

Abstract
A newborn's skull is highly malleable and rapidly expanding. As a result, any restrictive or constrictive forces applied to a baby's head can result in dramatic distortions. These changes can be mild, reversible deformations or severe, irreversible malformations that can result in brain injury. This paper reviews the anatomy and physiology of normal and abnormal brain and skull growth, the etiology of cranial deformation, the types of craniosynostosis most commonly seen in infants, and the importance of early diagnosis and treatment.
At birth, the shape of a newborn's skull is highly variable due to its inherent plasticity, intrauterine constraint, and the tortuous journey through the birth canal. Variations from the typical oval shape that usually result from the vaginal delivery process will generally return to normal in a relatively short period of time. If this does not occur, the possibility of a rapidly progressive, irreversible, and, in rare circumstances, life threatening cranial malformation needs to be considered.
Historical Perspective: “Intentional Cranial Deformation”
Man's fascination with misshapen heads dates back to prehistoric times. Archaeologists have found artistic renderings of the imposing heads of Neanderthals who lived 45 000 years ago. Hippocrates described in detail a people referred to as the “Macrocephales.” Even in modern times, the television series “Saturday Night Live” entertained viewers with their comic series “The Coneheads.”
Of particular historical interest is the practice of intentional cranial deformation, “the process of dynamic distortion of the normal vectors of infantile neurocranial growth through the agency of externally applied forces”(1). Taking advantage of the rapid head growth and malleable skull unique to the newborn period, individuals have applied constrictive devices (wooden boards, stones placed in a crib, ties, manual molding) over the past centuries to intentionally and permanently deform a child's skull. In 1805, the Lewis and Clark expedition encountered the Chinook tribe at the mouth of the Columbia River. Infants of the tribal leaders were noted to have their heads constrained by wooden sticks and rope (Figure 1). These devices were placed soon after birth and kept in place for months to years to create a permanent cranial deformity that was interpreted as a mark of distinction. Similarly, it was the practice in the House of Este in the 1400s to place restrictive ties known as “bandeau” at birth on the heads of the royal newborns. A portrait of a princess who underwent this process hangs in the Louvre Museum (Figure 2).
Figure 1.
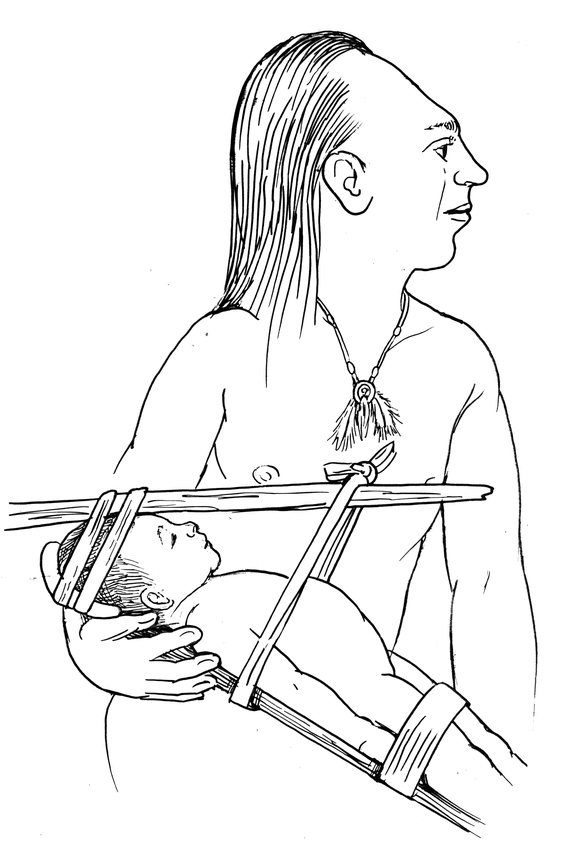
Chinook tribal leader with infant in wooden constraint
Figure 2.

Princess of the House of Este (circa 1400) in “bandeau.”
The rationale for this dramatic intervention was to create class and tribal distinction. In other societies, similar practices were felt to enhance intellectual and sexual abilities and to provide intimidation in battle through the imitation of beasts. The excavation of skulls that underwent this type of intentional deformation has led to a better understanding of growth and development of the human skull.
Normal Development of the Human Skull
The study of human head growth must begin with a basic understanding of the normal skull, brain, and cranial sutures. The skull is formed from embryonic mesoderm, which differentiates into the mesenchymal neurocranium and viscerocranium. The average occipital-frontal circumference (OFC) is 35 cm in the term newborn, 45 cm at 1 year, and 55 cm in an adult. These measurements illustrate the extremely rapid growth in the first years; in fact, a newborn's head circumference is larger than the chest circumference at birth. The OFC increases by 2 cm per month for the first 3 months of life, 1 cm per month for the second 3 months of life, and 0.5 cm per month from 6–12 months. The volume of the cranial vault is 65% of adult size at birth and 95% of the adult size at age 10 years. In contrast, facial size is 40% of adult size at birth and 65% at 10 years.
