

Abstract
The dolastatin series of unique peptides, originally discovered as constituents of the sea hare Dolabella auricularia, is of increasing importance in providing biological leads, especially to new and useful anticancer drugs. Dolastatin 10 and three analogues, minor structural modifications designated auristatins, are currently in human cancer clinical trials. The present study was undertaken to explore delivery to the cancer sites by way of phosphate or quinoline modifications. The initial objectives, auristatin TP as sodium phosphate 3b (GI50 10−2–10−4 μg/mL), auristatin 2-AQ (4, GI50 10−2–10−3 μg/mL), and auristatin 6-AQ (5, GI50 10−4 μg/mL), exhibited superior cancer cell growth inhibitory properties.
The remarkable anticancer properties of dolastatin 10 (1), a unique pentapeptide that we isolated from the sea hare Dolabella auricularia,2a,b has led to intense interest in closely related derivatives (auristatins) that are suitable for clinical trial. Such structural modifications have provided a number of potential clinical candidates with enhanced efficacy and pharmacological characteristics.2c,d Replacement of the dolaphenine (Doe) unit with phenethylamides, to give auristatins PE2c,3,4 (2a), PHE2d,3,5 (2b), and E,3,6 and with pyridylethylamide (auristatin PYE, 2c)2d,3,7 has led to active analogues that are undergoing preclinical7 and clinical development.3 Dolastatin 10 and three of the auristatins are in human cancer clinical trials, ranging from phase I to phase III.
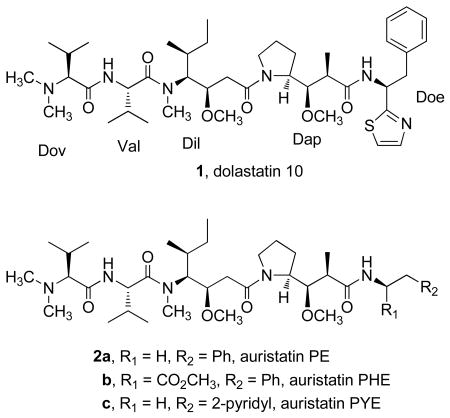
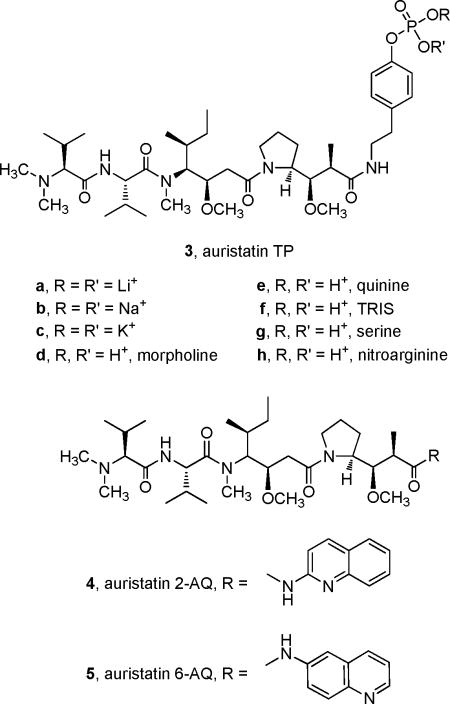
We now report the synthesis of auristatin TP (3), a tyramide phosphate modification of dolastatin 10 in the form of water-soluble salts. The synthesis of auristatins suitable for formulation of such salts is of considerable interest because the use of water-soluble phosphate derivatives has increased the bioavailability of a number of anticancer drugs, including combretastatins A-18 and A-4,8b,9 pancratistatin,10 taxol,11 and etoposide.12 The salts are dephosphorylated by serum phosphatases to yield the active drug, which is then transported intracellularly. We also report the syntheses of aminoquinoline (AQ) auristatin modifications 4 and 5. A number of 4- and 8-AQ derivatives have been used historically as antimalarial agents,13 and various biological activities have been reported for 2-AQ itself14a and for derivatives of the 3, 4-, 5-, 6-, and 8-AQ isomers.14b–f Use of the readily available isomers 2- and 6-AQ led to new auristatins 4 and 5, respectively. Each of the new auristatins displayed very strong cancer cell growth inhibition against a panel of murine and human cancer cell lines.
Results and Discussion
The synthesis of 3 was carried out as shown in Scheme 1. Reaction of the γ-amino acid Boc-Dap (6)15 with tyramine in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCl) and 1-hydroxybenzotriazole (HOBT) gave the protected amide 7a. Removal of the Boc group with bromotrimethylsilane (TMSBr) yielded the hydrobromide salt (7b), which was coupled with Dov-Val-Dil.TFA (8)16 in the presence of EDCI and HOBT to give the parent auristatin tyramide (9). The doubling of signals in the 1H and 13C NMR spectra of 9 indicated the presence of two isomers, a pattern similar to that of dolastatin 10 and due to conformational isomers arising from cis–trans isomerism at the Dil-Dap bond.16
Scheme 1.

Formation of phosphate diester 10a was achieved via in situ generation of dibenzyl chlorophosphate, from reaction of dibenzyl phosphite and carbon tetrachloride, and was followed by removal of the benzyl ester groups by hydrogenolysis to provide the free phosphoric acid 10b. The pure 10b was quite unstable but could be stored for short periods as a methanolic solution (< 0.01 M at 4 °C) and was generally used immediately as follows to provide compounds 3a–h. The passage of acid 10b through a Dowex cation exchange resin (Na+ form) provided the sodium salt (3b), and compounds 3a,c,d were similarly produced by ion exchange of the free acid or of either the sodium or potassium salts in the appropriate Dowex resin. The remaining salts (3e–h) were prepared directly from the free acid 10b by treatment with the appropriate base or amino acid. The solubilities of each salt and of precursor 9 were measured in distilled water at room temperature. The most soluble were the sodium (3b) and potassium (3c) salts (Table 1).
Table 1. Solubility of Compounds 3a–h and 10a.
| compound no. | mg/mL | compound no. | mg/mL |
|---|---|---|---|
| 3a | >65 | 3f | >51 |
| 3b | >236 | 3g | 7 |
| 3c | >120 | 3h | 7 |
| 3d | >72 | 9 | <1 |
| 3e | <5 |
In distilled water at 23 °C.
A similar convergent synthesis was planned for the preparation of the auristatin aminoquinoline modifications (4, 5), that is, formation of the Dap–aminoquinoline unit, followed by condensation with tripeptide 8. As shown in Scheme 2, Boc-Dap (6) and 2-aminoquinoline (2-AQ) were condensed to give Boc-Dap-2-AQ (11), diethylcyanophosphonate (DEPC) being used as coupling reagent, followed by deprotection to give the amine TFA salt (12). Coupling of 12 and 8 with use of DEPC gave the desired auristatin 2-AQ (4). The doubling of signals in the 1H NMR spectrum of 4 because of conformational changes was also noted.
Scheme 2.

Preparation of auristatins from other aminoquinoline isomers proved more difficult. First, the coupling of 5-aminoquinoline (5-AQ) with compound 6 was attempted, but use of DEPC failed to give the desired product. The coupling agent PyBroP was next used under standard conditions, but only starting material (6 and 5-AQ) was detected after 100 h. The activity of the aminoquinolines varies with the position of the amino group,13a,17 and they are in general poor nucleophiles. Therefore, we considered a route involving an activated intermediate preformed from the amino acid. Pozdnev18a used di-tert-butyl dicarbonate (Boc anhydride, Boc2O), in the presence of pyridine, to form activated esters of a number of protected amino acid derivatives, which were then condensed successfully with 6-aminoquinoline (6-AQ). Use of this method to couple 5-AQ and compound 6 failed, and was not further pursued, and the condensation of 6-aminoquinoline (6-AQ) with 6, via mixed anhydride 13 (Scheme 3), was next attempted.
