regulated in infected cells. We believe these findings represent the first demonstration of granzyme B inhibition by a cellular pathogen and indicate a new modality for host cell protection by T. gondii that may contribute to parasite immune evasion.
Keywords: Apicomplexa, Toxoplasma gondii, Apoptosis, Granzymes, Host-pathogen interaction, Immune evasion
1. Introduction
Toxoplasma gondii is a ubiquitous apicomplexan parasite that infects an estimated one-third of the global population (Montoya and Liesenfeld, 2004; Kim and Weiss, 2008). In acute stages of infection, the parasite expands via the rapid proliferation of tachyzoite forms. Immunocompetent hosts can mount a T cell-mediated defense that limits this expansion, allowing the differentiation of tachyzoites to slower-growing bradyzoites which form intracellular cysts that persist for the life of the host. While infections are normally asymptomatic, immune deficiency of the host can result in reactivated disease in which latent bradyzoites transform to proliferating tachyzoites (Joynson and Wreghitt, 2001), implying that continuous T cell surveillance is required to limit tachyzoite emergence in encysted tissues. Indeed, studies of chronically infected brain have revealed the presence of persistent CD8+ T cells recognizing parasite-encoded antigen (Schluter et al., 2002; Lutjen et al., 2006), and have shown that whilst these antigen-specific cells do not associate with cysts, they cluster in the vicinity of isolated parasites that may be derived from cyst rupture (Schaeffer et al., 2009).
CD8+ T cells, as well as natural killer (NK) cells, contribute to host defense against intracellular pathogens in large part via the induction of cell death in infected target cells. This function is accomplished primarily via the release of cytotoxic granule contents including perforin, which disrupts the membrane of target cells and granzymes, a family of death-inducing serine proteases that enter target cells in a perforin-dependent manner and are essential for the optimal function of cytotoxic lymphocytes in vivo (Bolitho et al., 2007; Chowdhury and Lieberman, 2008). Cytotoxic cells can also induce death by the activation of death receptors such as Fas on target cells. Toxoplasma gondii-infected cells have been shown to be resistant to apoptosis mediated by Fas (Vutova et al., 2007; Hippe et al., 2008), as well as by irradiation and various chemical inducers (Sinai et al., 2004; Carmen et al., 2006; Kim and Denkers, 2006). However, it remains undetermined whether the prevention of host cell apoptosis is of physiological significance in the pathogenesis of toxoplasmosis. Since CD8+ cells play a vital role both in the control of acute T. gondii infection and in the maintenance of latency (Suzuki and Remington, 1988; Brown and Mcleod, 1990; Suzuki and Remington, 1990; Parker et al., 1991; Gazzinelli et al., 1992; Khan et al., 1999), we decided to examine the effect of the parasite on granzyme-induced apoptosis.
The role of the perforin/granzyme pathway in toxoplasmosis is still uncertain. In the acute phase of T. gondii infection, successful host defense does not require perforin (Denkers et al., 1997), although the perforin-mediated cytotoxic action of NK cells appears to be a significant process at this stage (Persson et al., 2009). In chronically infected mice of the susceptible C57BL/6 strain, which fail to maintain latency and eventually succumb to toxoplasmic encephalitis, the absence of perforin increases brain cyst burden and accelerates mortality (Denkers et al., 1997). In contrast, in resistant BALB/c mice, perforin-deficient T cells are able to maintain latency, although this may be due to a compensatory up-regulation of IFN-γ production (Wang et al., 2004). A recent study demonstrated that chronically infected BALB/c mice in fact contain CD8+ T cells that are able to clear established cysts from the brain in a perforin-dependent manner (Suzuki et al., 2010), providing a potential explanation for the earlier observation of a perforin role in chronically infected C57BL/6 mice (Denkers et al., 1997).
These studies do not clarify the ability of the perforin/granzyme pathway to mediate host defense against tachyzoite-infected cells, either in acute toxoplasmosis or in recrudescent infection following cyst rupture. Toxoplasma gondii-infected animals generate cytotoxic T cells (CTLs) that recognize and kill tachyzoite-infected targets in vitro (Subauste et al., 1991). However, it is not clear whether this cell death represents granzyme-mediated apoptosis or a necrotic or egress response to high local concentrations of perforin, which can result in granzyme-independent cytotoxicity (Waterhouse et al., 2006b). CD8+ T cells can trigger T. gondii egress in vitro via perforin in the absence of caspase function, suggesting that this egress is independent of granzyme-induced apoptosis (Persson et al., 2007). In addition, NK cell-derived perforin may elicit egress in vivo (Persson et al., 2009). These findings may account for an earlier observation that treatment of infected cells with CTLs results in cell lysis without the formation of apoptotic DNA fragments (Nash et al., 1998). Since perforin-mediated immune function displays granzyme-dependence in vivo (Chowdhury and Lieberman, 2008), observations of granzyme-independent perforin action in vitro may not be relevant to host defense to T. gondii. The question of whether, in the absence of perforin-elicited lysis or egress, T. gondii can modulate granzyme-dependent cell death is still unanswered.
Granzyme B (GrB) is the most extensively characterized member of the granzyme family. GrB possesses caspase-like proteolytic activity and shares multiple substrates with caspases, including lamin B, tubulin and poly ADP-ribose polymerase. Human GrB, unlike the mouse enzyme, cleaves additional caspase substrates, including inhibitor of caspase-activated DNase and the BH3 protein Bid (Chowdhury and Lieberman, 2008). The Bid cleavage product, tBid, induces oligomerization of the pro-apoptotic Bcl-2 family members members Bax/Bak and consequent activation of the mitochondrial apoptosis pathway (Lalier et al., 2007). While some studies have shown a dependence of GrB-mediated apoptosis on Bid (Sutton et al., 2000), in other settings GrB can activate the mitochondrial apoptotic pathway in a Bid-independent, Bcl-2-insensitive manner (Goping et al., 2008). Furthermore, GrB can also directly cleave caspase 3 to a p20 form, which can then generate the active p17 form by autocatalysis, most likely as a result of mitochondrial release of Smac/Diablo and consequent relief of IAP-mediated caspase inhibition (Goping et al., 2003). This direct contribution of GrB to caspase 3 activation may account for the independence of GrB-induced apoptosis from caspase 9 (Pardo et al., 2008), which is normally required for apoptosome formation and caspase 3 activation in mitochondria-mediated apoptosis. Finally, while certain apoptotic manifestations in target cells are caspase-dependent, GrB can induce target cell death in a caspase-independent manner (Trapani et al., 1998).
Toxoplasma gondii can protect host mitochondria from apoptotic insults (Sinai et al., 2004; Carmen et al., 2006; Hippe et al., 2008). A recent study has shown that the parasite can prevent Bax/Bak activation downstream of BH3 protein signaling (Hippe et al., 2009). It is unclear whether this protective function of the parasite would suffice to counter the multiple apoptotic mechanisms triggered by GrB. We have found that T. gondii can protect host cells from GrB-induced apoptosis, and that, remarkably, this protection involves the abrogation of GrB activity in infected cells.
2. Materials and methods
2.1. Cell lines and parasites
YT-1 cells (a kind gift from Dr. Z. Nagy, University of Texas El Paso, USA) and the T-leukemic cell line Jurkat were maintained in RPMI medium supplemented with 10% FCS. HeLa cells were maintained in DMEM supplemented with 10% FCS (HyClone). The RH strain of T. gondii was maintained in human foreskin fibroblasts. Some experiments employed strains, derived from RH, that express yellow fluorescent protein (YFP) (a kind gift of B. Striepen, University of Georgia, USA) (Gubbels et al., 2003) or mCherry (a kind gift of M.-J. Gubbels, Boston University, USA).
2.2. Recombinant GrB
GrB was prepared from yeast because commercially available GrB derived from bacteria is not glycosylated and is not taken up efficiently by mammalian cells (Giesubel et al., 2006). The plasmid pPIC9-GrB, consisting of the yeast expression vector pPIC9 (Invitrogen) inserted with mature human GrB was a kind gift of Dr. W. Wels (Chemotherapeutisches Forschungsinstitut, Frankfurt am Main, Germany) and was used to prepare recombinant GrB as previously described (Giesubel et al., 2006). Briefly, the yeast Pichia pastoris, strain GS115 (Invitrogen), was transformed with the plasmid and positive clones were selected as per the manufacturer’s protocol. GrB-expressing clones were verified by Western blot using the monoclonal antibody 2C5 (Santa Cruz). High-expression clones were grown in medium containing 2% methanol to induce expression of GrB. After 5 days, the cells were centrifuged at 7,500 g and the supernatants were passed through a nickel column (GE Healthcare). GrB was eluted using 250 mM imidazole, pH 8.0. Purity was verified by Coomassie Blue staining and immunoblot, which identified a unique 37 kD species (Supplementary Fig. S1). GrB was dialyzed against PBS, pH 7.4 and stored at −80° C until use. Activity was assessed in colorimetric assays containing 200 μM of the synthetic GrB substrate N-acetyl-Ile-Glu-Thr-Asp-nitroaniline in reaction buffer (10 mM HEPES, pH 7.4, 140 mM NaCl and 2.5 mM CaCl2) in a total volume of 100 μl per sample. Substrate cleavage was determined at an absorbance of 490 nm. The activity was comparable with that of a commercially available GrB prepared from Escherichia coli (Supplementary Fig. S2).
2.3. Delivery of recombinant GrB
Target cells were treated with 60 nM of GrB in the presence of sublytic doses of the endosomolytic agents listeriolysin O (LLO) (ProSpec, USA) or streptolysin O (SLO) (Sigma). Sublytic doses were determined in pilot experiments and were defined as doses resulting in less than 10% cell lysis as measured by flow cytometry after staining with propidium iodide (PI). Alternatively, Influx Pinocytic loading reagent (Molecular Probes, USA) was used according to the supplier’s instructions to direct GrB to the cytosolic compartment by osmotic lysis of pinosomes.