Appreciation of normal brain growth guides our understanding and management of many forms of skull anomalies. The rapid growth in neuronal cell number during the 10th through 18th weeks of gestation achieves near adult cell numbers; this is followed by dramatic increases in dendritic growth and arborization then myelinization. At 15 months of age the brain is roughly 65% adult size while the cerebellum has achieved adult proportion. The majority of myelinization is complete by 2 years of age.
The cranial sutures are unique structures composed of dense connective tissue membranes that function as fibrous, or “syndesmotic,” joints, which permit growth. The most important sutures are the sagittal, coronal, lambdoidal, and metopic sutures (the standard method of viewing and illustrating skull shapes is the vertex view — Figure 3); importantly, these growth plates do not normally calcify and close until adolescence.
Figure 3.
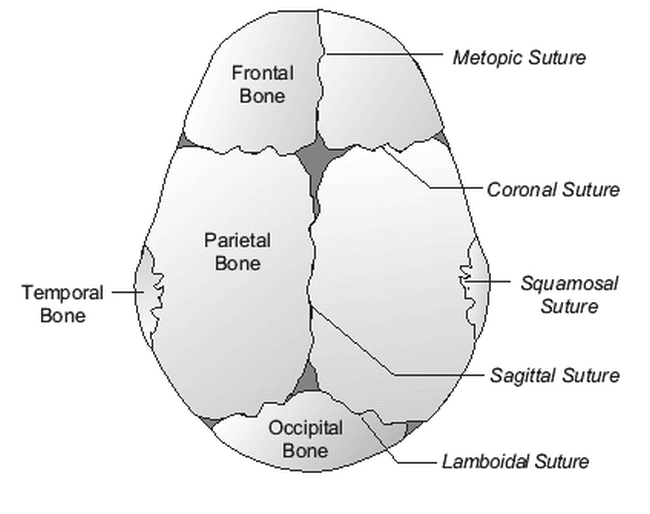
Vertex view of normal skull
Abnormal Development: Malformation vs. Deformation
There are two very distinct processes of morphogenesis through which the human skull is misshapen: malformation and deformation. Malformation refers to an intrinsically altered developmental process that interferes with cell migration and differentiation through genetically programmed biochemical processes or through extrinsic chemical interference (teratogens). In essence, this process represents an error in the normal development of a part. Examples would include congenital heart disease due to chromosome 22q11 deletion or a cleft lip secondary to fetal alcohol exposure. In contradistinction, a deformation is an alteration of a body part that is developing normally until a mechanical force is applied. This type of birth defect occurs after a part is fully formed and is then physically altered due to extrinsic pressures. Examples would be dislocated hips in a newborn, due to compression in the breech position during gestation, or a calcaneovalgus foot deformity due to in utero positioning. The most common abnormal cranial shapes are shown in Figure 4.
Figure 4.
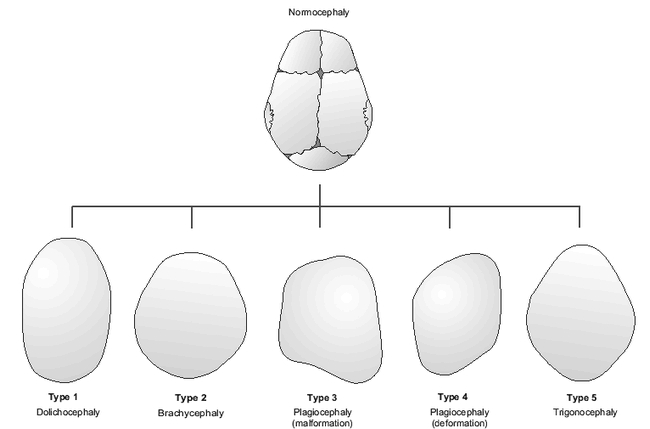
Abnormal cranial shapes
Cranial Deformations
In general, cranial deformations are common, mild, and typically reversible, while cranial malformations are relatively rare, progressive, and often irreversible anomalies which, if not aggressively treated in a timely manner, can result in severe cosmetic and functional impairment. Deformations, which occur in one out of three newborns, result from three major mechanisms—peripartal molding, supine sleep position, and torticollis (Table 1) (2).
Table 1.
Etiology of Cranial Deformations

Molding
One out of three infants will have some degree of deformational molding. Fetal head constraint is more common in primigravidas, large for gestational age babies, and when there is cephalopelvic disproportion, oligohydramnios, multiple births, or prolonged courses of labor. Caput succedaneum (Figure 5) is due to edema of the skin and subcutaneous tissues of the scalp resulting in a “conehead” appearance, which normally resolves in less than 6 days. A cephalohematoma is a traumatic subperiosteal hemorrhage that does not cross a suture line. This deformity is initially soft and, with time, becomes firm as it calcifies; it generally requires up to 4 months to resolve entirely.