Scheme 3.

A mixture of Boc2O and 6 in pyridine and dimethylformamide (DMF) was allowed to stir for 10 min, and 6-AQ was then added.18a After isolation of products, the reaction was found to have given the desired Boc-Dap-6-AQ (14), along with Boc-6-AQ (at least half of the 6-AQ was used in formation of this product). When 6 and Boc2O were allowed to stir in base for an hour so that formation of ester 13 could go to completion (with evolution of CO2) before addition of 6-AQ,18b formation of Boc-6-AQ was avoided, and in isolation of the desired product a citric acid wash was found useful for removal of unreacted aminoquinoline. However, the yield of product was still quite low, at 25%, and another method was sought.
Among the most reactive of the common activated intermediates are the amino acid fluorides,19 which have been shown to be very efficient reagents for peptide bond formation.20 With a sample of Boc-Dap-6-AQ (14) in hand for comparison, the condensation of the acid fluoride of 6 with 6-AQ was next attempted (Scheme 3). Reaction of cyanuric fluoride (15) with Boc-Dap (6) to give Boc-Dap-C(O)F (16) was carried out under mild conditions, and the crude product was used immediately in a condensation reaction with 6-AQ, in the presence of pyridine. The reaction did not go to completion (there was no detectable reduction in the amounts of unreacted compounds from 6 h to 20 h later), and the desired Boc-Dap-6-AQ (14) was isolated in 26% yield. Compound 14 was then treated with TFA to give the Dap-6-AQ.TFA salt (17), which was condensed with Dov-Val-Dil.TFA (8) to give auristatin-6-AQ (5).
In a repeat of the synthesis of acid fluoride 16, diisopropylethylamine (DIEA) was used as base, and purification of 16 was carried out on silica gel before condensation with 6-AQ, in the presence of DIEA, to give Boc-Dap-6-AQ (14). Reaction was slow, and at 44 h no change was apparent compared to the mixture at 32 h. The colorless oil that was isolated contained both product 14 and unreacted 16 (by tlc). According to the literature, the reaction of Fmoc amino acid fluorides with amines is often very slow and is not dependent on base20b,c (the presence of base can increase the reaction rate but lack of it can result in a cleaner reaction). Of the two methods to synthesize Boc-Dap-6-AQ (5), use of Boc2O to form active intermediate 13 led consistently to a yield of about 24%, whereas the yield from the Boc-Dap-C(O)F (16) method varied from 26% (using pyridine) to 6.6% (using DIEA and purifying the intermediate).
Compounds 3b, 3c, 4, 5 and 9 were evaluated against the murine P388 lymphocytic leukemia cell line and showed exceptional activity; auristatins 3b, 4, and 5 were also tested against a minipanel of human cancer cell lines in our laboratories, with similarly strong activity evident (Table 2), especially from compounds 3b and 5. These in vitro data are quite comparable to those of dolastatin 10 (1) and auristatin PE (2a), each of which had GI50 values of 10−5–10−6 μg/mL (10−2–10−3 nM) against a similar minipanel of human cell lines.2b,2c,3 Further biological testing of the new auristatins is under way.
Table 2. Murine and Human Cancer Cell Line Results [ED50 and GI50, μg/mL (nM)]a.
| compound number | cell lineb | ||||||
|---|---|---|---|---|---|---|---|
| P388 | NCI-H460 | KM20L2 | DU-145 | BXPC-3 | MCF-7 | SF-268 | |
| 3b | <0.001 (<1.2) |
0.00088 (1.05) |
0.00061 (0.72) |
0.00054 (0.64) |
0.046 (54.6) |
0.00068 (0.81) |
0.00125 (1.48) |
| 3c | 0.0076 (8.7) |
||||||
| 4 | 0.031 (42.8) |
0.016 (22.1) |
0.0077 (10.6) |
0.023 (31.8) |
0.029 (40.1) |
0.0046 (6.35) |
0.029 (40.1) |
| 5 | 0.0026 (3.59) |
0.00036 (0.50) |
0.00025 (0.35) |
0.00030 (0.41) |
0.00031 (0.43) |
0.00014 (0.19) |
0.00016 (0.22) |
| 9 | 0.0036 (5.0) |
||||||
Cytotoxicity concentrations as nanomolar values are given in parentheses.
Cancer cell lines in order: murine lymphocytic leukemia (P388); lung (NCI-H460); colon (KM20L2); prostate (DU-145); pancreas (BXPC-3); breast (MCF-7); CNS (SF-268).
Experimental Section
General Experimental Procedures
N-Boc-Dolaproine and Dov-Val-Dil.TFA were synthesized as described earlier.15,16 Reagents and anhydrous solvents were purchased from Acros Organics (Fisher Scientific), Sigma–Aldrich Chemical Company, and Lancaster Synthesis and were used as received. Diisopropylethylamine (DIEA) was redistilled over potassium hydroxide. Dibenzylphosphite was redistilled before use (bp 160 °C at 0.1 mm Hg). For thin-layer chromatography, Analtech silica gel GHLF Uniplates were used and visualized with short-wave UV irradiation and use of a permanganate dip followed by heating. Solvent extracts of aqueous solutions were dried over magnesium sulfate. For column chromatography, silica gel (230–400 mesh ASTM) from E. Merck (Darmstadt, Germany) was used. For ion-exchange chromatography, Dowex 50W×8-400 hydrogen form resin (Sigma–Aldrich) was washed with MeOH, hydrochloric acid (1 M), and deionized H2O before use. The cation forms of the resin were prepared by elution of an aqueous solution (1 M) of the corresponding base followed by deionized H2O.
Melting points are uncorrected and were determined with a Fischer–Johns melting point apparatus. Optical rotations were measured by use of a Perkin–Elmer 241 polarimeter, and the [α]D values are given in 10−1 deg cm2 g−1. The 1H, 13C and 31P NMR spectra were recorded using Varian Gemini 300 and Unity 400 and 500 instruments with deuterated solvents. The 31P spectra were referenced to 80% phosphoric acid or to the corresponding 1H spectra. High-resolution mass spectra were obtained with a Jeol JMS-LCmate mass spectrometer. Elemental analyses were determined by Galbraith Laboratories, Inc.