2.4. Apoptosis assays
To assess externalization of phosphatidylserine, cells were stained with annexin V-Cy5 (BD Bioscience) and PI according to the supplier’s protocol and analyzed by flow cytometry. Annexin V-positive, PI-negative cells were considered apoptotic. Volume normalization to measure cell yields was achieved using PeakFlow flow cytometry reference beads (Molecular Probes). The number of parasites per infected cell was determined by comparison of YFP or mCherry fluorescence with that of extracellular parasites, as previously described (Tomita et al., 2009). To assess mitochondrial depolarization, cells were stained with 100 nM tetramethylrhodamine, ethyl ester (TMRE) for 30 min at 37° C, and the mitochondrial retention of dye was determined by flow cytometry. To measure caspase 3 cleavage by flow cytometry, cells were fixed with 2% buffered paraformaldehyde for 10 min and then permeabilized in 90% methanol for 30 min on ice. Cells were then washed three times in PBS, blocked (0.5% BSA in PBS) for 1 h at room temperature, and then incubated for 1 h at room temperature with an antibody recognizing cleaved caspase 3 (Cell Signaling Technology, USA), followed by detection with a Cy5-coupled secondary antibody. Cytochrome c release was measured essentially as previously described (Waterhouse et al., 2006a). Briefly, pelleted cells were treated with 50 μg/ml digitonin in cold PBS containing 100 mM KCl, in order to permeabilize the plasma membrane while leaving mitochondria intact. Cells were then fixed in 4% paraformaldehyde, blocked for 1 h at room temperature with 1% BSA in PBS, stained with anti-cytochrome c overnight at 4°C, washed, stained with a Cy5 secondary antibody and analyzed by flow cytometry.
2.5. Correction for cell loss
For conservative estimation of apoptosis in infected cells, GrB-dependent reduction in the number of heavily infected Jurkat cells (> 2 parasites/cell) was attributed to apoptosis. In cultures of uninfected cells, we observed that high rates of GrB-induced apoptosis (> 50%) were associated with high rates of cell loss. Since Jurkat cells are prone to form aggregates (Munn et al., 1993), it is likely that high rates of loss reflect the formation of aggregates that may include healthy as well as apoptotic cells. We therefore excluded from the analysis experiments in which parallel uninfected cultures displayed mean GrB-dependent cell loss > 20%. From each infected culture, we collected the following parameters, each normalized to fluorescent bead number, total uninfected cells (NUN), apoptotic uninfected cells (AUN), total heavily infected cells (NINF) and apoptotic heavily infected cells (AINF). Values from GrB-treated and untreated cultures are denoted, respectively, NUN[GrB] and NUN[Con], etc. The frequency of GrB-dependent apoptosis in uninfected cells, FUN, is calculated by forming 100(AUN[GrB])/NUN[GrB]) and subtracting the mean value obtained for 100(AUN[Con])/NUN[Con]). The frequency of GrB-dependent apoptosis in heavily infected cells (FINF) is calculated similarly. The conservative estimate of GrB-dependent apoptosis in heavily infected cells (CINF) is then derived as follows:
where NINF[Con] and AINF[Con] denote mean values over all samples.
2.6. Immunoblot analysis
Cells were infected overnight at a multiplicity of infection (MOI) of 4. After treatment, cells were washed in PBS and lysed with ice cold lysis buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS). Cell lysates were centrifuged at 10,000 g at 4° C and supernatants (100 μg or lysate from 100,000 cells) was subjected to SDS-PAGE and transferred to polyvinylidene fluoride membranes. Alternatively, for assays of GrB substrates, cells were boiled in 2% SDS in order to avoid adventitious cleavage reactions (Metkar et al., 2003). Membranes were blocked with 5% non-fat milk in TPBS (0.5% Tween-20 in PBS), probed overnight at 4°C with primary antibody in TPBS containing 5% non-fat milk, washed with TPBS, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (KPL) in 5% milk in TPBS for 1 h, and imaged using enhanced chemiluminescence (Pierce) followed by exposure to X-ray film. The primary antibodies used were anti-Bcl-XL, anti-Bcl-2, anti-Mcl-1, anti-caspase 3 (Cell Signaling Technology), anti-Bid (R&D Systems), anti-PI-9, anti-early endosome antigen 1 (EEA1) and anti-GrB (Santa Cruz Biotechnology, USA).
2.7. GranToxiLux assay
Cells were treated with GrB and an endosomolytic agent for 30 min at 37° C, washed in PBS to remove extracellular granzyme, resuspended in GranToxiLux solution (OncoImmunin, USA), either undiluted or diluted four-fold in medium, and incubated for another 2 h. Cells were then washed with GranToxiLux washing buffer supplied by the manufacturer and assayed for GranToxiLux cleavage on a flow cytometer. The GranToxiLux cleavage product fluoresces in the FL1 channel. In assays employing the Influx Pinocytic Loading reagent, the last wash step was replaced with the washes in the manufacturer’s protocol.
2.8. IFA
Jurkat cells, infected at a MOI of 1 for 16 h, were treated for 1 h with 6 ng/ml LLO and 60 nM GrB. To remove membrane-bound GrB, as previously described (Shi et al., 2005), some cells were washed three times with PBS, suspended in 0.25% trypsin- EDTA (Invitrogen) and incubated for 5 min at room temperature. Cells were then washed, fixed with 4% buffered paraformaldehyde for 10 min at room temperature, washed, suspended in PBS supplemented with 0.2% BSA (Sigma) and 5 × 104 cells were deposited on a slide with a cytocentrifuge. Cells were permeabilized in 0.1% Triton-X-100 in PBS for 5 min, blocked for 1 h with blocking buffer (3% BSA in PBS), incubated for 1 h with anti-GrB clone 2C5 (Santa Cruz) (1,400 in blocking buffer), washed, incubated for 30 min with Cy5-labeled secondary antibody (Jackson, USA) and mounted with DAPI in ProLong Gold antifade (Invitrogen). Images were collected on an Olympus IX 81 microscope with a 60X N.A. 1.4 objective. For quantitation of GrB+ cells, between 28 and 47 cells were scored for each data point.
2.9. Cytotoxic lymphocyte-induced apoptosis
For assessment of caspase-3 activation, YT-1 effectors were co-cultured with HeLa targets (effector:target ratio of 5) for 6 h and then removed by rinsing. In a pilot experiment, the HeLa cells were prelabeled with Hoechst 33342 and flow cytometry performed to verify that after rinsing only target cells remained. HeLa cells were then detached and processed for detection of cleaved caspase-3 by flow cytometry. For analysis of GrB activity, YT-1 were co-cultured for 1 h with Jurkat target cells that had been prelabeled with Hoechst 33342 (0.2 μg/ml) for 30 min (effector:target ration of 1). The cells were pelleted, suspended in undiluted GranToxiLux, incubated for 1 h and analyzed by flow cytometry.
2.10. Statistics
Statistical significance was determined using a Student’s t test. P values less than 0.05 were considered significant.
3. Results
3.1. Inhibition of GrB-induced apoptosis in T. gondii-infected cells
The Jurkat cell line has been widely used for studies of GrB-mediated apoptosis and in preliminary experiments we observed that induction of apoptosis by GrB was particularly effective in these cells (data not shown). We examined the protective capacity of T. gondii in Jurkat cells that had been infected for 16 h with parasites expressing YFP (YFP-RH). The parasite was used at a MOI of 1 to generate a mixture of infected and uninfected cells in the same culture that could be analyzed simultaneously for apoptosis. Infected and uninfected cultures were treated for varying times with 60 nM GrB in the presence of either LLO or SLO, endosomolytic agents that provide effective substitutes for perforin (Browne et al., 1999). The endosomolytic agents were preferred for these studies, since perforin can induce substantial T. gondii egress responses (Persson et al., 2007) and is difficult to prepare in a recombinant form due to instability. Apoptotic cells were defined as annexin V-positive and PI-negative.
In the presence of GrB and either LLO or SLO, 60 to 70% of the uninfected population, either within the infected culture (Fig. 1A, B) or in uninfected control cell cultures (Supplementary Fig. S3) entered apoptosis by 2.5 h. Only low frequencies of necrosis (PI-positive cells) were observed. A time-course study demonstrated that apoptotic cells accumulated between 1 and 2.5 h, and no further initiation of apoptosis was observed at 5 h (Fig. 1C). No GrB-induced apoptosis was observed in the absence of LLO (Supplementary Fig. S3). As expected from previous studies in Jurkat cells (Waterhouse et al., 2006a), the apoptotic response to GrB is caspase-dependent (Supplementary Fig. S4). Compared with uninfected cells, the infected population displayed a reduced apoptotic frequency, in the range of 10 to 20%, regardless of which endosomolytic agent was used. This frequency did not increase between 2.5 and 5 h (Fig. 1C), indicating that the apparent protective effect of the parasite did not simply reflect a delay in apoptosis onset.
Fig. 1.

Reduced frequency of granzyme B (GrB)-induced apoptosis in Toxoplasma gondii – infected cells. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein (YFP-RH) at a multiplicity of infection of 1 for 16 h. (A, B) Cells were incubated for 2.5 h with 6 ng/ml listeriolysin O (LLO) (A) or 10 ng/ml streptolysin O (B) in the presence or absence of 60 nM GrB, stained with annexin V and propidium iodide (PI) and analyzed by flow cytometry. Uninfected and infected gates within the same sample were analyzed separately as indicated. The mean infection frequency for the experiments shown is 36%. (C) Cells were incubated for the indicated times with 6 ng/ml LLO in the presence or absence of 60 nM GrB, stained with annexin V and PI and analyzed by flow cytometry. Within each sample, gates of heavily infected cells (> 2 parasites/cell)(black bars), total infected cells (open bars), and uninfected cells (hatched bars) were separately analyzed. Data represent mean ± S.E.M. (n = 3). * P < 0.01 compared with uninfected. The data are representative of at least three similar experiments.
While these data were suggestive of a parasite blockade of GrB-induced apoptosis, several alternative interpretations had to be addressed. First, it was possible that lysis of infected cells, followed by a preferential re-infection of healthy cells, led to an overestimate of protection due to the scoring of these reinfected cells as parasite-protected. Re-infected cells would be expected to contain one to two parasites/cell. We have previously demonstrated that, using the YFP-RH strain, YFP intensity can be used to quantitate intracellular parasite numbers (Tomita et al., 2009). We therefore separately analyzed gates that contained all infected cells or only heavily infected cells (> 2 parasites/cell) (Supplementary Fig. S5). Similar data were obtained for both of these populations, ruling out this explanation (Fig. 1C). Second, it was possible that apoptotic infected cells were lost as a result of parasite egress or cell aggregation. Fluorescent beads were therefore included in all samples to normalize analyzed volumes so that cell recovery could be quantified. This analysis did reveal a differential cell loss in GrB-treated compared with control cultures that was variable among experiments (Table 1). We therefore recalculated the data with the conservative assumption that all of the excess cell loss of heavily infected cells in GrB-treated, compared with untreated, cultures represents undetected apoptosis of infected cells. The analysis of two representative experiments is displayed in Table 1 which shows that, even with the conservative cell loss correction, a parasite protective effect of approximately two-fold was observed. Over a total of 12 similar experiments, the mean fold protection by T. gondii was 19 ± 1.5 without cell loss correction and 1.8 ± 0.1 with correction.