Figure 5.
Caput succedaneum
Babies born breech (Figure 6) typically have craniofacial and limb deformations resulting from their in utero position. These babies characteristically have a long, narrow head, (“dolichocephaly” or “type 1”), with a prominent occipital shelf, redundant skin over the neck, overlapping lambdoidal sutures, and an indentation below their ears (from shoulder compression). These babies are also more likely to have a head-tilt, or torticollis, after birth due to fetal constraint. Developmental dysplasia of the hips and calcaneovalgus foot deformity are also more commonly seen in this population and may or may not be reversible without intervention.
Figure 6.
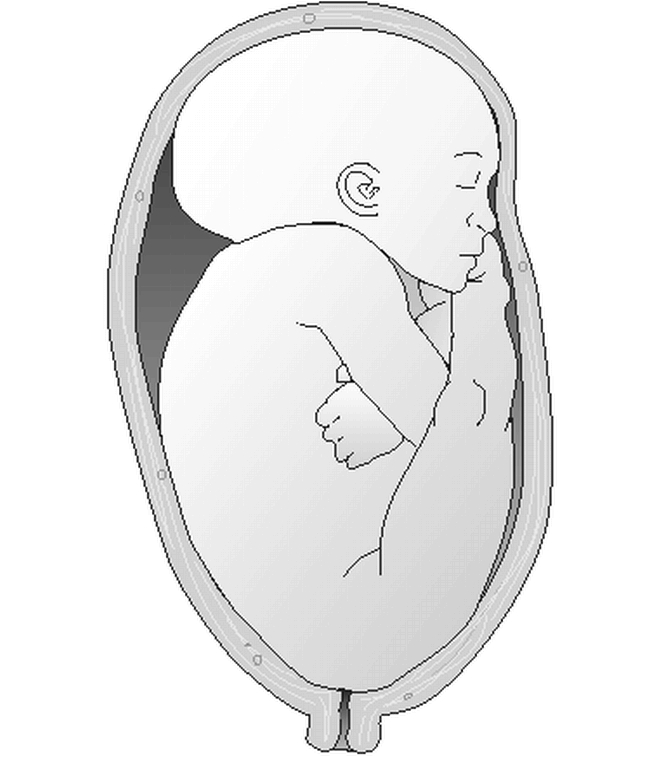
In utero breech position
Supine Sleep Position
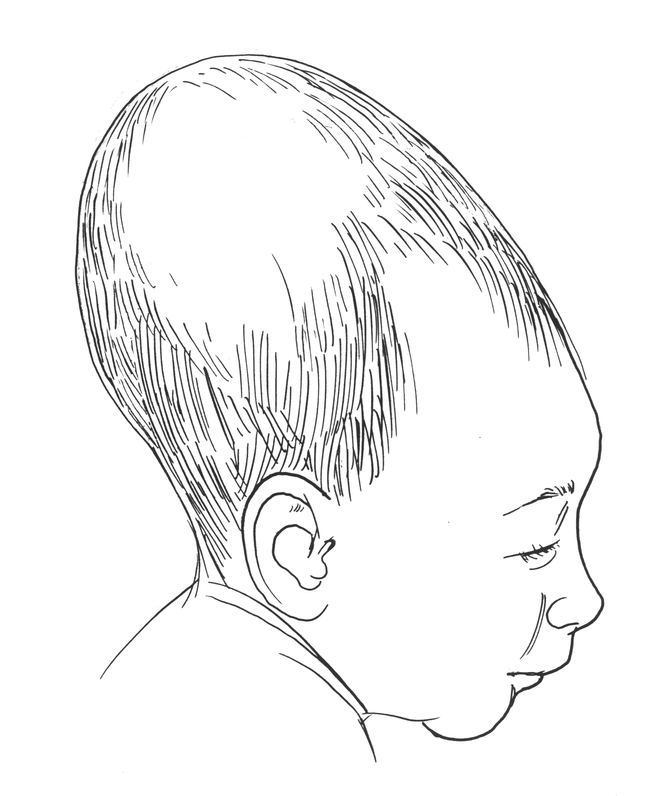
In 1992, the American Academy of Pediatrics published guidelines recommending that all healthy infants be placed to sleep in the supine position during the first 6 months of life in an attempt to reduce the incidence of Sudden Infant Death Syndrome (SIDS). This “Back-to-Sleep” campaign has resulted in a dramatic 50% reduction in the incidence of SIDS in the United States. An unanticipated, and relatively minor sequelae of this program has been the dramatic increase in the numbers of babies with acquired posterior cranial deformities, specifically occipital flattening with brachycephaly (Figures 4 & 7)(3). If recognized early, simply placing the baby prone while awake or alternating the point of contact between the occiput and bed during sleep can smooth out this deformation.