N-Boc-Dap-4-hydroxyphenethylamide (7a)
To a solution of Boc-Dap15 (6, 0.49 g, 1.71 mmol) in dry DMF (3 mL) that was stirring at 20 °C was added 1-hydroxybenzotriazole (HOBT, 0.37 g, 2.74 mmol). Diisopropylethylamine (DIEA, 0.95 mL, 5.48 mmol) was added, followed by N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI), and the reaction mixture was stirred for 10 min before the addition of tyramine (0.28 g, 2.05 mmol). The mixture was stirred at 20 °C for 16 h before termination of reaction by addition of saturated NaHCO3 solution (5 mL) and extraction with EtOAc (4 × 5 mL). The combined organic extract was washed with brine (20 mL) and dried. Removal of solvent yielded a yellow oil (0.89 g) that was fractionated by column chromatography (eluent: 2.5– 6.0% CH3OH in CH2Cl2) to provide 7a as a colorless oil (0.57 g, 82%) that crystallized from 1:1 CH2Cl2–hexane: mp 163–164 °C; [α]23D −30.4 (c 1.9, CHCl3); IR (neat) νmax 3305, 2975, 2935, 1650, 1515, 1168, 755 cm−1; 1H NMR (CD3OD, 300 MHz) δ 6.92 (d, J = 8.4 Hz, 2H), 6.57 (d, J = 8.4 Hz, 2H), 3.55 (br m, 1H), 3.49− 3.33 (m, 3H), 3.29 (s, 3H), 3.26−3.19 (m, 2H), 3.19−3.03 (m, 2H), 2.70− 2.53 (m, 2H), 2.11 (m, 1H), 1.81−1.70 (m, 2H), 1.61−1.50 (m, 2H), 1.40 (br s, 9H), 1.05 (d, J = 6.3 Hz, 3H); 13C NMR (CDCl3, 100.5 MHz) (two conformers observed) δ 174.7, 174.1, 155.5, 155.3, 154.8, 154.4, 129.5, 129.3, 115.5, 115.4, 83.7, 82.0, 80.1, 79.5, 60.6, 59.1, 58.6, 46.9, 46.5, 44.2, 43.8, 40.9, 40.7, 34.3, 28.5, 28.4, 25.7, 25.1, 24.4, 24.0, 14.3, 14.0; HRMS (FAB) m/z 407.2565 [M + H]+ (calcd for C22H35N2O5, 407.2546).
Dap-4-hydroxyphenethylamide Hydrobromide (7b)
Bromotrimethylsilane (0.46 mL, 3.5 mmol) was added to a stirred solution of 7a (0.57 g, 1.4 mmol) in dry CH2Cl2 at 20 °C, and stirring was continued for 18 h. Water (5 mL) was added, and the mixture was stirred vigorously for 30 min. The aqueous layer was removed, and the organic phase was extracted again with H2O (5 mL). Freeze-drying of the combined aqueous phase provided the hydrobromide salt 7b as a colorless solid (0.54 g, 99%), which was used without further purification. A sample was crystallized from CH2Cl2–hexane: mp 79− 81 °C; IR (neat) νmax 3275, 2980, 1640, 1515, 1235, 830 cm−1; 1H NMR (CD3OD, 300 MHz) δ 6.95 (d, J = 8.4 Hz, 2H), 6.60 (d, J = 8.4 Hz, 2H), 3.47 (m, 1H), 3.41 (s, 3H), 3.25−3.02 (m, 5H), 2.72–2.62 (m, 2H), 2.33 (m, 1H), 1.88−1.76 (m, 2H), 1.72–1.64 (m, 2H), 1.13 (d, J = 7.2 Hz, 3H); 13C NMR (CD3OD, 125.5 MHz) δ 175.6, 157.0, 131.0, 130.8, 116.2, 81.6, 62.9, 61.8, 46.5, 45.5, 41.3, 35.2, 24.1, 23.9, 15.5; HRMS (APCI) m/z 307.2026 [(M −HBr) + H]+ (calcd for C17H27N2O3, 307.2022).
Dov-Val-Dil-Dap-4-hydroxyphenethylamide (9)
To a solution of 816 (0.78 g, 1.43 mmol) that was stirring in dry DMF (2 mL) at 20 °C was added HOBT (0.31 g, 2.29 mmol). Next was added DIEA (0.96 mL, 5.50 mmol), followed by EDCI (0.44g, 2.29 mmol), and the reaction mixture was stirred for 15 min before the addition of a solution of 7b (0.50 g, 1.30 mmol) in DMF (4 mL). The mixture was stirred at 20 °C for 6 h, and then reaction was terminated by addition of saturated NaHCO3 solution (10 mL), followed by extraction of the mixture with EtOAc (4 × 10 mL). The combined organic extract was washed with brine (50 mL) and dried. Removal of solvent yielded a viscous brown oil (0.83 g) that was fractionated by column chromatography (eluent: 5−10% MeOH in CH2Cl2) to provide 9 as a viscous colorless oil (0.61 g, 65%): [α]23D −44.0 (c 2.2, CHCl3); IR (neat) νmax 3295, 2965, 1620, 1515, 1100, 755 cm−1; 1H NMR (CD3OD, 400 MHz) δ 7.25 (m, 1H), 7.05−7.01 (m, 4H), 6.68 (t, J = 8.5 Hz, 4H), 4.74 (d, J = 8.4 Hz, 1H), 4.71 (d, J = 8.4 Hz, 1H), 4.63 (d, J = 8.8 Hz, 1H), 4.15 (m, 1H), 4.07 (m, 1H), 3.90−3.83 (m, 2H), 3.78 (m, 1H), 3.68 (m, 1H), 3.57 (m, 6H), 3.40−3.32 (m, 2H), 3.38 (s, 3H), 3.36 (s, 3H), 3.29 (s, 6H), 3.26 (s, 3H), 3.13 (s, 3H), 2.81−2.68 (m, 4H), 2.65−2.62 (m, 2H), 2.48 (d, J = 6.6 Hz, 2H), 2.31 (s, 6H), 2.29 (s, 6H), 2.27−2.19 (m, 2H), 2.08−1.86 (m, 10H), 1.78−1.63 (m, 4H), 1.44−1.35 (m, 2H), 1.16 (t, J = 7.1 Hz, 6H), 1.05−0.95 (m, 28H), 0.90−0.84 (m, 12H); 13C NMR (CD3OD, 100.5 MHz) δ 176.5, 176.4, 175.3, 173.3, 173.2, 171.9, 157.0, 156.9, 136.5, 131.2, 130.9, 130.8, 130.7, 116.3, 116.2, 87.2, 83.6, 79.8, 76.0, 75.8, 62.1, 61.4, 61.0, 60.8, 58.6, 58.3, 57.8, 56.2, 56.0, 45.9, 45.7, 42.5, 42.4, 41.8, 41.4, 38.2, 35.3, 33.8, 33.1, 31.8, 31.7, 28.8, 27.0, 26.8, 25.8, 24.5, 20.2, 20.2, 19.9, 19.5, 19.3, 16.3, 16.0, 15.8, 15.1, 10.9, 10.8; HRMS (FAB) m/z 718.5084 [M + H]+ (calcd for C39H68N5O7, 718.5119).