Table 1.
Granzyme B (GrB)-dependent apoptosis and cell loss in Toxoplasma gondii-infected cultures.
| Experiment No. | FUN (%) | FINF (%) | Fold protection | % Cell loss (uninfected) | % Cell loss (infected) | CINF (%) | Fold protection (cell loss corrected) | P |
|---|---|---|---|---|---|---|---|---|
| 1 | 40 ± 2 | 10 ± 1 | 4.0 ± 0.1 | 10 ± 4 | 20 ± 2 | 28 ± 1 | 1.5 ± 0.1 | 0.005 |
| 2 | 28 ± 2 | 1.6 ± 0.4 | 19 ± 4 | 0.6 ± 1.1 | 11 ± 3 | 12 ± 3 | 2.5 ± 0.5 | 0.013 |
Fold protection by T. gondii was determined by dividing the frequency of apoptotic cells in the uninfected fraction of infected cultures (FUN) by the frequency in heavily infected cells (FINF). GrB-dependent cell loss, measured by comparing cell recovery in GrB-treated and control cultures, was determined both for uninfected cultures and for heavily infected cells in infected cultures. The latter value was used to generate a conservative estimate of apoptosis in heavily infected cells (CINF) by correcting for cell loss as described in section 2.5. Corrected fold protection was measured as the ratio of FUN to CINF and the significant difference between CINF and FUN was determined by a Student’s t test.
3.2. Inhibition by T. gondii of mitochondrial apoptotic signaling elicited by GrB
Since the mitochondrial apoptotic pathway is a major mechanism of GrB-induced apoptosis, we examined the effect of T. gondii on mitochondrial events in GrB-treated cells. Activation of the mitochondrial pathway leads to the loss of mitochondrial transmembrane potential, an event that is conveniently measured by the reduced retention of the cationic lipophilic dye TMRE. Treatment with GrB led to mitochondrial depolarization in approximately half of uninfected cells, but in less than 10% of T. gondii-infected cells (Fig. 2). The magnitude of this protective effect was similar to that observed with respect to phophatidylserine exposure.
Fig. 2.

Toxoplasma gondii infection prevents granzyme B (GrB)-induced mitochondrial depolarization. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein at a multiplicity of infection of 1 for 16 h, treated for 2.5 h with 6 ng/ml listeriolysin O in the presence or absence of 60 nM GrB, and then incubated with tetramethylrhodamine (TMRE) to assess mitochondrial polarization. The cells were then stained with annexin V and analyzed by flow cytometry. (A) Representative scatter plots. The heavily infected, total infected and uninfected gates are from the same culture. (B) Depolarization (mean ± S.E., n = 3) of heavily infected (black bars), total infected (open bars) and uninfected (hatched bars) gates. The data represent the percentage of cells found in the two left quadrants within each scatter plot as shown in (A). * P < 0.01 compared with uninfected. The results are representative of two experiments.
The induction of apoptosis through mitochondria involves the release of several pro-apoptotic factors from the mitochondrial intermembrane space, including cytochrome c, Smac/Diablo and Omi/HtrA2, which then contribute to the activation of executioner caspases (Bao and Shi, 2007). We examined the release of cytochrome c by performing flow cytometry to analyze cytochrome c content in cells that had been permeabilized with digitonin to permit the loss of cytochrome c that had entered the cytosol (Fig. 3A). The loss of cytochrome c was detected as the appearance of a bimodal distribution of cytochrome c intensity following GrB treatment of uninfected cells. As expected from previous studies (Pinkoski et al., 2001), GrB induced cytochrome c release in the uninfected cell population in a caspase-independent manner, implying that this event represents a proximal effect of GrB on mitochondria, rather than an amplification event subsequent to caspase activation. In contrast, in the T. gondii-infected population, GrB treatment did not produce any detectable alteration in the cytochrome c profile. Similar data were obtained when infected cells were compared with cells in uninfected cultures (Supplementary Fig. S6). These results imply that the inhibitory effect of T. gondii occurs at a point coincident or prior to the initial activation of mitochondria, rather than during the amplification loop.
Fig. 3.

Inhibition of granzyme B (GrB)-induced cytochrome c release and caspase activation in Toxoplasma gondii-infected cells. (A) Jurkat cells were infected with T. gondii expressing yellow fluorescent protein (YFP-RH) at a multiplicity of infection (MOI) of 1 for 16 h and treated for 2.5 h with 6 ng/ml listeriolysin O (LLO) in the presence or absence of 60 nM GrB. Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD) 1 h before LLO addition. Cytochrome c release was measured by flow cytometry following digitonin permeabilization and cytochrome c immunostaining to determine remaining cell-bound cytochrome c. Infected (Inf.) and uninfected (Uninf.) fractions of the same culture were analyzed. The frequency of infection was 16.3%. (B) Jurkat cells were infected with YFP-RH at a MOI of 1 for 16 h and treated for the indicated times with 6 ng/ml LLO and 60 nM GrB. Fixed cells were immunostained to detect cleaved caspase-3 and analyzed by flow cytometry. Infected and uninfected fractions of the same culture were analyzed. The frequency of infection was 81%. (C) HeLa cells were infected with YFP-RH at a MOI of 4 for 16 h, treated with 2.25 μg/ml streptolysin O and 60 nM GrB for 1.5 h and then analyzed as in (A). The frequency of infection was 41.5%. The data in the figure are representative of two to three experiments.
We next examined the activity of caspase-3, the chief executioner caspase in apoptosis. Caspase-3 is activated by cleavage of a 32 kD procaspase to a p20 form that further matures via autocatalysis to an active p17 form (Pop and Salvesen, 2009). Flow cytometric analysis, using an antibody that recognizes a neo-epitope generated by these cleavage events, revealed that caspase-3 activation is an early event in GrB-induced apoptosis. In the uninfected cell population within an infected culture, activation was detectable by 45 min of GrB treatment and reached a maximal level at 90 min (Fig. 3B). Incubation with LLO in the absence of GrB did not lead to caspase 3 cleavage above basal levels (Supplementary Fig. S7). In contrast, infected cells in the same culture failed to activate caspase-3 even after 2.5 h of GrB treatment. Similar data were obtained upon treatment of infected HeLa cells with GrB and SLO (Fig. 3C), indicating that the protective effect of T. gondii is not specific to a single cell line or endosomolytic agent. Induction of HeLa apoptosis by GrB was more efficient with SLO than LLO (data not shown).
A potential mechanism for parasite intervention at the level of mitochondrial activation is the regulation of the expression of Bcl-2 family members. The anti-apoptotic effect of T. gondii in fibroblasts treated with TNF or staurosporine is associated with up-regulation of anti-apoptotic Bcl2s (Molestina et al., 2003), which include Bcl-2, Bcl-xL, Mcl-1 and A1/Bfl1. Mcl-1 is a substrate for GrB (Han et al., 2005). We detected no up-regulation of Bcl-2, Bcl-xL or Mcl-1 expression by T. gondii (Fig. 4). No signal for A1/Bfl1 was detected (data not shown). Toxoplasma gondii has also been reported to induce degradation of the pro-apoptotic Bcl-2, Bax, in fibroblasts (Carmen et al., 2006). However, we detected no alteration in Bax protein expression (Fig. 4).
Fig. 4.
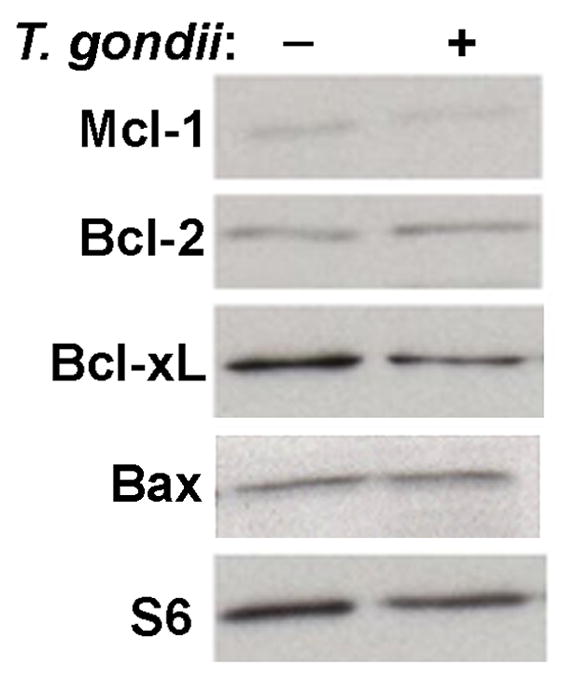
Expression of the Bcl-2 family of apoptotic regulatory proteins in Toxoplasma gondii- infected cells. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein at a multiplicity of infection of 4 for 16 h. The frequency of infection was 81%. Lysates (20 μg) were separated by 12% SDS-PAGE and immunoblots analyzed for the Bcl-2 family proteins Mcl-1, Bcl-2, Bcl-xL and Bax, as well as ribosomal protein S6 as a loading control. The results are representative of two experiments.
3.3. GrB activity is prevented in T. gondii-infected cells
We next sought to determine whether the inhibition of GrB-induced apoptosis could be explained by an affect of the parasite on GrB activity. GrB can activate the mitochondrial apoptotic pathway by cleavage of Bid to form the proapoptotic fragment tBid (Sutton et al., 2000; Goping et al., 2008); therefore we initially attempted to determine whether this event was inhibited by T. gondii. However, Bid can also be cleaved by caspase-3 and we observed that Bid cleavage in GrB-treated Jurkat cells was attributable to caspases (Supplementary Fig. S8), obviating this approach to the assessment of GrB activity. Our next approach exploited the observation that GrB can directly process procaspase-3 to the intermediate p20 form (Martin et al., 1996), resulting in the accumulation of p20 in cells treated with GrB under conditions that inhibit caspase activity, thereby preventing the autocatalytic maturation of p20 to p17 (Goping et al., 2003). We therefore treated cells with GrB in the presence of n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD-fmk), in order to ensure that p20 generation was the direct result of GrB activity rather than an intermediate caspase. Cells were treated with GrB for 1.5 h, the time at which maximal caspase activation was expected (Fig. 3B). In uninfected GrB-treated cells, we observed the zVAD-fmk-dependent accumulation of p20, as expected (Fig. 5). In contrast, p20 accumulation was barely detectable in T. gondii-infected cells, implying that GrB activity towards procaspase-3 is reduced by infection.