Figure 7.
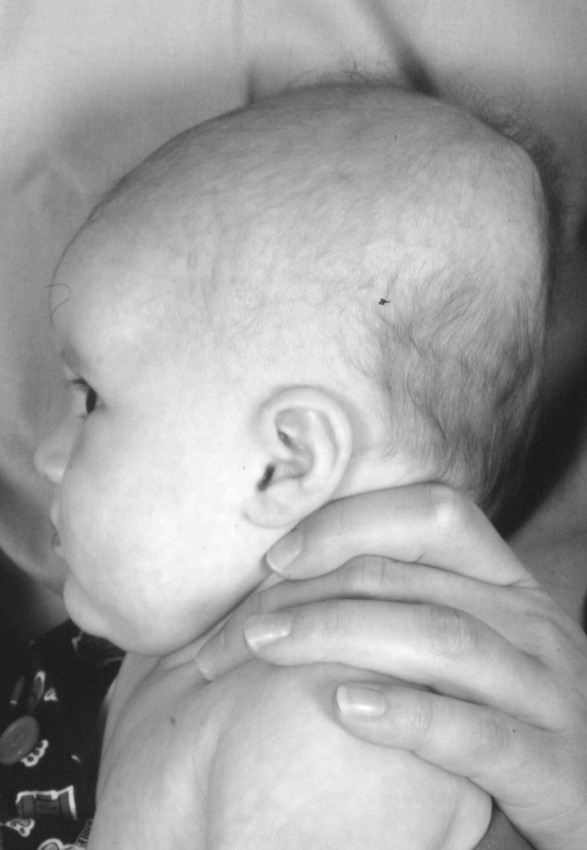
Newborn with flattened occiput (positional deformation)
Postnatal deformation can also occur in the neonatal intensive care nursery when high-risk babies are kept paralyzed and intubated on their side for extended periods. These babies have long and narrow heads due to their relatively large heads and poor neck muscle tone; the skull bones are soft and thin and the skull is flattened by gravity alone. It is also important to note that neurologically impaired infants with hypo- or hypertonia may have a greater degree of positional deformity of their heads due to limited mobility when prone.
Toricollis
Torticollis is a head tilted to one side with the occiput rotated towards the shoulder and the chin rotated in the opposite direction and elevated (Figure 8a). The most common etiologies in newborns are congenital muscular torticollis (CMT) and anomalies of the cervical spine. CMT is due to stretching of the neck during difficult deliveries, often in large babies, with resultant hemorrhage into the sternocleidomastoid muscle. With time, fibrous changes occur in the muscle and it contracts resulting in torticollis. Since babies are initially somewhat floppy and because this contraction is progressive during the first few weeks, it may not be noted for several weeks after birth.

Figure 8A. Torticollis
Among cervical spine anomlies, Klippel-Feil Syndrome is due to a segmentation abnormality of two or more cervical vertebrae. Other causes, such as cranial nerve IV palsy in which the infant tilts the head to achieve normal vision, are listed in Table 1.
Whatever mechanism results in the head rotating or tilting, the side of the occiput that is posteriorly positioned is flattened since the child will preferentially lie on it. The contralateral occiput and the ipsilateral forehead will become more prominent due to gravitational forces resulting in a parallelogram shape to the head. This is an example of posterior positional (or, deformational) plagiocephaly (Figures 4 & 8b). This is generally a subtle cosmetic defect, which may be cleverly masked by hair. In the more severe forms of this deformation, normal cranial ossification will lead to a permanently misshapen skull if action is not taken to even out the positional forces.
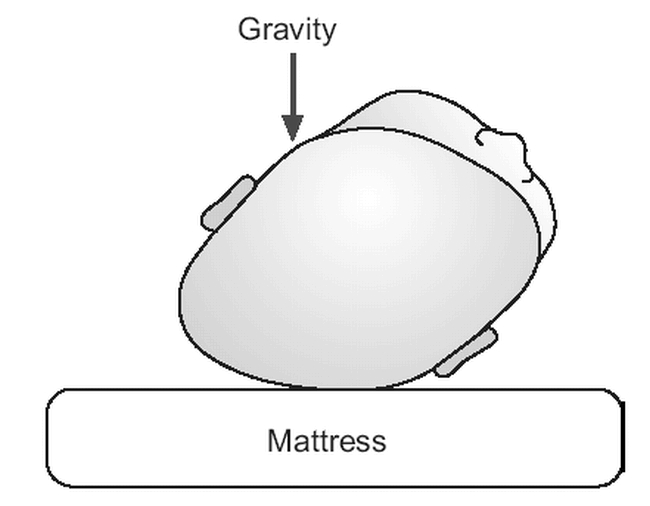
Figure 8B. Positional plagiocephaly due to supine lie in infant with torticollis
Cranial Malformations
The other, and far more ominous, type of abnormal cranial development is craniosynostosis, or premature fusion of one or more cranial sutures. This malformation occurs in 1 in 2500 neonates as opposed to the 1 in 3 babies with a deformational anomaly. Craniosynostosis is classified as simple (1 suture) versus compound (2 or more sutures), and isolated (no other major malformations) versus syndromic (one of multiple associated anomalies).