Dov-Val-Dil-Dap-4-(dibenzylphosphoryloxy)phenethylamide (10a)
To a solution of 9 (0.51 g, 0.70 mmol) in dry CH3CN (4 mL) at −15 °C (ice/salt) was added carbon tetrachloride (0.34 mL, 1.02 mmol), followed by DIEA (0.26 mL, 1.50 mmol) and 4-dimethylaminopyridine (9 mg, 0.07 mmol). Dibenzylphosphite (0.23 mL, 1.02 mmol) was next added over a 20-min period to the mixture, the temperature being maintained between −15 and −18 °C. After addition, the mixture was cooled to −20 °C and then allowed to warm to 5 °C over 90 min, and reaction was terminated by addition of saturated NaHCO3 solution (10 mL). The mixture was extracted with EtOAc (3 × 10 mL), and the combined organic extract was washed with brine (50 mL) and dried. Removal of solvent yielded a viscous pale yellow oil (0.60 g) that was fractionated by column chromatography (eluent: 5–10% MeOH in CH2Cl2) to provide 10a as a colorless oil (0.34 g, 49%): IR (neat) νmax 3305, 2965, 1620, 1455, 1215, 1015, 955 cm−1; 1H NMR (CD3OD, 500 MHz) δ 7.36−7.31 (m, 20H), 7.21 (d, J = 6.6 Hz, 2H), 7.20 (d, J = 6.6 Hz, 2H), 7.07 (d, J = 6.6 Hz, 2H), 7.03 (d, J = 6.6 Hz, 2H), 5.13−5.10 (m, 8H), 4.81−4.71 (m, 2H), 4.71 (d, J = 8.0 Hz, 1H), 4.62 (d, J = 8.0 Hz, 1H), 4.14 (m, 1H), 4.06 (m, 1H), 3.91−3.86 (m, 2H), 3.80 (m, 1H), 3.69 (m, 1H), 3.56−3.47 (m, 4H), 3.43−3.32 (m, 2H), 3.38 (s, 3H), 3.36 (s, 3H), 3.28 (s, 6H), 3.26 (s, 3H), 3.11 (s, 3H), 2.86−2.77 (m, 4H), 2.65−2.61 (m, 3H), 2.51 (m, 1H), 2.46 (d, J = 6.5 Hz, 2H), 2.30 (s, 6H), 2.29 (s, 6H), 2.28−2.18 (m, 2H), 2.08−1.86 (m, 9H), 1.76−1.64 (m, 5H), 1.43−1.36 (m, 2H), 1.16 (d, J = 7.5 Hz, 3H), 1.15 (d, J = 7.5 Hz, 3H), 1.02−0.92 (m, 28H), 0.87−0.81 (m, 12H); 13C NMR (CD3OD, 125.5 MHz) δ 175.2, 175.1, 171.9, 170.6, 149.0 (d, JC–P = 7.0 Hz), 148.9 (d, JC–P = 7.0 Hz), 136.6, 136.4, 129.9, 129.8, 128.5, 128.3, 127.9 (d, JC–P = 2.6 Hz), 119.8 (d, JC–P = 4.4 Hz), 119.6 (d, JC–P = 4.4 Hz), 85.7, 82.2, 78.5, 78.4, 78.4, 74.5, 74.4, 70.1, 70.1, 60.7, 60.0, 59.6, 57.3, 56.9, 54.8, 54.6, 46.7, 44.5, 44.3, 41.1, 41.1, 40.0, 39.7, 36.8, 35.6, 34.1, 32.3, 32.2, 31.7, 30.4, 30.3, 27.4, 25.7, 25.5, 24.4 (d, JC–P = 3.6 Hz), 23.1, 18.8 (d, JC–P = 4.4 Hz), 18.5, 18.2, 18.0, 14.9, 14.6, 14.5, 13.7, 9.5; 31P NMR (CD3OD, 202.5 MHz) δ −6.51, −6.54; HRMS (FAB) m/z 978.5811 [M + H]+ (calcd for C53H81N5O10P 978.5721).
Dov-Val-Dil-Dap-4-(dihydrophosphoryloxy)phenethylamide (10b)
To a solution of dibenzyl phosphate 10a (38 mg, 0.04 mmol) in MeOH (5 mL) was added palladium on activated carbon (10 wt% Pd, 10 mg), and hydrogen gas (balloon) was bubbled through the suspension for 1 h. The mixture was filtered through a plug of Celite, and the filter was washed with MeOH (2 × 5 mL). Removal of solvent from the filtrate yielded the free phosphoric acid 10b as a glassy solid (32 mg, quantitative): mp 168− 170 °C; IR (neat) νmax 3400, 2970, 1635, 1460, 1095, 910 cm−1; 1H NMR (CD3OD, 500 MHz) δ 7.20− 7.12 (m, 8H), 4.77−4.71 (m, 2H), 4.67 (d, J = 8.5 Hz, 1H), 4.62 (d, J = 9.0 Hz, 1H), 4.11 (m, 1H), 4.05 (m, 1H), 3.93−3.89 (m, 2H), 3.73−3.68 (m, 2H), 3.61−3.48 (m, 4H), 3.44−3.33 (m, 2H), 3.41 (s, 3H), 3.37 (s, 3H), 3.29 (s, 6H), 3.28 (s, 3H), 3.15 (s, 3H), 2.90 (s, 6H), 2.79−2.73 (m, 4H), 2.66−2.49 (m, 4H), 2.42−2.26 (m, 4H), 2.08−1.64 (m, 14H), 1.46−1.38 (m, 2H), 1.23 (d, J = 7.0 Hz, 3H), 1.17 (d, J = 7.0 Hz, 3H), 1.07−1.00 (m, 20H), 0.97−0.84 (m, 20H); 31P NMR (CD3OD, 202.5 MHz) δ -4.11.
Sodium Auristatin TP (3b)
Ion-exchange chromatography of free acid 10b (32 mg) with aqueous NaOH led to 3b as a colorless solid (24 mg, 71 %): mp 170–171 °C; IR (neat) νmax 3305, 2965, 1625, 1510, 1105, 990 cm−1; 1H NMR (CD3OD, 400 MHz) δ 7.17−7.15 (m, 4H), 7.09 (d, J = 8.0 Hz, 4H), 4.78−4.72 (m, 2H), 4.72 (d, J = 8.0 Hz, 1H), 4.64 (d, J = 8.4 Hz, 1H), 4.12 (m, 1H), 4.07 (m, 1H), 3.98− 3.94 (m, 2H), 3.91 (dd, J = 9.1, 2.3 Hz, 2H), 3.70 (m, 1H), 3.59 (m, 1H), 3.51−3.41 (m, 4H), 3.39 (s, 3H), 3.37 (s, 3H), 3.36−3.32 (m, 2H), 3.30 (s, 6H), 3.27 (s, 3H), 3.14 (s, 3H), 2.81−2.70 (m, 4H), 2.65 (d, J = 7.2 Hz, 1H), 2.63 (d, J = 7.2 Hz, 1H), 2.49 (d, J = 6.4 Hz, 2H), 2.31 (s, 6H), 2.29 (s, 6H), 2.29− 2.22 (m, 2H), 2.08−1.87 (m, 9H), 1.81−1.68 (m, 5H), 1.43−1.36 (m, 2H), 1.17 (t, J = 5.3 Hz, 6H), 1.03− 0.95 (m, 28H), 0.85 (q, J = 7.2 Hz, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −3.42.
Lithium Auristatin TP (3a)
Ion-exchange chromatography of sodium salt 3b (12 mg, 0.014 mmol) with aqueous LiOH led to 3a as a colorless solid (11 mg, 96 %): mp 263 °C (dec); IR (neat) νmax 3315, 2965, 1630, 1105, 1005, 920 cm−1; 1H NMR (CD3OD, 400 MHz) δ 7.20 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 7.04 (d, J = 8.0 Hz, 4H), 4.74−4.70 (m, 2H), 4.72 (d, J = 8.0 Hz, 1H), 4.64 (d, J = 8.8 Hz, 1H), 4.14−4.00 (m, 4H), 3.95 (m, 1H), 3.91 (dd, J = 9.0, 2.2 Hz, 1H), 3.71 (m, 1H), 3.58 (m, 1H), 3.52− 3.34 (m, 6H), 3.39 (s, 3H), 3.38 (s, 3H), 3.30 (s, 6H), 3.27 (s, 6H), 3.13 (s, 3H), 2.75−2.68 (m, 4H), 2.64 (d, J = 4.8 Hz, 1H), 2.63 (d, J = 4.8 Hz, 1H), 2.51 (d, J = 6.4 Hz, 2H), 2.30 (s, 6H), 2.29 (s, 6H), 2.27− 2.23 (m, 2H), 2.07–1.94 (m, 9H), 1.82−1.71 (m, 5H), 1.45−1.37 (m, 2H), 1.18 (t, J = 6.2 Hz, 6H), 1.03− 0.95 (m, 28H), 0.85 (q, J = 6.9 Hz, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −0.58.