Fig. 5.

Toxoplasma gondii inhibits granzyme B (GrB)-mediated cleavage of caspase-3. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein at a multiplicity of infection of 4 for 16 h and treated for 1.5 h with 6 ng/ml listeriolysin O in the presence or absence of 60 nM GrB. The frequency of infection was 83%. Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD-fmk) 1 h prior to GrB addition. Lysates representing 1 × 105 cells were immunoblotted for the detection of the p32, p20 and p17 forms of caspase 3 and then reprobed for the detection of ribosomal protein S6. Cells treated with 2 μM staurosporine (STS) for 1.5 h served as a positive control for caspase cleavage. The results are representative of two experiments.
In a second approach, we made use of an artificial cell-permeable fluorogenic GrB substrate, GranToxiLux, that is not cleaved by caspases (Packard et al., 2007). After a 30 min incubation to permit GrB entry, excess extracellular GrB was removed and GranToxiLux added to generate a cumulative measure of intracellular GrB activity over the ensuing 2 h. As expected, the majority of cells in an uninfected culture contained cleaved substrate (Fig. 6). In contrast, GrB activity was present in only a small proportion of cells in a T. gondii RH-infected culture. Similar effects of T. gondii were observed using either LLO or SLO as endosomolytic agents (Fig. 6B). Taken together with the caspase-3 processing data, these results imply that T. gondii prevents GrB activity. The GranToxiLux assay further suggests that residual GrB activity in infected cultures is not distributed through the culture but is concentrated in a subset of cells with active GrB, implying that GrB activity is reduced to undetectable levels in the majority of T. gondii-infected cells.
Fig. 6.

Toxoplasma gondii inhibits granzyme B (GrB)-mediated cleavage of an exogenous GrB substrate. Jurkat cells, uninfected or infected with T. gondii at a multiplicity of infection of 4 for 16 h, were treated with 10 ng/ml listeriolysin O (LLO) in the presence or absence of 60 nM GrB for 30 min. The cells were washed to remove extracellular GrB and then incubated in the presence of GranToxiLux solution (diluted four-fold in medium) for an additional 2 h prior to analysis by flow cytometry. (A) Representative histograms of GranToxiLux cleavage in uninfected (Uninf.) and infected (Inf.) cultures. (B) Proportion of cells in infected (black) or uninfected (gray) cultures displaying GranToxiLux cleavage mediated by GrB in the presence of LLO or streptolysin O (SLO) (mean ± S.E, n = 3). * P < 0.001 compared with uninfected. The data are representative of two experiments.
3.3.1. Normal accumulation of GrB in T. gondii-infected cells
The prevention of GrB activity in infected cells could be explained by three potential mechanisms. First, uptake of GrB might be deficient in infected cells. Second, the parasite might impede the delivery of GrB from a vesicular compartment to the host cytosol. Finally, GrB activity in the cytosolic compartment might be inhibited in infected cells. To address these questions, we first examined GrB uptake (Fig. 7). A preliminary immunoblot analysis indicated similar levels of cell-associated GrB in infected and uninfected cultures (data not shown). However, this analysis did not distinguish between internalized and cell surface-associated GrB. We then examined GrB-treated cells by immunofluorescence microscopy. Previous studies of GrB uptake, in the presence or absence of perforin or LLO, have shown that internalized GrB is distributed in a novel vesicular compartment distinct from typical endosomes or lysosomes (Pinkoski et al., 1998; Bird et al., 2005; Giesubel et al., 2006; Pipkin and Lieberman, 2007). Consistent with these studies, we observed a clumpy, irregular distribution of GrB (Fig. 7B) that was distinct from distribution observed for EEA1-bearing endosomes (Supplementary Fig. S9). Similar staining patterns and intensities of internalized GrB were observed in both uninfected and T. gondii-infected cells within infected cultures (Fig. 7B), as well as in T. gondii-infected cells treated with trypsin to remove cell surface-associated GrB (Fig. 7C). The frequency of cells positive for GrB by microscopic examination was similar for infected and uninfected cells, regardless of trypsin treatment (Table 2). In addition, infected cells did not display any diminution in normal endocytosis of transferrin by immunofluorescence (Supplementary Fig. S10). Therefore it is unlikely that the prevention of GrB activity in infected cells is due to an alteration in GrB uptake.
Fig. 7.

Uptake of granzyme B (GrB) in Toxoplasma gondii-infected cells. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein for 16 h and treated for 1 h with 6 ng/ml listeriolysin O in the absence (A) or presence (B,C) of 60 nM GrB. Cells were fixed either immediately (A,B) or after trypsin treatment (C) and stained to detect GrB (red), followed by counterstaining with DAPI (blue). Infection is indicated by the presence of parasitophorous vacuoles detected as clusters of green parasites. Scale bar = 5 μm. The results are representative of three similar experiments.
Table 2.
Granzyme B (GrB) uptake in Toxoplasma gondii-infected cells.
| Experiment No. | Infected | Uninfected | Infected (+ trypsin) | Uninfected (+ trypsin) |
|---|---|---|---|---|
| 1 | 64 | 53 | 61 | 59 |
| 2 | 67 | 68 | 71 | 74 |
Jurkat cells were infected with T. gondii RH strain expressing yellow fluorescent protein for 16 h and treated for 1 h with 6 ng/ml listeriolysin O and 60 nM GrB. Cells were fixed either immediately or after trypsin treatment and stained to detect GrB. The percentage of GrB-positive cells was determined for infected and uninfected cells within the same sample.
3.4. Inhibition by T. gondii of GrB delivered to host cell cytosol
We next sought to distinguish between the two remaining possible mechanisms, alteration of GrB delivery to the cytosol, and inhibition of enzyme activity. For this purpose, we treated cells with GrB delivered directly to the cytosol by osmotic lysis of pinosomes, in order to bypass the intracellular vesicular trafficking steps. In this method, GrB is pinocytosed under hyperosmotic conditions and the cells are then returned to isoosmotic medium, resulting in lysis of the pinosomes and release of their contents to the cytosol (Okada and Rechsteiner, 1982). The experiment is conducted in the absence of LLO, so that apoptosis only occurs as a result of pinocytic lysis. Direct cytosolic delivery of GrB by several methods has been previously reported to induce apoptosis (Pinkoski et al., 1998; Hostetter et al., 2007).
As expected, GrB delivery by pinocytic lysis efficiently induced apoptosis in the uninfected population within an infected culture (Fig. 8A). In infected cells, apoptosis was reduced by 42%, indicating that the parasite can exert an inhibitory effect downstream of GrB entry into the cytosol. GrB-induced cell loss was not observed in this experiment (Supplementary Fig. S11). Furthermore, a similar experiment performed in the presence of GranToxiLux showed that most of the enzymatic activity of GrB delivered by pinocytic lysis was inhibited in T. gondii-infected cells (Fig. 8B). These results imply that parasite inhibition of GrB-mediated apoptosis is most likely due to inhibition of GrB activity.
Fig. 8.

Toxoplasma gondii inhibits apoptosis and granzyme B (GrB) activity after delivery by pinocytic lysis. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein (A) or expressing mCherry (B) at a multiplicity of infection of 1 for 16 h. The frequencies of infection were 26% and 45%, respectively. GrB (60 nM), in the absence of endosomolytic agents, was loaded into cell cytosol by osmotic lysis of pinosomes. (A) At 2.5 h after GrB loading, the cells were analyzed by flow cytometry for apoptosis by annexin staining. Uninfected and infected fractions of the same culture were compared. * P < 0.001 compared with uninfected. (B) After pinocytic loading, cells were washed, suspended in medium and allowed to recover for 30 min. GranToxiLux (1/3 volume) was added and after 2 h the cleavage of GranToxiLux was analyzed by flow cytometry. Uninfected (Uninf.) and infected (Inf.) fractions of the same culture were compared. * P < 0.05 compared with uninfected. The data are representative of two experiments.
3.5. Toxoplasma gondii GrB inhibition is not due to PI-9 up-regulation
We next considered the possibility that T. gondii might inhibit GrB by up-regulating an endogenous GrB inhibitor. The serpin protease inhibitor 9 (PI-9) is the only known naturally occurring inhibitor of GrB to be expressed in human cells (Sun et al., 1996). However, immunoblot analysis revealed that PI-9 expression is in fact slightly reduced following T. gondii infection (Fig. 9), ruling out this mechanism.
Fig. 9.
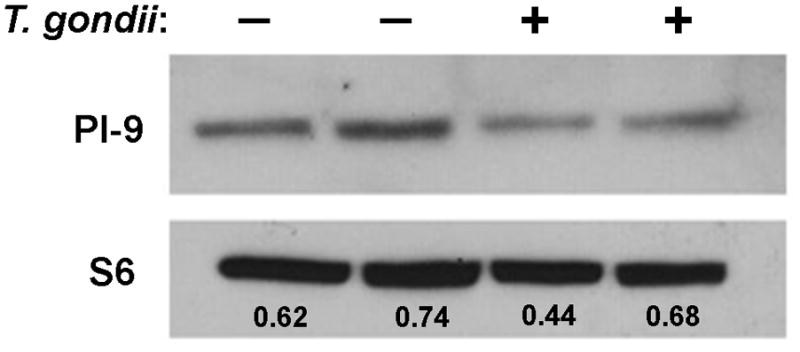
Expression of the granzyme B inhibitor, PI-9, in Toxoplasma gondii-infected cells. Jurkat cells were infected with T. gondii expressing yellow fluorescent protein at a multiplicity of infection of 4 for 16 h. The frequency of infection was 77%. Immunoblots of cells lysates (100 μg) were probed as indicated. Densitometry was performed in ImageJ. The ratio of PI-9 intensity to the intensity of loading control, ribosomal protein S6, is displayed in each lane. The results are representative of two experiments.