The mechanism of skull malformation caused by a fused suture(s) in a developing skull was initially described by Virchow in 1851 (4). He pointed out that cranial growth restriction will occur in the plane parallel to a prematurely fused suture and enhanced in the perpendicular planes (Figure 9). Thus, if the sagittal suture were fused early, one would expect the skull to be restricted in the transverse dimension and to overcompensate in the anterior-posterior dimension in response to the growing brain resulting in dolichocephaly (type 1).
Figure 9.
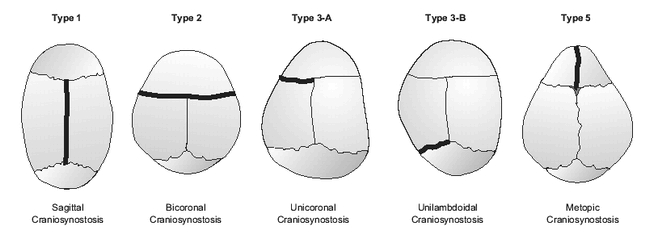
Mechanisms of head malformation in craniosynotonosis. Darkened lines represent synostotic sutures
Craniosynostosis is also classified based on the mechanism of malformation; namely, primary versus secondary. Primary craniosynostosis represents an intrinsic abnormality in the bone or suture, such as fibroblast growth factor deficiency, which results in premature fusion of a suture. Recent research has shown that craniosynostosis can be induced in animals through in utero compression alone, a deformational change resulting in a malformation (5). Suture patency is also dependent on a growing brain, which exerts pressure against the developing skull. Failure of the brain to enlarge and expand properly results in secondary craniosynostosis and, characteristically, all sutures would be involved (Table 2).
Table 2.
Etiology of craniosynostosis

Of the children with primary craniosynostosis caused by single gene defects, the most common type would be isolated (i.e., no other defects) craniosynostosis involving a single suture (Table 3). Less than 5% of the cases would be syndromic (a collection of malformations which aggregate together) (Table 4). Infants with syndromic forms are most likely to appear markedly dysmorphic with abnormalities of their limbs, heart, genitourinary system, and multisutural craniosynostosis (Figure 10) (6).
Table 3.
Primary Craniosynostosis; Isolated (95%)
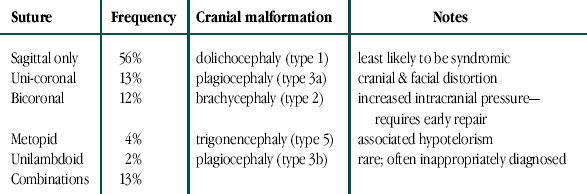
Table 4.
Examples of Primary Craniosynostosis/Monogenic etiology; syndromic (5%)

Figure 10.
Infant with Pfeiffer Syndrome. An example of multisutural craniosynostosis
Differentiation of Malformation vs. Deformation: Position vs. Pathology
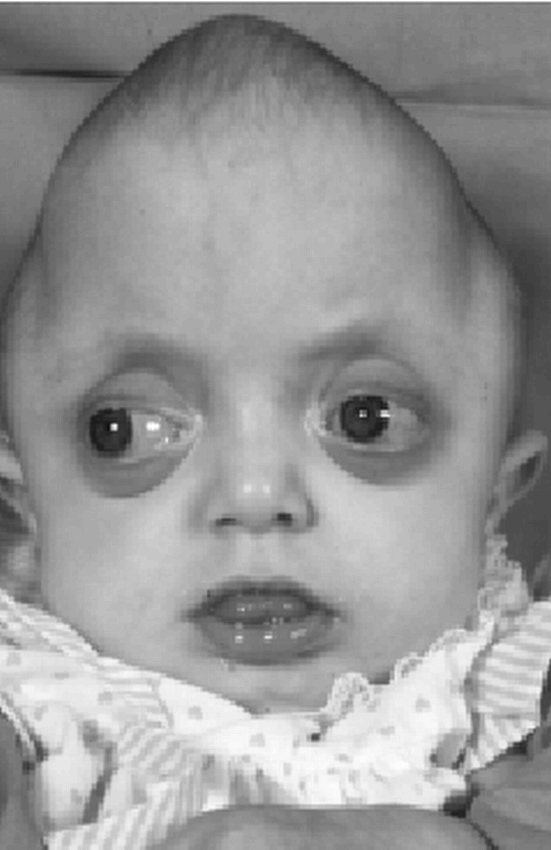
Children with syndromic forms of craniosynostosis are readily detected, though the specific syndrome often requires the expertise of a dysmorphologist. The greatest diagnostic challenge often involves the differentiation of a malformation from a deformation, because it usually determines whether the child will undergo major craniofacial surgery, with its significant risks, versus conservative management. Proper diagnosis can often be achieved through a careful history and physical examination. If questions remain, radiographic procedures may be useful. Plain films of the skull may show perisutural sclerosis, and indistinctness of the suture. Facial views may reveal distinctive uplifting of the orbital roof (“harlequin eye deformity”), which represents a classic finding in coronal craniosynostosis (Figures 11A & 11B). Plain CT scans of the head are of limited help in the evaluation of craniosynostosis. The best current imaging study would be a CT scan of the head with 3D computer reconstruction (Figure 11C). There are three major cranial shape abnormalities for which one must differentiate positional deformation from craniosynostosis.