Potassium Auristatin TP (3c)
Ion-exchange chromatography of acid 10b with aqueous KOH led to 3c as a colorless solid (4 mg, 64 %): mp 198 °C; IR (neat) νmax 3230, 2965, 1620, 1100, 980, 885 cm−1; 1H NMR (CD3OD, 400 MHz) δ 7.21 (d, J = 8.2 Hz, 2H), 7.19 (d, J = 8.2 Hz, 2H), 7.04 (d, J = 8.2 Hz, 4H), 4.75−4.72 (m, 2H), 4.72 (d, J = 8.0 Hz, 1H), 4.64 (d, J = 6.6 Hz, 1H), 4.14−4.01 (m, 4H), 3.95 (m, 1H), 3.91 (dd, J = 8.8, 2.4 Hz, 1H), 3.71 (m, 1H), 3.58 (m, 1H), 3.52−3.35 (m, 6H), 3.39 (s, 3H), 3.38 (s, 3H), 3.30 (s, 6H), 3.26 (s, 3H), 3.13 (s, 3H), 2.75−2.68 (m, 4H), 2.64 (d, J = 8.8 Hz, 1H), 2.63 (d, J = 9.2 Hz, 1H), 2.49 (d, J = 5.6 Hz, 2H), 2.30 (s, 6H), 2.29 (s, 6H), 2.27−2.23 (m, 2H), 2.07−1.94 (m, 9H), 1.83−1.72 (m, 5H), 1.45−1.37 (m, 2H), 1.18 (t, J = 6.2 Hz, 6H), 1.03−0.95 (m, 28H), 0.84 (q, J = 6.9 Hz, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −0.42.
Morpholine Auristatin TP (3d)
Ion-exchange chromatography of potassium salt 3c with aqueous morpholine led to 3d as a colorless solid: mp 148−150 °C; IR (neat) νmax 3295, 2965, 1620, 1455, 1105, 880 cm−1; 1H NMR (CD3OD, 500 MHz) δ 7.18−7.11 (m, 8H), 4.82−4.74 (m, 2H), 4.71 (d, J = 8.5 Hz, 1H), 4.65 (d, J = 8.5 Hz, 1H), 4.13 (m, 1H), 4.07 (m, 1H), 3.97 (m, 1H), 3.91 (dd, J = 9.3, 2.3 Hz, 1H), 3.80 (br s, 16H), 3.71 (m, 1H), 3.60 (m, 1H), 3.52 (d, J = 8.5 Hz, 1H), 3.49−3.43 (m, 3H), 3.40 (s, 3H), 3.39 (s, 3H), 3.37−3.34 (m, 2H), 3.31 (s, 6H), 3.28 (s, 3H), 3.15 (s, 3H), 3.06 (br s, 16H), 2.82 (m, 1H), 2.77 (q, J = 7.2 Hz, 4H), 2.69 (d, J = 8.5 Hz, 1H), 2.67 (m, 1H), 2.54 (d, J = 9.0 Hz, 1H), 2.50 (d, J = 6.0 Hz, 2H), 2.42 (s, 6H), 2.34 (s, 6H), 2.32−2.14 (m, 2H), 2.13−1.88 (m, 9H), 1.81−1.71 (m, 5H), 1.46− 1.38 (m, 2H), 1.20 (d, J = 6.5 Hz, 3H), 1.18 (d, J = 7.5 Hz, 3H), 1.04−0.95 (m, 28H), 0.91−0.87 (m, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −3.43.
General Procedure for the Synthesis of 3e–h
The amine or amino acid (25.0 μmol) was added to a stirred solution of acid 10b (10 mg, 12.5 μmol) in either MeOH (300 μL) or deionized H2O (for 3h), and the mixture was stirred for 15 h. Removal of solvent yielded the desired salt.
Quinine Auristatin TP (3e)
Colorless solid: mp 118−120 °C; 1H NMR (CD3OD, 500 MHz) δ 8.67 (d, J = 4.8 Hz, 4H), 7.93 (d, J = 9.3 Hz, 4H), 7.72 (d, J = 4.8 Hz, 4H), 7.43 (d, J = 2.3 Hz, 4H), 7.40 (dd, J = 9.3, 2.3 Hz, 4H), 7.17 (t, J = 7.3 Hz, 4H), 7.03 (d, J = 7.3 Hz, 4H), 5.93 (s, 4H), 5.73 (m, 4H), 5.05 (d, J = 17.5 Hz, 4H), 4.95 (d, J = 11 Hz, 4H), 4.82−4.71 (m, 2H), 4.72 (d, J = 8.0 Hz, 1H), 4.65 (d, J = 8.5 Hz, 1H), 4.12 (m, 1H), 4.07 (m, 1H), 3.98 (s, 12H), 3.91 (d, J = 2.0 Hz, 1H), 3.89 (d, J = 2.0 Hz, 1H), 3.71−3.65 (m, 2H), 3.56 (m, 1H), 3.50 (d, J = 10.0 Hz, 1H), 3.45−3.23 (m, 12H), 3.39 (s, 3H), 3.38 (s, 3H), 3.30 (s, 3H), 3.29 (s, 3H), 3.27 (s, 3H), 3.14 (s, 3H), 3.00−2.92 (m, 8H), 2.71−2.65 (m, 6H), 2.56−2.48 (m, 8H), 2.34 (s, 6H), 2.31 (s, 6H), 2.30−2.21 (m, 2H), 2.08−1.90 (m, 24H), 1.78−1.71 (m, 10H), 1.45−1.40 (m, 6H), 1.17 (t, J = 7.0 Hz, 6H), 1.04−0.95 (m, 28H), 0.98−0.80 (m, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −1.82.
TRIS Auristatin TP (3f)
Colorless solid: mp 122−123 °C; 1H NMR (D2O, 500 MHz) δ 7.21−7.12 (m, 1H), 4.73−4.64 (m, 4H), 4.17 (m, 1H), 4.11 (m, 1H), 3.92−3.86 (m, 2H), 3.74−3.62 (m, 2H), 3.67 (s, 24H), 3.59−3.38 (m, 6H), 3.44 (s, 3H), 3.42 (s, 3H), 3.33 (s, 3H), 3.33 (s, 3H), 3.26 (s, 3H), 3.18 (s, 3H), 3.12 (d, J = 8.5 Hz, 3H), 3.02 (d, J = 9.5 Hz, 1H), 2.87−2.75 (m, 2H), 2.67−2.62 (m, 2H), 2.58−2.54 (m, 2H), 2.52 (s, 6H), 2.45 (s, 6H), 2.36−2.27 (m, 2H), 2.22−2.09 (m, 2H), 2.08−2.01 (m, 2H), 1.99−1.72 (m, 9H), 1.68−1.61 (m, 1H), 1.39−1.30 (m, 2H), 1.20 (d, J = 6.5 Hz, 3H), 1.16 (d, J = 7.0 Hz, 3H), 1.05− 0.97 (m, 28H), 0.88−0.84 (m, 12H); 31P NMR (CD3OD, 162.0 MHz) δ −0.01.