3.6. Toxoplasma gondii protects against cytotoxic lymphocyte-mediated apoptosis
The preceding results indicate that T. gondii can inhibit the activity of GrB delivered by multiple methods, including two endosomolytic agents and pinocytic lysis. Nevertheless, it remained to be determined whether similar inhibition occurs when GrB is delivered in a more physiological manner via perforin. To address this issue, we initially examined apoptosis induction in ovalbumin peptide-bearing peritoneal macrophages exposed to antigen-specific CTLs prepared from OT-1 T cell receptor (TCR)-transgenic mice. While we were able to show clear parasite-mediated protection against apoptosis in some of these experiments, the results were inconsistent due to a variable sensitivity of the parasite egress response to effector:target ratio (data not shown). We therefore turned to the use of the NK-like tumor cell line YT-1 as a source of cytotoxic effector lymphocytes, using HeLa and Jurkat cells as targets.
Exposure of uninfected HeLa cells to YT-1 led to caspase-3 activation in the majority of target cells (Fig. 10A). In contrast, after similar treatment of T. gondii-infected cells, no clear peak of caspase-activated cells could be identified in flow cytometry histograms. We did not observe YT-1-induced cell loss in these experiments: the recovery of YT-treated cells was 99% of the untreated control (mean value of two experiments). Therefore T. gondii is able to protect host cells from cytotoxic lymphocyte-mediated apoptosis. To more specifically determine whether T. gondii can suppress the activity of GrB delivered by these effector cells, we assessed GranToxiLux cleavage in Jurkat target cells (Fig. 10B). As expected, the presence of YT-1 cells generated a population of uninfected target cells displaying GranToxiLux cleavage. In contrast, in T. gondii-infected cells the GranToxiLux signal remained similar to the background levels observed in the absence of YT-1 cells (Fig. 10C). These data imply that T. gondii-mediated inhibition of GrB activity is independent of the mode of GrB entry.
Fig. 10.

Toxoplasma gondii inhibits granzyme B (GrB) activity and apoptosis induced by cytotoxic lymphocytes. (A) Inhibition of apoptosis. HeLa cells, infected with T. gondii expressing yellow fluorescent protein at a multiplicity of infection (MOI) of 4 for 16 h, were co-cultured for 6 h with YT-1 effector lymphocytes, rinsed to remove effector cells, and processed for detection of caspase-3 cleavage by flow cytometry. Uninfected (Uninf.) and infected (Inf.) fractions of the same culture were compared; the frequency of infection was 54%. (B) Inhibition of GrB activity. Jurkat cells, infected with T. gondii expressing mCherry at a MOI of 1 for 16 h and stained with Hoechst 33342, were co-cultured for 1 h with YT-1 cells, incubated 1 h in GranToxiLux, and analyzed for GranToxiLux cleavage by flow cytometry. Uninf. and inf. fractions of the same culture were compared; the frequency of infection was 40%. (C) Quantitation of the experiment shown in (B). * P = 0.01 by Student’s t test (n = 4). The data in the figure are representative of two experiments.
4. Discussion
The key conclusion of our study is that T. gondii is able to protect its host cell from GrB-induced apoptosis by the inhibition of GrB activity. The basis for this conclusion is two-fold. First, the protective activity of the parasite includes the suppression of two functions that represent the direct action of GrB, the caspase-independent cleavage of caspase-3 and the cleavage of a GrB-specific synthetic substrate. Similarly the GrB-mediated, caspase-independent release of cytochrome C was suppressed by T. gondii. Second, the suppression of these functions cannot be accounted for by an alteration in the uptake or trafficking of GrB in T. gondii-infected cells. Since GrB trafficking is still poorly understood, our results do not rule out the possibility that T. gondii elicits subtle, significant alterations in this trafficking. However, this mechanism would not be sufficient to account for our results, since the suppression of GrB activity is observed even when GrB is delivered to host cytosol by pinocytic lysis.
In recent years, host defense against an increasing number of intracellular pathogens, including bacterial and fungal pathogens as well as T. gondii and other protozoa, has been shown to involve the perforin/granzyme-mediated action of cytotoxic lymphocytes (Kagi et al., 1994; Zhou et al., 2001; Muller et al., 2003; Ma et al., 2004; Tsagozis et al., 2005). Immune evasion strategies to counter this host function could conceivably operate either by preventing the generation of CTLs, for example via the suppression of antigen presentation, or by negating the effector functions of these cells. While evasive strategies of the first kind have been widely documented for both cellular and viral pathogens, strategies that counter effector functions have received relatively less attention. Only a single pathogen, adenovirus 5, has been demonstrated to protect its host cell from granzyme-induced apoptosis (Andrade et al., 2001). Cowpox virus also encodes a GrB inhibitor, CrmA (Tewari et al., 1995); however, this protein may not provide significant protection against cytotoxic granule-mediated killing of host cells (Quan et al., 1995). We believe our study is the first to provide a demonstration of a comparable ability of a cellular pathogen to subvert the granzyme-induced apoptosis of infected cells.
The dearth of examples of pathogen mechanisms that counter granzyme action may simply reflect limited investigation, since such mechanisms are not likely to be revealed by conventional CTL assays, which measure granzyme-independent target cell lysis (Simon et al., 1997). Alternatively, GrB antagonism as an evasive strategy may be specifically suited to T. gondii rather than other pathogens. For example, while granzyme inhibition can prevent apoptosis, it is not clear how efficiently it can prevent ultimate host cell death in response to cytotoxic lymphocytes, and therefore this mechanism may be of limited value to many pathogens for which maintenance of host cell viability is essential. In contrast, a notable feature of T. gondii is the ability of the parasite to egress and maintain viability at any time during its intracellular residence. We have shown that egress after short periods of residence, followed by re-invasion, is a natural occurrence during acute infection of mice (Tomita et al., 2009). Toxoplasma gondii egress is rapidly triggered by host plasma membrane compromise (Moudy et al., 2001) and can be elicited by perforin (Persson et al., 2007). Intracellular T. gondii have been shown to survive CTL-induced host cell lysis (Yamashita et al., 1998), and the encounter of T cells with infected target cells in vivo results in egress and invasion of the T cells (Chtanova et al., 2009). For T. gondii it may be less important, therefore, to prevent host cell demise than to prevent apoptosis, which may generate a non-supportive intracellular environment and may lead to host cell opsonization and engulfment by phagocytes equipped for parasite destruction.
These considerations suggest two mechanisms by which T. gondii might benefit from anti-apoptosis following the encounter of infected cells with cytotoxic lymphocytes. The first is that anti-apoptosis, via GrB inhibition, may provide interim support for the parasite until host cell damage is sufficient to trigger egress. The second is that the parasite may tailor its response, choosing either an anti-apoptosis or an egress strategy, with the choice influenced by factors such as the kinetics and pathway of cytotoxic granule release, which vary depending on the avidity of TCR-antigen interaction (Beal et al., 2009) and may affect the local concentration of perforin or other cytotoxic mediators. These scenarios, which are not mutually exclusive, underline the importance of experimentally distinguishing between egress and anti-apoptosis, since both of these events can generate a similar reduction in the number of infected apoptotic cells. In an early study of the fate of CTL-challenged T. gondii-infected cells, this distinction was not examined and it is consequently not clear whether the results, which showed a high frequency of lysed cells but not DNA fragmentation in infected cultures, are reflective of egress or anti-apoptosis (Nash et al., 1998). In the current study, the impact of egress was accounted for by quantification of the recovery of infected cells and anti-apoptosis could be demonstrated.
In addition to the perforin/granzyme pathway, CTL can also induce target cell apoptosis by stimulation of the death receptor pathway through CD95/Fas. Several studies by Luder and coworkers have demonstrated that T. gondii can inhibit Fas-mediated apoptosis (Vutova et al., 2007; Hippe et al., 2008). The parasite was shown to block apoptosis in target cells expressing either the type 1 or type 2 Fas-responsive death pathways via, respectively, the degradation of caspase-8 or the suppression of mitochondrial amplification of the apoptotic signal. Several groups have described T. gondii modulation of host Bcl-2s as a potential mechanism of suppressing mitochondrial amplification. These events include up-regulated expression of anti-apoptotic Bcl-2s (Molestina et al., 2003), down-regulation of pro-apoptotic Bax (Carmen et al., 2006), and the prevention of Bax/Bak activation by BH3-only family members (Hippe et al., 2009). Such mechanisms may not be adequate to counter GrB-induced apoptosis, which can proceed via multiple pathways that are both dependent and independent of Bcl-2 family expression (Sutton et al., 2000; Goping et al., 2008). In fact, we failed to observe Bcl-2 family modulations consistent with anti-apoptosis. In preliminary studies using inhibitors of phosphatidylinositol 3-kinase, we also failed to observe a role for the Akt pathway (data not shown), which was reported to support T. gondii-mediated survival of staurosporine-treated host cells (Kim and Denkers, 2006). Our results are therefore most consistent with the view that T. gondii suppression of GrB-induced apoptosis is primarily due to parasite-mediated inhibition of GrB activity.
Two naturally occurring mechanisms have been identified for the inhibition of human GrB. Inhibition by adenovirus 5 is mediated by the viral L4-100K protein (Andrade et al., 2001), while the serpin PI-9 is the sole known endogenous GrB inhibitor in human cells (Sun et al., 1996). In each case, the inhibitor forms a stable complex with GrB and contains a pseudosubstrate loop that occupies the catalytic site, resulting in steric hindrance of substrate association (Huntington et al., 2000; Andrade et al., 2003). We did not observe PI-9 up-regulation in T. gondii-infected cells; nor have we detected predicted proteins homologous to either L4-100K or PI-9 in the T. gondii genome. However, GrB recognition sequences are highly variable and therefore pseudosubstrate sequences are likely to be diverse; indeed, L4-100K and PI-9 have no detectable sequence similarity. It is therefore possible that T. gondii encodes and secretes a novel GrB inhibitor. We have as yet been unable to detect inhibitory activity in extracts of T. gondii-infected Jurkat cells (data not shown); however, these assays are limited by the concentration and stability of any inhibitor that may be present. In addition, parasite extracts were unable to inhibit GrB activation of caspase-3, although the same extracts were sufficient to inhibit activation of the caspase by cytochrome c (Keller et al., 2006). It is evident that additional genetic and biochemical studies will be needed to elucidate the mechanism of this novel inhibitory function of T. gondii. As GrB activity, extra- as well as intracellular, is increasingly implicated in a variety of pathologies (Boivin et al., 2009), the delineation of a new inhibitory mechanism will be useful for the development of this granzyme as a therapeutic target.