Figure 11A. Photo of infant with left coronal craniosynostosis
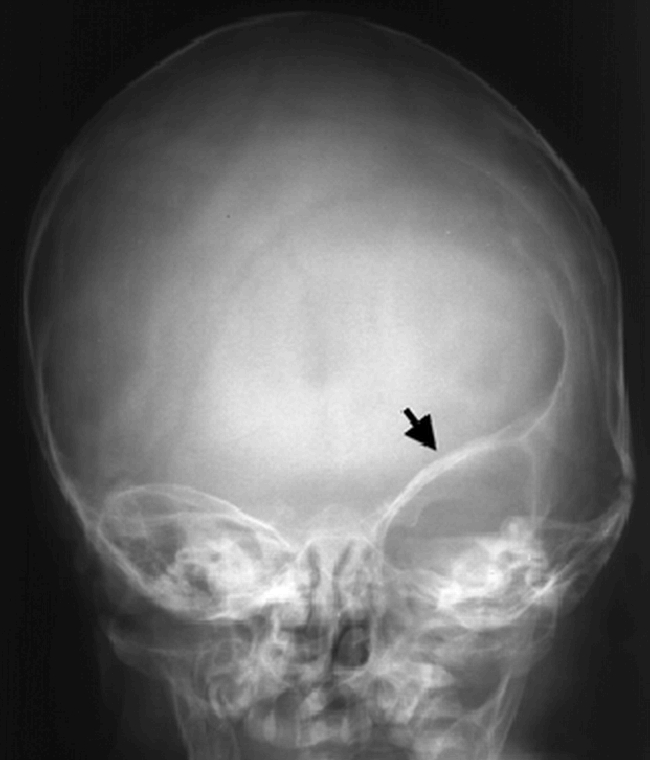
Figure 11B. Facial x-ray demonstrating “Harlequin eye deformity”
Figure 11C. Three dimensional computed tomography
Dolichocephaly (type 1)
In the presence of a prolonged positional insult (a premie lying on the side for an extended period of time) one should suspect a dolichocephalic cranial shape. If other physical findings of craniosynostosis (ridged sutures, progression over time) are noted, radiographic studies should be ordered. It is important to note that only portions of the suture may be stenotic (Figures 12a & 12b), so a patent fontanel does not rule out this anomaly.
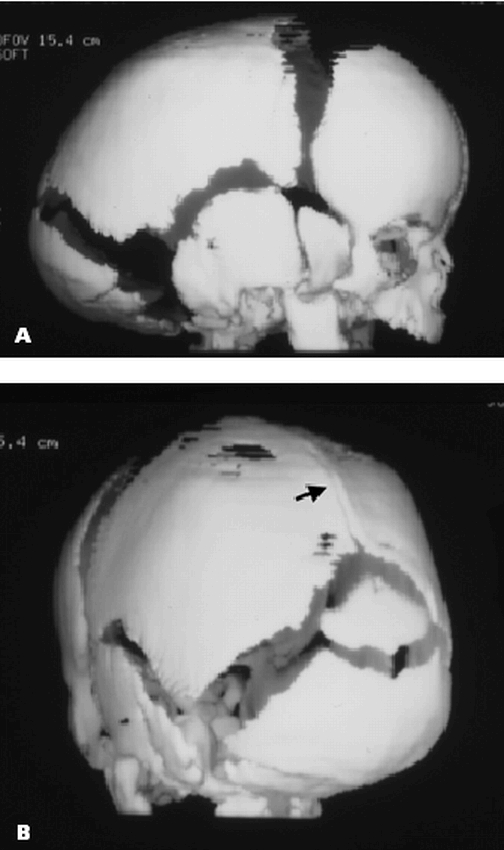
Figure 12A & B. Posterior sagittal craniosynostosis
Brachycephaly (type 2)
A baby with brachycephaly due to a positional problem is more likely to have had a normal head at birth and subsequently spent most of her time in the supine position. Parents often report that the baby moves little when asleep thus flattening the occiput more than the child that rolls side-to-side. It is important to be wary of neurodevelopmental problems in this group since hypotonic infants are less likely to rotate and elevate their heads when supine. In contrast, babies with bicoronal craniosynostosis were noted to have an abnormal skull at birth that has rapidly changed regardless of sleep position. On examination, infants with isolated positional flattening have normal facial features while those with bicoronal craniosynostosis will have restriction of facial growth since the synostotic coronal suture, which may be palpable, extends from the anterior fontanel to the cranial base.