Serine Auristatin TP (3g)
Colorless solid: mp 158 °C (dec); 1H NMR (D2O, 500 MHz) 7.26 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H), 4.75 (d, J = 9.5 Hz, 1H), 4.73−4.68 (m, 2H), 4.66 (d, J = 9.5 Hz, 1H), 4.18 (m, 1H), 4.11 (m, 1H), 4.01−3.93 (m, 10H), 3.86−3.83 (m, 6H), 3.79 (t, J = 5.8 Hz, 2H), 3.74−3.68 (m, 2H), 3.62−3.51 (m, 4H), 3.47−3.36 (m, 4H), 3.44 (s, 3H), 3.39 (s, 3H), 3.33 (s, 3H), 3.32 (s, 3H), 3.24 (s, 3H), 3.18 (s, 3H), 2.97 (s, 6H), 2.95 (s, 6H), 2.92−2.79 (m, 4H), 2.67−2.44 (m, 6H), 2.33 (m, 1H), 2.24 (m, 1H), 2.12−2.02 (m, 2H), 1.97−1.66 (m, 9H), 1.51 (m, 1H), 1.38−1.32 (m, 2H), 1.20 (d, J = 7.0 Hz, 3H), 1.14 (d, J = 7.0 Hz, 3H), 1.05− 0.95 (m, 30H), 0.92−0.82 (m, 10H); 31P NMR (CD3OD, 162.0 MHz) δ −4.07.
Nitroarginine Auristatin TP (3h)
Colorless solid: mp 157−158 °C (dec); IR (neat) νmax 3295, 2965, 1625, 1360, 1270, 1095 cm-1; 1H NMR (D2O, 500 MHz) δ 7.21 (d, J = 7.8 Hz, 2H), 7.18 (d, J = 7.8 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 7.05 (d, J = 7.8 Hz, 2H), 4.71 (d, J = 9.0 Hz, 1H), 4.70−4.64 (m, 2H), 4.62 (d, J = 8.5 Hz, 1H), 4.14 (m, 1H), 4.06 (m, 1H), 3.81−3.72 (m, 4H), 3.74 (t, J = 6.5 Hz, 4H), 3.69−3.63 (m, 2H), 3.59−3.45 (m, 4H), 3.43−3.33 (m, 2H), 3.40 (s, 3H), 3.35 (s, 3H), 3.30 (t, J = 6.5 Hz, 8H), 3.29 (s, 3H), 3.28 (s, 3H), 3.20 (s, 3H), 3.14 (s, 3H), 2.93 (s, 6H), 2.90 (s, 6H), 2.88−2.75 (m, 4H), 2.63−2.48 (m, 5H), 2.44−2.39 (m, 1H), 2.29 (m, 1H), 2.20 (m, 1H), 2.08−1.97 (m, 2H), 1.95−1.61 (m, 25H), 1.47 (m, 1H), 1.34−1.27 (m, 2H), 1.16 (d, J = 6.5 Hz, 3H), 1.10 (d, J = 7.0 Hz, 3H), 1.01−0.96 (m, 18H), 0.93−0.90 (m, 12H), 0.86−0.81 (m, 10H); 31P NMR (CD3OD, 162.0 MHz) δ −3.56.
N-Boc-Dap-2-aminoquinoline (11)
To a solution of Boc-Dap15 (6, 0.172 g; 0.6 mmol) in CH2Cl2 (3 mL) was added 2-aminoquinoline (82.8 mg; 0.57 mmol), and the mixture was stirred and cooled to 0 °C under argon. Triethylamine (TEA, 0.3 mL; 2.1 mmol) and diethylcyanophosphonate (DEPC; 0.2 mL; 1.2 mmol) were added, and the resultant yellow solution was allowed to warm to room temperature (rt) and was stirred under argon for 6 h. Removal of solvent yielded a dark orange-brown residue that was fractioned under pressure on silica gel [eluent: hexane–acetone (7:2 to 2:3)] to give the product as a colorless solid (90.8 mg, 0.22 mmol, 36.6%, based on recovery of starting material): 1H NMR (CDCl3, 300 MHz) δ 8.43 (1H, dd, J = 8.7. 1.5 Hz), 8.16 (1H, d, J = 8.7 Hz), 7.83 (1H, d, J = 8.7 Hz), 7.72 (1H, d, J = 8.4 Hz), 7.66 (1H, t, J = 7.5 Hz), 7.44 (1H, t, J = 7.5 Hz), 4.05–3.92 (2H, m, NCH, OCH), 3.53 (3H, s, OCH3), 3.44 (2H, br d, J = 13 Hz, NCH2), 2.60−2.80 (1H, m, CHCH3), 1.74−1.98 (4H, m, 2 × CH2), 1.52 (9H, s, C(CH3)3), 1.45 (3H, d, J = 9.3 Hz, CHCH3) ); MS (APCI+) m/z 414.2373 [M + H]+ (calcd for C23H32N3O4, 414.2393).
Dap-2-aminoquinoline Trifluoroacetate (12)
To a solution of N-Boc-Dap-2-AQ (11, 68.0 mg, 0.16 mmol) in CH2Cl2 (4 mL) that was stirring at 0 °C under argon was added trifluoroacetic acid (TFA, 2 mL), and stirring was continued for 2 h with warming to rt. The solvent was removed under vacuum, toluene being used to form an azeotrope with the remaining TFA. The residue, a yellow oil, was allowed to stand under diethyl ether for 1 h. Removal of the ether left a yellowish oily solid to which hexane was added and removed under vacuum until a constant weight was reached (99.4 mg; quantitative), and this material was used immediately in the next reaction.
Dov-Val-Dil-Dap-2-aminoquinoline (Auristatin 2-AQ, 4)
The Dap-2-AQ salt 12 and Dov-Val-Dil.TFA16 (8, 87.0 mg; 0.16 mmol) were dissolved in CH2Cl2 (5 mL), and the solution was stirred under argon and cooled to 0 °C. Next, TEA (0.12 mL; 0.86 mmol) and DEPC (0.035 mL; 0.21 mmol) were added, and the mixture was stirred under argon for 7 h with warming to rt. Removal of solvent yielded a yellowish oil (310 mg) that was separated on silica gel under pressure [eluent: hexane–acetone (5:2 to 3:2)] to give the product as a colorless glass (powder when scratched) (64 mg; 0.09 mmol): 1H NMR (CDCl3, 300 MHz) δ 8.43 (1H, dd, J = 8.7. 1.5 Hz), 8.14 (1H, d, J = 8.7 Hz), 7.80−7.41 (4H, m), 6.90 (1H, t, J = 9.3 Hz), 6.73 (1H, d, J = 9.0 Hz), 4.86 (1H, m), 4.75 (1H, m), 4.26 (1H, m), 4.14 (1H, m), 4.04 (1H, m), 3.51 and 3.44 (3H, s), 3.35 and 3.32 (3H, s), 3.38−3.19 (2H, m), 3.02 (3H, s), 2.42 (3H, m), 2.23 (6H, s), 2.23 (1H, m), 2.08−1.98 (5H, m), 1.95−1.74 (1H, m), 1.43−1.33 (2H, m), 0.80−1.06 (22H, m); MS (APCI+) m/z 725.4997 [M + H]+ (calcd for C40H65N6O6, 725.4966).
N-Boc-Dap-6-aminoquinoline (14)
Method A
To a stirring solution of Boc-Dap15 (6, 87.2 mg; 0.3 mmol) in DMF (2 mL) and pyridine (0.1 mL) was added Boc2O (0.183 g; 0.84 mmol). After 10 min, 6-aminoquinoline (6-AQ; 50.4 mg; 0.35 mmol) was added to the solution, and stirring was continued for 64 h, at which time starting material was still present. Solvent was removed from the mixture, and the residue was fractionated by column chromatography in hexane–acetone (5:1 to 2:1 gradient). The first fractions to elute contained Boc-6-AQ (35 mg): 1H NMR (CDCl3, 300 MHz) δ 8.79 (1H, dd, J = 4.5, 1.5 Hz), 8.01−8.12 (3H, m), 7.48 (1H, dd, J = 9, 2.7 Hz), 7.36 (1H, dd, J = 7.1, 4.2 Hz), 7.03 (1H, br s), 1.55 (9H, s).