The growing repertoire of T. gondii – host cell interactions now includes multiple mechanisms of anti-apoptosis, including parasite interventions at the level of initiating caspases and mitochondrial apoptotic events. Our study identifies the parasite-mediated inhibition of GrB as an additional mechanism and demonstrates that this inhibition is associated with the protection of infected host cells from cytotoxic lymphocyte-mediated apoptosis. The physiological significance of these findings remains to be determined. We were able to observe GrB inhibition in multiple cell types, using GrB delivered via multiple routes, including two endosomolytic agents, pinocytic lysis and, importantly, physiological delivery by an NK-like cell line. These results argue against experimental artifact as the basis for GrB inhibition. The elucidation of the functional significance of this anti-apoptotic mechanism may shed light on immune evasion in T. gondii pathogenesis and lead to an improved understanding of the relative roles of the different effector mechanisms of the host immune response.
Supplementary Material
Immunoblot of recombinant granzyme B (GrB). GrB was prepared from Pichia pastoris as described in Materials and methods (section 2.2), and 0.4 ng was analyzed by probing an immunoblot with anti-GrB. Molecular weight markers are indicated to the left.
Activity of granzyme B (GrB) purified from Pichia pastoris. GrB was prepared and assayed as described in Materials and methods (section 2.2) and compared with commercial Escherichia coli-derived GrB (EMD Biosciences).
Apoptosis induced by recombinant granzyme B (GrB) requires an endosomolytic agent. Jurkat cells (uninfected) were incubated for 2.5 h in the presence or absence of 6 ng/ml listeriolysin O (LLO) and 60 nM GrB, stained with annexin V and propidium iodide (PI), and analyzed by flow cytometry. The ordinate indicates the percent of cells positive for annexin V and negative for PI.
Granzyme B (GrB)-induced apoptosis is caspase-dependent. Jurkat cells were infected with yellow fluorescent protein-expressing Toxoplasma gondii at a multiplicity of infection of 1 for 16 h and treated for 2.5 h with 6 ng/ml listeriolysin O in the absence (open bar) or presence (closed, hatched bars) of 60 nM GrB. To inhibit caspases, cells were treated with 100 μM of n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD-fmk) 1 h prior to addition of GrB (hatched bar). The frequency of cells stained with annexin V but not propidium iodide was determined by flow cytometry. Only cells in the uninfected gate are displayed in the figure.
Illustration of flow cytometric gating of Toxoplasma gondii-infected cells. Jurkat cells were infected with yellow fluorescent protein-expressing T. gondii (YFP-RH) for 16 h, fixed and analyzed by flow cytometry. Uninfected, total infected and heavily infected gates were drawn as indicated. We have previously demonstrated that the fluorescence of YFP-RH is linearly related to intracellular parasite content (Tomita et al., 2009). Cells in the heavily infected gate have a parasite content > 2.
Cytochrome c release in uninfected and Toxoplasma gondii-infected cultures. Jurkat cells, uninfected or infected with yellow fluorescent protein-expressing T. gondii at a multiplicity of infection of 1 for 16 h, were treated for 2.5 h with 6 ng/ml listeriolysin O (LLO) and 60 nM GrB. Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD) 1 h before LLO addition. Cytochrome c release was measured by flow cytometry following digitonin permeabilization and cytochrome c immunostaining to determine remaining cell-bound cytochrome c. The distribution for a control untreated and uninfected culture is shown in grey.
Listeriolysin O (LLO) alone does not induce caspase 3 activation. Samples of Jurkat cells treated for the indicated times with 6 ng/ml LLO in the absence of granzyme B were obtained in the experiment displayed in Fig. 3B and analyzed for caspase 3 cleavage.
Bid cleavage in granzyme B (GrB)-treated cells is caspase-dependent. Jurkat cells were infected with Toxoplasma gondii at a multiplicity of infection of 4 for 16 h and treated for 1.5 h with 6 ng/ml listeriolysin O in the presence or absence of 60 nM GrB, or alternatively with 2 μM staurosporine (STS). Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD) 1 h prior to GrB addition. The immunoblot was probed with anti-Bid and exposed for 1 s or 10 s as indicated in the figure. The arrow indicates the cleaved tBid band. Note that the Bid cleavage generated by GrB is abolished by the caspase inhibitor. Molecular weight markers are indicated at the left.
Distinct localizations of intracellular granzyme B (GrB) and early endosome antigen 1 (EEA1). Jurkat cells were infected for 16 h with Toxoplasma gondii RH strain (non-fluorescent), treated with 60 nM GrB for the indicated times, and immunostained with anti-GrB (fluorescein isothiocyanate secondary antibody) and anti-EEA1 (cyanine 5 secondary antibody). Arrows indicate parasitophorous vacuoles within infected cells. The merged panels display superimposed EEA1, GrB and DAPI signals. Scale bar, 5 μm.
Transferrin uptake is undiminished in Toxoplasma gondii-infected cells. (A) Jurkat cells were infected overnight with yellow fluorescent protein-expressing-T. gondii (YFP-RH) and treated with 5 μg/ml transferrin for 1 h at either 37° C or 4° C. Cells were fixed and immunostained with Alexa647-anti-transferrin. The arrow and arrowhead indicate, respectively, examples of infected and uninfected cells. (B) HeLa cells were infected overnight with YFP-RH, treated with 5 μg/ml transferrin for 1 h and immunostained with Alexa647-anti-transferrin. The image displays an overlay of YFP, transferrin and DAPI signals. (C) The whole-cell Alexa647 intensities were collected in ImageJ from a total of 22 cells imaged in the experiment displayed in (B). Backgrounds were subtracted for each cell and the mean intensities determined for infected and uninfected cells.
Absence of cell loss during apoptosis induced by granzyme B (GrB) delivered by pinocytic lysis. Control and GrB-treated samples from the experiment described in Fig. 8A were analyzed by flow cytometry to determine the ratio of total cells to the fluorescent beads added for volume normalization. The cell/bead ratio is proportional to cell recovery. Uninfected and heavily infected populations within the infected cultures were analyzed separately.
Acknowledgments
We wish to thank Zsuzsanna Nagy (University of Texas at El Paso, USA), Winfried Wels (Chemotherapeutisches Forschungsinstitut Georg-Speyer-Haus, Germany), Boris Striepen (University of Georgia, USA) and Marc-Jan Gubbels (Boston College, USA) for cells and reagents.
We also thank Yanfen Ma for assistance with parasite preparation. This study was supported by funds from National Institutes of Health, USA grant AI-55358 to A.O and AI-39454 to L.M.W., and by the Flow Cytometry Core of the Center for AIDS Research (Albert Einstein College of Medicine, USA) (AI-51519).
Footnotes
Note: Supplementary data associated with this article
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade F, Bull HG, Thornberry NA, Ketner GW, Casciola-Rosen LA, Rosen A. Adenovirus L4–100K assembly protein is a granzyme B substrate that potently inhibits granzyme B-mediated cell death. Immunity. 2001;14:751–761. doi: 10.1016/s1074-7613(01)00149-2. [DOI] [PubMed] [Google Scholar]
- Andrade F, Casciola-Rosen LA, Rosen A. A novel domain in adenovirus L4–100K is required for stable binding and efficient inhibition of human granzyme B, Possible interaction with a species-specific exosite. Mol Cell Biol. 2003;23:6315–6326. doi: 10.1128/MCB.23.17.6315-6326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q, Shi Y. Apoptosome, a platform for the activation of initiator caspases. Cell Death Differ. 2007;14:56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- Beal AM, Anikeeva N, Varma R, Cameron TO, Vasiliver-Shamis G, Norris PJ, Dustin ML, Sykulev Y. Kinetics of Early T Cell Receptor Signaling Regulate the Pathway of Lytic Granule Delivery to the Secretory Domain. Immunity. 2009;31:632–642. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CH, Sun J, Ung K, Karambalis D, Whisstock JC, Trapani JA, Bird PI. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol Cell Biol. 2005;25:7854–7867. doi: 10.1128/MCB.25.17.7854-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease, challenging the dogma. Lab Invest. 2009;89:1195–1220. doi: 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol. 2007;19:339–347. doi: 10.1016/j.coi.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Brown CR, Mcleod R. Class-I Mhc Genes and Cd8+ T-Cells Determine Cyst Number in Toxoplasma gondii Infection. J Immunol. 1990;145:3438–3441. [PubMed] [Google Scholar]
- Browne KA, Blink E, Sutton VR, Froelich CJ, Jans DA, Trapani JA. Cytosolic delivery of granzyme B by bacterial toxins: Evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol Cell Biol. 1999;19:8604–8615. doi: 10.1128/mcb.19.12.8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen JC, Hardi L, Sinai AP. Toxoplasma gondii inhibits ultraviolet light-induced apoptosis through multiple interactions with the mitochondrion-dependent programmed cell death pathway. Cell Microbiol. 2006;8:301–15. doi: 10.1111/j.1462-5822.2005.00622.x. [DOI] [PubMed] [Google Scholar]
- Chowdhury D, Lieberman J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu Rev Immunol. 2008;26:389–420. doi: 10.1146/annurev.immunol.26.021607.090404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Han SJ, Schaeffer M, van Dooren GG, Herzmark P, Striepen B, Robey EA. Dynamics of T Cell, Antigen-Presenting Cell, and Pathogen Interactions during Recall Responses in the Lymph Node. Immunity. 2009;31:342–355. doi: 10.1016/j.immuni.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkers EY, Yap G, SchartonKersten T, Charest H, Butcher BA, Caspar P, Heiny S, Sher A. Perforin-mediated cytolysis plays a limited role in host resistance to Toxoplasma gondii. J Immunol. 1997;159:1903–1908. [PubMed] [Google Scholar]