Plagiocephaly (type 3 & type 4)
This skull abnormality represents the most common source of confusion. The clinician must differentiate between a positional abnormality and unilateral coronal or lambdoidal craniosynostosis. Babies with positional problems are reported to lie almost exclusively on one side of their occiput. This is particularly dramatic in babies with torticollis who are essentially fixed in this position when placed down. An interesting observation can assist the clinician in differentiating positional plagiocephaly from unilateral coronal or lambdoidal craniosynostosis. In positional changes, the babies lie preferentially on their right or left occiput. Based on the laws of physics, one would expect gravitational forces to result in widening of the plane drawn from the contralateral (non-flattened) occiput to the ipsilateral forehead and shortening of the plane connecting the involved (flattened) occiput to the contralateral forehead. The resulting geometric shape (type 4) would be a parallelogram (Figure 13a). Conversely, early closure of a unicoronal (type 3a) or unilambdoidal (type 3b) suture would result in restricted growth in the vertical plane perpendicular to the involved suture and compensatory overgrowth in the parallel plane perpendicular to the normal coronal suture. This would result in a trapezoidal shape to the skull (Figure 13b). If one were to draw a line down the center of the skull, the volume of side A=B in positional plagiocephaly, while A>B in unicoronal or unilambdoidal craniosynostosis (7). Facial features are also distinctive (8). Radiographic studies may also be utilized as previously outlined.
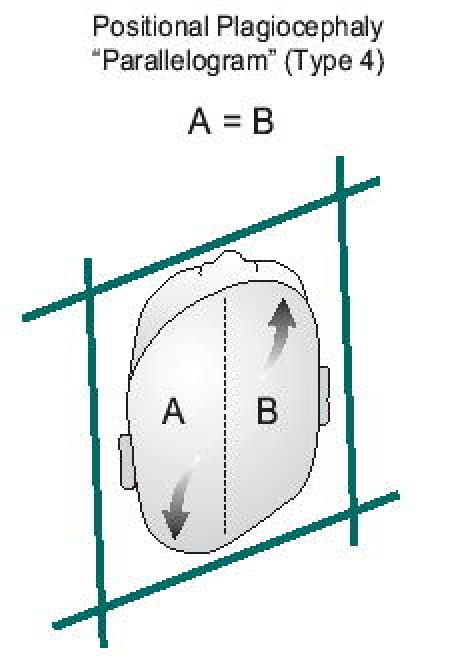
Figure 13A
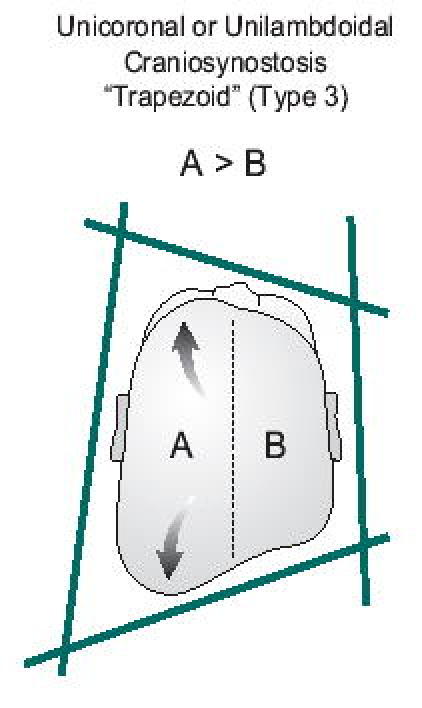
Figure 13B
Management and Prognosis
Surgical management of craniosynostosis dates back to the late 19th century, but it was not until 1967 that a more comprehensive intracranial approach to syndromic craniosynostosis was first published by Tessier (9). He noted that simple craniectomy or morcellization of the involved suture in early infancy resulted frequently in reossification of the suture. As a result, he performed a frontal-orbital advancement with cranial vault remodeling between 6–12 months. This resulted in decompression of the cranial vault, provided protection of the globes and dramatically improved aesthetics. In bicoronal craniosynostosis, a strip craniectomy often needs to be performed before 3 months of age due to increased intracranial pressure. Subsequent surgery includes a midface advancement procedure (LeFort III osteotomy) at 9–12 years and secondary orthognathic surgery to improve orthodontic-facial deformities (LeFort I, mandibular osteotomies) at age 14–18 years. Several excellent textbooks provide detailed surgical discussions (10, 11).