Following the elution of the remaining Boc-Dap (6), compound 14 (29.7 mg, 0.07 mmol, 23% yield, or 28% based on 14.9-mg recovery of 6-AQ) was collected: 1H NMR (CDCl3, 300 MHz) δ 8.82 (2H, m), 8.45 (1H, br s), 8.11 (1H, d, J = 8.1 Hz), 8.04 (1H, d, J = 9.3 Hz), 7.72 (1H, br), 7.37 (1H, dd, J = 8.4, 4.2 Hz), 4.15−3.90 (2H, m, NCH, OCH), 3.55 (3H, s, OCH3), 3.43 (m, 1H), 3.27 (m, 1H), 2.72 (1H, m), 2.06−1.76 (4H, m), 1.50 (9H, s), 1.39 (3H, m); MS (APCI+) m/z 414.2408 [M + H]+ (calcd for C23H32N3O4, 414.2393).
Method B
Intermediate acid fluoride 16 was first prepared by successive addition of pyridine (0.05 mL) and cyanuric fluoride (15, 0.15 mL, 1.75 mmol) to a solution of Boc-Dap (6, 76.3 mg, 0.27 mmol) that was stirring under argon at 0 °C, with continued stirring for 20 h and warming to rt. Next, CH2Cl2 (10 mL) and ice were added, followed by cold H2O. The organic phase was removed and the aqueous layer was further extracted with CH2Cl2. The combined organic extract was washed with cold H2O and dried to give a dark orange oily solid (65.6 mg) that by tlc comprised product 16 and a trace of Boc-Dap (6). Without further purification, the crude product was dissolved in CH2Cl2 and was treated with pyridine (0.1 mL) followed by 6-AQ (34.8 mg, 0.24 mmol). The mixture was stirred for 21 h and was then extracted with CH2Cl2 (10 mL). The solution was washed with 10% citric acid solution, followed by H2O. Drying over Na2SO4 and removal of solvent yielded a pale brown oil (52.8 mg) that was separated by column chromatography [eluent: toluene–acetone (2:1)] to give product 14 (30.3 mg, 0.07 mmol, 26%).
Dap-6-aminoquinoline Trifluoroacetate (17)
To a solution of N-Boc-Dap-6-AQ (14, 49.3 mg, 0.12 mmol) in CH2Cl2 (2 mL) that was stirring at 0 °C under argon was added trifluoroacetic acid (TFA, 2 mL). Stirring was continued for 3 h with warming to rt. The solvent was removed under vacuum, toluene being used to form an azeotrope with the remaining TFA, to give a green-tinged oily solid (17; quantitative) that was used immediately in the next reaction.
Dov-Val-Dil-Dap-6-aminoquinoline (Auristatin 6-AQ, 5)
Dap-6-AQ salt 17 (0.12 mmol) and Dov-Val-Dil.TFA16 (8, 70.0 mg; 0.13 mmol) were dissolved in CH2Cl2 (2 mL), and the solution was stirred under argon and cooled to 0 °C. Next were added TEA (0.11 mL; 0.79 mmol) and DEPC (0.03 mL; 0.18 mmol), and the mixture was stirred under argon for 18 h with warming to rt. Removal of solvent and separation on silica gel under pressure [eluent: hexane–acetone (5:2 to 2:3)] gave the crude product 5 (48.6 mg), of which a 19.1-mg sample was further purified by column chromatography in CH2Cl2–MeOH (19:1) to give auristatin 6-AQ (5) as a pale yellow glassy oil (powder when scratched): 1H NMR (CDCl3, 300 MHz) δ 9.04 (1H, br s), 8.81 (1H, br d, J = 3 Hz), 8.47 (1H, s), 8.11 (1H, d, J = 8.4 Hz), 8.02 (1H, d, J = 9.3 Hz), 7.76 (1H, br d, J = 8.7 Hz), 7.36 (1H, dd, J = 8.4, 3.8 Hz), 6.96 (1H, br), 4.79 (2H, m), 4.30−4.07 (3H, m), 3.51 (3H, s), 3.50−3.26 (2H, m), 3.35 (3H, s), 3.05 (s, 3H), 2.71 (1H, m), 2.54−2.42 (2H, m), 2.32−2.22 (1H, m), 2.28 (6H, s), 2.12–2.05 (2H, m), 1.82 (2H, m), 1.42– 1.26 (5H, m), 1.08–0.80 (21H, m); MS (APCI+) m/z 725.4907 [M + H]+ (calcd for C40H65N6O6, 725.4966).
Supplementary Material
Acknowledgments
We are pleased to acknowledge the very necessary financial support from grants R01 CA 90441-01-05, 2R56 CA 090441-06A1, and 5-R01 CA 90441-07-08 from the Division of Cancer Treatment and Diagnosis, NCI, DHHS; the Arizona Disease Control Research Commission; the Fannie E. Rippel Foundation; and the Robert B. Dalton Endowment Fund. Other very helpful assistance was provided by Drs. J.–C. Chapuis and J. C. Knight and by M. Dodson and L. Williams.
Footnotes
Dedicated to Dr. Koji Nakanishi of Columbia University for his pioneering work on bioactive natural products.
Supporting Information Available: 1H NMR spectra of compounds 3b, 4, and 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.For Antineoplastic Agents Part 591 refer to; Bai R, Nguyen TL, Burnett JC, Atasoylu O, Munro MHG, Pettit GR, Smith AB, III, Gussio R, Hamel E. J Chem Inf Model. doi: 10.1021/ci200077t. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. J Am Chem Soc. 1987;109:6883–6885. [Google Scholar]; (b) Pettit GR. In: Progress in the Chemistry of Organic Natural Products. Herz W, Kirby GW, Moore RE, Steglich W, Tamm C, editors. Vol. 70. Springer; Vienna: 1997. pp. 1–79. [DOI] [PubMed] [Google Scholar]; (c) Pettit GR, Srirangam JK, Barkoczy J, Williams MD, Durkin KPM, Boyd MR, Bai R, Hamel E, Schmidt JM, Chapuis JC. Anti-Cancer Drug Des. 1995;10:529–544. [PubMed] [Google Scholar]; (d) Pettit GR, Srirangam JK, Barkoczy J, Williams MD, Boyd MR, Hamel E, Pettit RK, Hogan F, Bai R, Chapuis JC, McAllister S, Schmidt JM. Anti-Cancer Drug Des. 1998;13:243–277. [PubMed] [Google Scholar]
- 3.Pettit GR. Dolastatin anticancer drugs. In: Chabner B, Cortés-Funes H, editors. International Oncology Updates: Marine anticancer compounds in the era of targeted therapies. Permanyer Publications; Barcelona: 2009. [Google Scholar]