- Gazzinelli R, Xu YH, Hieny S, Cheever A, Sher A. Simultaneous Depletion of Cd4+ and Cd8+ Lymphocytes-T Is Required to Reactivate Chronic Infection with Toxoplasma gondii. J Immunol. 1992;149:175–180. [PubMed] [Google Scholar]
- Giesubel U, Dalken B, Mahmud F, Wels WS. Cell binding, internalization and cytotoxic activity of human granzyme B expressed in the yeast Pichia pastoris. Biochem J. 2006;394:563–573. doi: 10.1042/BJ20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping IS, Barry M, Liston P, Sawchuk T, Constantinescu G, Michalak KM, Shostak I, Roberts DL, Hunter AM, Korneluk R, Bleackley RC. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity. 2003;18:355–365. doi: 10.1016/s1074-7613(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Goping IS, Sawchuk T, Rieger A, Shostak I, Bleackley RC. Cytotoxic T lymphocytes overcome Bcl-2 inhibition, target cells contribute to their own demise. Blood. 2008;111:2142–2151. doi: 10.1182/blood-2007-08-105221. [DOI] [PubMed] [Google Scholar]
- Gubbels MJ, Li C, Striepen B. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob Agents Ch. 2003;47:309–316. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Goldstein LA, Gastman BR, Rabinovitz A, Rabinowich H. Disruption of Mcl-1 center dot Bim complex in granzyme B-mediated mitochondrial apoptosis. J Biol Chem. 2005;280:16383–16392. doi: 10.1074/jbc.M411377200. [DOI] [PubMed] [Google Scholar]
- Hippe D, Lytovchenko O, Schmitz I, Luder CGK. Fas/CD95-mediated apoptosis of type II cells is blocked by Toxoplasma gondii primarily via interference with the mitochondrial amplification loop. Infect Immun. 2008;76:2905–2912. doi: 10.1128/IAI.01546-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippe D, Weber A, Zhou LY, Chang DC, Hacker G, Luder CGK. Toxoplasma gondii infection confers resistance against Bim S.-induced apoptosis by preventing the activation and mitochondrial targeting of pro-apoptotic Bax. J Cell Sci. 2009;122:3511–3521. doi: 10.1242/jcs.050963. [DOI] [PubMed] [Google Scholar]
- Hostetter DR, Loeb CRK, Chu F, Craik CS. Hip is a pro-survival substrate of granzyme B. J Biol Chem. 2007;282:27865–27874. doi: 10.1074/jbc.M704312200. [DOI] [PubMed] [Google Scholar]
- Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- Joynson DH, Wreghitt TJ. Toxoplasmosis, A Comprehensive Clinical Guide. Cambridge: Cambridge University Press; 2001. [Google Scholar]
- Kagi D, Ledermann B, Burki K, Hengartner H, Zinkernagel RM. Cd8 +. T-Cell-Mediated Protection Against An Intracellular Bacterium by Perforin-Dependent Cytotoxicity. Eur J Immunol. 1994;24:3068–3072. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- Keller P, Schaumburg F, Fischer SF, Hacker G, Gross U, Luder CGK. Direct inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS Microbiol Lett. 2006;258:312–319. doi: 10.1111/j.1574-6968.2006.00241.x. [DOI] [PubMed] [Google Scholar]
- Khan IA, Green WR, Kasper LH, Green KA, Schwartzman JD. Immune CD8+ T cells prevent reactivation of Toxoplasma gondii infection in the immunocompromised host. Infect Immun. 1999;67:5869–5876. doi: 10.1128/iai.67.11.5869-5876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IA, Thomas SY, Moretto MM, Lee FS, Islam SA, Combe C, Schwartzman JD, Luster AD. CCR5 is essential for NK cell trafficking and host survival following Toxoplasma gondii infection. Plos Pathogens. 2006;2:484–500. doi: 10.1371/journal.ppat.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Weiss LM. Toxoplasma, the next 100 years. Microbes Infect. 2008;10:978–984. doi: 10.1016/j.micinf.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L, Denkers EY. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci. 2006;119:2119–26. doi: 10.1242/jcs.02934. [DOI] [PubMed] [Google Scholar]
- Lalier L, Cartron PF, Juin P, Nedelkina S, Manon S, Bechinger B, Vallette FM. Bax activation and mitochondrial insertion during apoptosis. Apoptosis. 2007;12:887–896. doi: 10.1007/s10495-007-0749-1. [DOI] [PubMed] [Google Scholar]
- Lutjen S, Soltek S, Virna S, Deckert M, Schluter D. Organ- and disease-stage-specific regulation of Toxoplasma gondii-specific CD8-T-cell responses by CD4 T cells. Infect Immun. 2006;74:5790–5801. doi: 10.1128/IAI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LL, Wang CLC, Neely GG, Epelman S, Krensky AM, Mody CH. NK cells use perforin rather than granulysin for anticryptococcal activity. J Immunol. 2004;173:3357–3365. doi: 10.4049/jimmunol.173.5.3357. [DOI] [PubMed] [Google Scholar]
- Martin SJ, AmaranteMendes GP, Shi LF, Chuang TH, Casiano CA, Obrien GA, Fitzgerald P, Tan EM, Bokoch GM, Greenberg AH, Green DR. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J. 1996;15:2407–2416. [PMC free article] [PubMed] [Google Scholar]
- Metkar SS, Wang B, Ebbs ML, Kim JH, Lee YJ, Raja SM, Froelich CJ. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J Cell Biol. 2003;160:875–885. doi: 10.1083/jcb.200210158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molestina RE, Payne TM, Coppens I, Sinai AP. Activation of NF-kappa B by Toxoplasmal gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated I kappa B to the parasitophorous vacuole membrane. J Cell Sci. 2003;116:4359–4371. doi: 10.1242/jcs.00683. [DOI] [PubMed] [Google Scholar]
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J Biol Chem. 2001;276:41492–41501. doi: 10.1074/jbc.M106154200. [DOI] [PubMed] [Google Scholar]
- Muller U, Sobek V, Balkow S, Holscher C, Mullbacher A, Museteanu C, Mossmann H, Simon MM. Concerted action of perforin and granzymes is critical for the elimination of Trypanosoma cruzi from mouse tissues, but prevention of early host death is in addition dependent on the FasL/Fas pathway. Eur J Immunol. 2003;33:70–78. doi: 10.1002/immu.200390009. [DOI] [PubMed] [Google Scholar]
- Munn LL, Glacken MW, Mcintyre BW, Zygourakis K. Analysis of Lymphocyte Aggregation Using Digital Image-Analysis. J Immunol Methods. 1993;166:11–25. doi: 10.1016/0022-1759(93)90324-z. [DOI] [PubMed] [Google Scholar]
- Nash PB, Purner MB, Leon RP, Clarke P, Duke RC, Curiel TJ. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J Immunol. 1998;160:1824–1830. [PubMed] [Google Scholar]
- Okada CY, Rechsteiner M. Introduction of macromolecules into cultured mammalian cells by osmotic lysis of pinocytic vesicles. Cell. 1982;29:33–41. doi: 10.1016/0092-8674(82)90087-3. [DOI] [PubMed] [Google Scholar]
- Packard BZ, Telford WG, Koxlloriya A, Henkart PA. Granzyme B activity in target cells detects attack by cytotoxic lymphocytes. J Immunol. 2007;179:3812–3820. doi: 10.4049/jimmunol.179.6.3812. [DOI] [PubMed] [Google Scholar]
- Pardo J, Wallich R, Martin P, Urban C, Rongvaux A, Flavell RA, Mullbacher A, Borner C, Simon MM. Granzyme B-induced cell death exerted by ex vivo CTL, discriminating requirements for cell death and some of its signs. Cell Death Differ. 2008;15:567–579. doi: 10.1038/sj.cdd.4402289. [DOI] [PubMed] [Google Scholar]
- Parker SJ, Roberts CW, Alexander J. Cd8+ T-Cells Are the Major Lymphocyte Subpopulation Involved in the Protective Immune-Response to Toxoplasma gondii in Mice. Clin Exp Immunol. 1991;84:207–212. doi: 10.1111/j.1365-2249.1991.tb08150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson EK, Agnarson AM, Lambert H, Hitziger N, Yagita H, Chambers BJ, Barragan A, Grandien A. Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol. 2007;179:8357–8365. doi: 10.4049/jimmunol.179.12.8357. [DOI] [PubMed] [Google Scholar]
- Persson CM, Lambert H, Vutova PP, Dellacasa-Lindberg I, Nederby J, Yagita H, Ljunggren HG, Grandien A, Barragan A, Chambers BJ. Transmission of Toxoplasma gondii from Infected Dendritic Cells to Natural Killer Cells. Infect Immun. 2009;77:970–976. doi: 10.1128/IAI.00833-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkoski MJ, Hobman M, Heibein JA, Tomaselli K, Li F, Seth P, Froelich CJ, Bleackley RC. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood. 1998;92:1044–1054. [PubMed] [Google Scholar]
- Pinkoski MJ, Waterhouse NJ, Heibein JA, Wolf BB, Kuwana T, Goldstein JC, Newmeyer DD, Bleackley RC, Green DR. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J Biol Chem. 2001;276:12060–12067. doi: 10.1074/jbc.M009038200. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Salvesen GS. Human Caspases: Activation, Specificity, and Regulation. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan LT, Caputo A, Bleackley RC, Pickup DJ, Salvesen GS. Granzyme-B Is Inhibited by the Cowpox Virus Serpin Cytokine Response Modifier-A. J Biol Chem. 1995;270:10377–10379. doi: 10.1074/jbc.270.18.10377. [DOI] [PubMed] [Google Scholar]
- Schaeffer M, Han SJ, Chtanova T, van Dooren GG, Herzmark P, Chen Y, Roysam B, Striepen B, Robey EA. Dynamic Imaging of T Cell-Parasite Interactions in the Brains of Mice Chronically Infected with Toxoplasma gondii. J Immunol. 2009;182:6379–6393. doi: 10.4049/jimmunol.0804307. [DOI] [PubMed] [Google Scholar]
- Schluter D, Meyer T, Kwok LY, Montesinos-Rongen M, Lutjen S, Strack A, Schmitz ML, Deckert M. Phenotype and regulation of persistent intracerebral T cells in murine Toxoplasma encephalitis. J Immunol. 2002;169:315–322. doi: 10.4049/jimmunol.169.1.315. [DOI] [PubMed] [Google Scholar]
- Shi L, Keefe D, Durand E, Feng HP, Zhang D, Lieberman J. Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J Immunol. 2005;174:5456–5461. doi: 10.4049/jimmunol.174.9.5456. [DOI] [PubMed] [Google Scholar]