Most infants with positional plagiocephaly improve spontaneously without intervention; yet, early detection and prevention of positional problems can result in more optimal outcomes. Placing a baby supine remains a priority to protect against SIDS; however, parents are encouraged to place their baby on their “nonpreferred” side using a wedge device and to attach interesting visuals to attract the baby's attention. For infants with torticollis, following clearance of any structural spine anomaly, referral to an experienced physical therapist is also beneficial. Recently, there has been considerable interest in the use of individually molded helmets in babies to remodel their deformed skulls. These expensive, cumbersome devices must be worn at all times for months with proper follow-up and adjustments. Such therapy should be initiated before 8 months of age to achieve benefit.
Conclusions
Early diagnosis is critical in optimizing treatment, or non-treatment, of the infant with a misshapen head. Timely identification of potentially silent associated malformations (such as congenital heart disease, sensorineural hearing loss, etc.) is also important. In addition, children with craniosynostosis are often at risk for learning disabilities and should be provided with early intervention services. It is essential that children with craniofacial abnormalities have a comprehensive evaluation. Referral to a multidisciplinary craniofacial program is advised. Craniofacial teams are affiliated with the American Cleft Palate/ Craniofacial Association (www.cleftline.org) and include specialists in pediatrics, neurosurgery, plastic and reconstructive surgery, otolaryngology, oral surgery, orthodontics, speech pathology, genetics, nursing, psychology, and social work. Financial and psychosocial considerations for these patients are considerable and complex (12). For all of these children and their families, the earlier the recognition and initiation of comprehensive treatment, the better the ultimate outcome.
Acknowledgments
Special thanks to Duane Superneau, MD and Barbara Siede for their assistance with this article.

Dr. Bronfin is the Head of Ochsner's General Pediatric Section and the Medical Director of the Ochsner Craniofacial Team
References
- Gerszten P. C., Gerszten E. Intentional cranial deformation: a disappearing form of self-mutilation. Neurosurgery. 1995;37:374–381. doi: 10.1227/00006123-199509000-00002. [DOI] [PubMed] [Google Scholar]
- Graham J. M. Smith's Recognizable Patterns of Human Deformation, 2nd edition. Philadelphia: W.B. Saunders, 1988. [Google Scholar]
- Kane A. A., Mitchell L. E., Craven K. P. Observations on a recent increase in plagiocephaly without synostosis. Pediatrics. 1996;97:877–885. [PubMed] [Google Scholar]
- Virchow R. Uber den cretinismus, namentlicht in franken, und uber pathologische schadelformen. Ver Phys Med Gesellsch Wurzburg. 1851;2:230–271. [Google Scholar]
- Hunenko O., Karmacharya J., Ong G. Toward an understanding of nonsyndromic craniosynostosis: altered patterns of TGF-beta receptor and FGF receptor expression induced by intrauterine head constraint. Ann Plast Surg. 2001;46:546–553. doi: 10.1097/00000637-200105000-00015. [DOI] [PubMed] [Google Scholar]
- Jones K. L. Smith's Recognizable Patterns of Human Malformation, 5th edition. Philadelphia: W.B. Saunders, 1997. [Google Scholar]
- Mulliken J. B., Vander Woude D. L., Hansen M. Analysis of posterior plagiocephaly: deformational versus synostotic. Plast Reconstr Surg. 1999;103:371–380. doi: 10.1097/00006534-199902000-00003. [DOI] [PubMed] [Google Scholar]
- Huang M. H., Mouradian W. E., Cohen S. R. The differential diagnosis of abnormal head shapes: separating craniosynostosis from positional deformities and normal variants. Cleft Palate Craniofac J. 1998;35:204–211. doi: 10.1597/1545-1569_1998_035_0204_tddoah_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Tessier P. Osteotomies totales de la face. Syndrome de Crouzon, syndrome d'Apert: oxycephalies, scaphocephalies, turricephalies. Ann Chir Plast. 1967;12:273–286. [PubMed] [Google Scholar]
- Bartlett S., MacKay G. Craniosynostosis Syndromes. In: Aston S J, Beasley RW, Thorne CHM (editors). Grabb and Smith's Plastic Surgery, 5th edition. Philadelphia: Lippincott-Raven Publishers, 1997. [Google Scholar]
- McCarthy J. G. (editor). Plastic Surgery. Volume 4. Philadelphia: W.B. Saunders, 1990. [Google Scholar]
- Charkins H. Children with Facial Difference. A Parents' Guide. Bethesda: Woodbine House, 1996. [Google Scholar]