- 4.(a) Kobayashi M, Natsume T, Tamaoki S, Watanabe J, Asano H, Mikami T, Miyasaka K, Miyazaki K, Gondo M, Sakakibara K, Tsukagoshi S. Jpn J Cancer Res. 1997;88:316–327. doi: 10.1111/j.1349-7006.1997.tb00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fennell B, Carolan S, Pettit GR, Bell A. J Antimicrob Chemother. 2003;51:833–841. doi: 10.1093/jac/dkg151. [DOI] [PubMed] [Google Scholar]; (c) Watanabe J, Minami M, Kobayashi M. Anticancer Res. 2006;26:1973–1981. [PubMed] [Google Scholar]; (d) Horti J, Juhasz E, Monostori Z, Maeda K, Eckhardt S, Bodrogi I. Cancer Chemother Pharmacol. 2008;62:173–180. doi: 10.1007/s00280-007-0665-7. [DOI] [PubMed] [Google Scholar]; (e) Banerjee S, Wang Z, Mohammad M, Sarkar FH, Mohammad RM. J Nat Prod. 2008;71:492–496. doi: 10.1021/np0705716. [DOI] [PubMed] [Google Scholar]
- 5.Woyke T, Berens ME, Hoelzinger DB, Pettit GR, Winkelmann G, Pettit RK. Antimicrob Agents Chemother. 2004;48:561–567. doi: 10.1128/AAC.48.2.561-567.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL, Senter PD. Nat Biotechnol. 2003;21:778–784. doi: 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]; (b) Temming K, Mayer DL, Zabinski R, Dijkers ECF, Poelstra K, Molema G, Kok RJ. Bioconjugate Chem. 2006;17:1385–1394. doi: 10.1021/bc060087z. [DOI] [PubMed] [Google Scholar]; (c) Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G, Kok RJ. Mol Pharmaceut. 2007;4:686–694. doi: 10.1021/mp0700312. [DOI] [PubMed] [Google Scholar]
- 7.Shnyder SD, Cooper PA, Millington NJ, Pettit GR, Bibby MC. Int J Oncol. 2007;31:353–360. [PubMed] [Google Scholar]
- 8.(a) Pettit GR, Lippert JW., III Anti-Cancer Drug Des. 2000;15:203–216. [PubMed] [Google Scholar]; (b) Kirwan IG, Loadman PM, Swaine DJ, Anthoney DA, Pettit GR, Lippert JW, III, Shnyder SD, Cooper PA, Bibby MC. Clin Cancer Res. 2004;10:1446–1453. doi: 10.1158/1078-0432.ccr-0518-03. [DOI] [PubMed] [Google Scholar]; (c) Hokland SL, Horsman MR. Int J Hypothermia. 2007;23:599–606. doi: 10.1080/02656730701739554. [DOI] [PubMed] [Google Scholar]; (d) Dalal S, Burchill SA. Eur J Cancer. 2009;45:713–722. doi: 10.1016/j.ejca.2008.11.045. [DOI] [PubMed] [Google Scholar]
- 9.(a) Pettit GR, Rhodes MR. Anti-Cancer Drug Des. 1998;13:183–191. [PubMed] [Google Scholar]; (b) Patterson DM, Rustin GJS, Serradell N, Rosa E, Bolos J. Drugs of the Future. 2007;32:1025–1032. [Google Scholar]
- 10.Pettit GR, Melody N, Herald DL. J Nat Prod. 2004;67:322–327. doi: 10.1021/np030299+. [DOI] [PubMed] [Google Scholar]
- 11.Mamber SW, Mikkilineni AB, Pack EJ, Rosser MP, Wong H, Ueda Y, Forenza S. J Pharm Exp Ther. 1995;274:877–883. [PubMed] [Google Scholar]
- 12.Saulnier MG, Langley DR, Kadow JF, Senter PD, Knipe JO, Tun MM, Vyas DM, Doyle TW. Bioorg Med Chem Lett. 1994;4:2567–2572. [Google Scholar]
- 13.(a) Egan TJ, Hunter R, Kaschula CH, Marques HM, Misplon A, Walden J. J Med Chem. 2000;43:283–291. doi: 10.1021/jm990437l. [DOI] [PubMed] [Google Scholar]; (b) Nanayakkara NPD, Ager AL, Jr, Bartlett MS, Yardley V, Croft SL, Khan IA, McChesney JD, Walker LA. Antimicrob Agents Chemother. 2008;52:2130–2137. doi: 10.1128/AAC.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Vale N, Moreira R, Gomes P. Eur J Med Chem. 2009;44:937–953. doi: 10.1016/j.ejmech.2008.08.011. [DOI] [PubMed] [Google Scholar]; (d) Kouznetsov VV, Gómez-Barrio A. Eur J Med Chem. 2009;44:3091–3113. doi: 10.1016/j.ejmech.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 14.(a) Pfister JR. J Nat Prod. 1988;51:969–970. doi: 10.1021/np50059a027. [DOI] [PubMed] [Google Scholar]; (b) Sheridan RE, Deshpande SS, Nicholson JD, Adler M. Toxicon. 1997;35:1439–1451. doi: 10.1016/s0041-0101(96)00208-5. [DOI] [PubMed] [Google Scholar]; (c) Beresnevicius ZJ, Viliunas V, Kantminiene K. Chem Heterocyclic Compounds (Khim Geterotsikl Soedin) 2000;36:432–438. [Google Scholar]; (d) Goda FE, Abdel-Aziz AAM, Ghoneim HA. Bioorg Med Chem. 2005;13:3175–3183. doi: 10.1016/j.bmc.2005.02.050. [DOI] [PubMed] [Google Scholar]; (e) Shao Y, Zhang J, Tu C, Dai C, Xu Q, Guo Z. J Inorg Chem. 2005;99:1490–1496. doi: 10.1016/j.jinorgbio.2005.04.007. [DOI] [PubMed] [Google Scholar]; (f) DeVita RJ. Curr Topics Med Chem. 2007;7:1433–1439. doi: 10.2174/156802607782194789. [DOI] [PubMed] [Google Scholar]
- 15.(a) Pettit GR, Singh SB, Herald DL, Lloyd-Williams P, Kantoci D, Burkett DD, Barkóczy J, Hogan F, Wardlaw TR. J Org Chem. 1994;59:6287–6295. [Google Scholar]; (b) Pettit GR, Grealish MP. J Org Chem. 2001;66:8640–8642. doi: 10.1021/jo010530t. [DOI] [PubMed] [Google Scholar]; (c) Mordant C, Reymond S, Tone H, Lavergne D, Touati R, Ben Hassine B, Ratovelomanana-Vidal V, Genet JP. Tetrahedron. 2007;63:6115–6123. [Google Scholar]
- 16.Pettit GR, Srirangam JK, Singh SB, Williams MD, Herald DL, Barkóczy J, Kantoci D, Hogan F. J Chem Soc, Perkin Trans 1. 1996:859–863. [Google Scholar]
- 17.(a) Schulman SG, Abate K, Kovi PJ, Capomacchia AC, Jackman D. Anal Chim Acta. 1973;65:59–67. [Google Scholar]; (b) Abernethy JL, Kilday W. J Org Chem. 1960;25:1924–1928. [Google Scholar]
- 18.(a) Pozdnev VF. Int J Peptide Protein Res. 1994:36–48. doi: 10.1111/j.1399-3011.1994.tb00402.x. [DOI] [PubMed] [Google Scholar]; (b) Furlong ST, Mauger RC, Strimpler AM, Liu YP, Morris FX, Edwards PD. Bioorg Med Chem. 2002;10:3637–3647. doi: 10.1016/s0968-0896(02)00174-8. [DOI] [PubMed] [Google Scholar]
- 19.(a) Olah GA, Nojima M, Kerekes I. Synthesis. 1973:487–488. [Google Scholar]; (b) Bertho JN, Loffet A, Pinel C, Reuther F, Sennyey G. Tetrahedron Lett. 1991;32:1303–1306. [Google Scholar]
- 20.(a) Carpino LA, Mansour ESME, Sadat-Aalee D. J Org Chem. 1991;56:2611–2614. [Google Scholar]; (b) Wenschuh H, Beyermann M, El-Faham A, Ghassemi S, Carpino LA, Bienert M. J Chem Soc, Chem Commun. 1995:669–670. [Google Scholar]; (c) Carpino LA, Beyermann M, Wenschuh H, Bienert M. Acc Chem Res. 1996;29:268–274. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.