- Simon MM, Hausmann M, Tran T, Ebnet K, Tschopp J, ThaHla R, Mullbacher A. In vitro- and ex vivo-derived cytolytic leukocytes from granzyme A × B double knockout mice are defective in granule-mediated apoptosis but not lysis of target cells. J Exp Med. 1997;186:1781–1786. doi: 10.1084/jem.186.10.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinai AP, Payne TM, Carmen JC, Hardi L, Watson SJ, Molestina RE. Mechanisms underlying the manipulation of host apoptotic pathways by Toxoplasma gondii. Int J Parasitol. 2004;34:381–391. doi: 10.1016/j.ijpara.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Subauste CS, Koniaris AH, Remington JS. Murine Cd8+ Cytotoxic Lymphocytes-T Lyse Toxoplasma gondii-Infected Cells. J Immunol. 1991;147:3955–3959. [PubMed] [Google Scholar]
- Sun JR, Bird CH, Sutton V, McDonald L, Coughlin PB, Dejong TA, Trapani JA, Bird PI. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier a is present in cytotoxic lymphocytes. J Biol Chem. 1996;271:27802–27809. doi: 10.1074/jbc.271.44.27802. [DOI] [PubMed] [Google Scholar]
- Sutton VR, Davis JE, Cancilla M, Johnstone RW, Ruefli AA, Sedelies K, Browne KA, Trapani JA. Initiation of apoptosis by granzyme B requires direct cleavage of Bid, but not direct granzyme B-mediated caspase activation. J Exp Med. 2000;192:1403–1413. doi: 10.1084/jem.192.10.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Remington JS. Dual Regulation of Resistance Against Toxoplasma gondii Infection by Lyt-2+ and Lyt-1+, L3T4+ T-Cells in Mice. J Immunol. 1988;140:3943–3946. [PubMed] [Google Scholar]
- Suzuki Y, Remington JS. The Effect of Anti-Ifn-Gamma Antibody on the Protective Effect of Lyt-2+ Immune T-Cells Against Toxoplasmosis in Mice. J Immunol. 1990;144:1954–1956. [PubMed] [Google Scholar]
- Suzuki Y, Wang XS, Jortner BS, Payne L, Ni YY, Michie SA, Xu BH, Kudo T, Perkins S. Removal of Toxoplasma gondii Cysts from the Brain by Perforin-Mediated Activity of CD8 +. T Cells. Am J Pathol. 2010;176:1607–1613. doi: 10.2353/ajpath.2010.090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M, Telford WG, Miller RA, Dixit VM. Crma, A Poxvirus-Encoded Serpin, Inhibits Cytotoxic T-Lymphocyte-Mediated Apoptosis. J Biol Chem. 1995;270:22705–22708. doi: 10.1074/jbc.270.39.22705. [DOI] [PubMed] [Google Scholar]
- Tomita T, Yamada T, Weiss LM, Orlofsky A. Externally Triggered Egress Is the Major Fate of Toxoplasma gondii during Acute Infection. J Immunol. 2009;183:6667–6680. doi: 10.4049/jimmunol.0900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapani JA, Jans DA, Jans PJ, Smyth MJ, Browne KA, Sutton VR. Efficient nuclear targeting of granzyme B and the nuclear consequences of apoptosis induced by granzyme B and perforin are caspase-dependent, but cell death is caspase-independent. J Biol Chem. 1998;273:27934–27938. doi: 10.1074/jbc.273.43.27934. [DOI] [PubMed] [Google Scholar]
- Tsagozis P, Karagouni E, Dotsika E. Function of CD8 +. T lymphocytes in a self-curing mouse model of visceral leishmaniasis. Parasitol Int. 2005;54:139–146. doi: 10.1016/j.parint.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Vutova P, Wirth M, Hippe D, Gross U, Schulze-Osthoff K, Schmitz I, Luder CGK. Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell Microbiol. 2007;9:1556–1570. doi: 10.1111/j.1462-5822.2007.00893.x. [DOI] [PubMed] [Google Scholar]
- Wang XS, Kang H, Kikuchi T, Suzuki Y. Gamma interferon production, but not perforin-mediated cytolytic activity, of T cells is required for prevention of toxoplasmic encephalitis in BALB/c mice genetically resistant to the disease. Infect Immun. 2004;72:4432–4438. doi: 10.1128/IAI.72.8.4432-4438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse NJ, Sedelies KA, Sutton VR, Pinkoski MJ, Thia KY, Johnstone R, Bird PI, Green DR, Trapani JA. Functional dissociation of Delta Psi m and cytochrome c release defines the contribution of mitochondria upstream of caspase activation during granzyme B-induced apoptosis. Cell Death Differ. 2006a;13:607–618. doi: 10.1038/sj.cdd.4401772. [DOI] [PubMed] [Google Scholar]
- Waterhouse NJ, Sutton VR, Sedelies KA, Ciccone A, Jenkins M, Turner SJ, Bird PI, Trapani JA. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J Cell Biol. 2006b;173:133–144. doi: 10.1083/jcb.200510072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, Yui K, Ueda M, Yano A. Cytotoxic T-lymphocyte-mediated lysis of Toxoplasma gondii-infected target cells does not lead to death of intracellular parasites. Infect Immun. 1998;66:4651–4655. doi: 10.1128/iai.66.10.4651-4655.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Freidag BL, Caldwell CC, Seder RA. Perforin is required for primary immunity to Histoplasma capsulatum. J Immunol. 2001;166:1968–1974. doi: 10.4049/jimmunol.166.3.1968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunoblot of recombinant granzyme B (GrB). GrB was prepared from Pichia pastoris as described in Materials and methods (section 2.2), and 0.4 ng was analyzed by probing an immunoblot with anti-GrB. Molecular weight markers are indicated to the left.
Activity of granzyme B (GrB) purified from Pichia pastoris. GrB was prepared and assayed as described in Materials and methods (section 2.2) and compared with commercial Escherichia coli-derived GrB (EMD Biosciences).
Apoptosis induced by recombinant granzyme B (GrB) requires an endosomolytic agent. Jurkat cells (uninfected) were incubated for 2.5 h in the presence or absence of 6 ng/ml listeriolysin O (LLO) and 60 nM GrB, stained with annexin V and propidium iodide (PI), and analyzed by flow cytometry. The ordinate indicates the percent of cells positive for annexin V and negative for PI.
Granzyme B (GrB)-induced apoptosis is caspase-dependent. Jurkat cells were infected with yellow fluorescent protein-expressing Toxoplasma gondii at a multiplicity of infection of 1 for 16 h and treated for 2.5 h with 6 ng/ml listeriolysin O in the absence (open bar) or presence (closed, hatched bars) of 60 nM GrB. To inhibit caspases, cells were treated with 100 μM of n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD-fmk) 1 h prior to addition of GrB (hatched bar). The frequency of cells stained with annexin V but not propidium iodide was determined by flow cytometry. Only cells in the uninfected gate are displayed in the figure.
Illustration of flow cytometric gating of Toxoplasma gondii-infected cells. Jurkat cells were infected with yellow fluorescent protein-expressing T. gondii (YFP-RH) for 16 h, fixed and analyzed by flow cytometry. Uninfected, total infected and heavily infected gates were drawn as indicated. We have previously demonstrated that the fluorescence of YFP-RH is linearly related to intracellular parasite content (Tomita et al., 2009). Cells in the heavily infected gate have a parasite content > 2.
Cytochrome c release in uninfected and Toxoplasma gondii-infected cultures. Jurkat cells, uninfected or infected with yellow fluorescent protein-expressing T. gondii at a multiplicity of infection of 1 for 16 h, were treated for 2.5 h with 6 ng/ml listeriolysin O (LLO) and 60 nM GrB. Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD) 1 h before LLO addition. Cytochrome c release was measured by flow cytometry following digitonin permeabilization and cytochrome c immunostaining to determine remaining cell-bound cytochrome c. The distribution for a control untreated and uninfected culture is shown in grey.
Listeriolysin O (LLO) alone does not induce caspase 3 activation. Samples of Jurkat cells treated for the indicated times with 6 ng/ml LLO in the absence of granzyme B were obtained in the experiment displayed in Fig. 3B and analyzed for caspase 3 cleavage.
Bid cleavage in granzyme B (GrB)-treated cells is caspase-dependent. Jurkat cells were infected with Toxoplasma gondii at a multiplicity of infection of 4 for 16 h and treated for 1.5 h with 6 ng/ml listeriolysin O in the presence or absence of 60 nM GrB, or alternatively with 2 μM staurosporine (STS). Some samples received 100 μM n-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD) 1 h prior to GrB addition. The immunoblot was probed with anti-Bid and exposed for 1 s or 10 s as indicated in the figure. The arrow indicates the cleaved tBid band. Note that the Bid cleavage generated by GrB is abolished by the caspase inhibitor. Molecular weight markers are indicated at the left.
Distinct localizations of intracellular granzyme B (GrB) and early endosome antigen 1 (EEA1). Jurkat cells were infected for 16 h with Toxoplasma gondii RH strain (non-fluorescent), treated with 60 nM GrB for the indicated times, and immunostained with anti-GrB (fluorescein isothiocyanate secondary antibody) and anti-EEA1 (cyanine 5 secondary antibody). Arrows indicate parasitophorous vacuoles within infected cells. The merged panels display superimposed EEA1, GrB and DAPI signals. Scale bar, 5 μm.
Transferrin uptake is undiminished in Toxoplasma gondii-infected cells. (A) Jurkat cells were infected overnight with yellow fluorescent protein-expressing-T. gondii (YFP-RH) and treated with 5 μg/ml transferrin for 1 h at either 37° C or 4° C. Cells were fixed and immunostained with Alexa647-anti-transferrin. The arrow and arrowhead indicate, respectively, examples of infected and uninfected cells. (B) HeLa cells were infected overnight with YFP-RH, treated with 5 μg/ml transferrin for 1 h and immunostained with Alexa647-anti-transferrin. The image displays an overlay of YFP, transferrin and DAPI signals. (C) The whole-cell Alexa647 intensities were collected in ImageJ from a total of 22 cells imaged in the experiment displayed in (B). Backgrounds were subtracted for each cell and the mean intensities determined for infected and uninfected cells.
Absence of cell loss during apoptosis induced by granzyme B (GrB) delivered by pinocytic lysis. Control and GrB-treated samples from the experiment described in Fig. 8A were analyzed by flow cytometry to determine the ratio of total cells to the fluorescent beads added for volume normalization. The cell/bead ratio is proportional to cell recovery. Uninfected and heavily infected populations within the infected cultures were analyzed separately.