

Abstract
The P2Y1 receptor is a member of the P2Y family of nucleotide-activated G protein-coupled receptors, and it is an important therapeutic target based on its broad tissue distribution and essential role in platelet aggregation. We have designed a set of highly selective and diverse pharmacological tools for studying the P2Y1 receptor using a rational approach to ligand design. Based on the discovery that bisphosphate analogues of the P2Y1 receptor agonist, ADP, are partial agonists/competitive antagonists of this receptor, an iterative approach was used to develop competitive antagonists with enhanced affinity and selectivity. Halogen substitutions of the 2-position of the adenine ring provided increased affinity while an N6 methyl substitution eliminated partial agonist activity. Furthermore, various replacements of the ribose ring with symmetrically branched, phosphorylated acyclic structures revealed that the ribose is not necessary for recognition at the P2Y1 receptor. Finally, replacement of the ribose ring with a five member methanocarba ring constrained in the Northern conformation conferred dramatic increases in affinity to both P2Y1 receptor antagonists as well as agonists. These combined structural modifications have resulted in a series of selective high affinity antagonists of the P2Y1 receptor, two broadly applicable radioligands, and a high affinity agonist capable of selectively activating the P2Y1 receptor in human platelets. Complementary receptor modeling and computational ligand docking have provided a putative structural framework for the drug-receptor interactions. A similar rational approach is being applied to develop selective ligands for other subtypes of P2Y receptors.
Keywords: Adenosine-3′,5′-bisphosphate; MRS2179; MRS2279; MRS2500; P2Y receptors; P2Y1 receptor; platelet aggregation; radioligand
INTRODUCTION
The concept that adenine and uridine nucleotides function as extracellular signaling molecules has expanded markedly in the last decade. At least fifteen nucleotide-activated cell surface receptors exist in mammals, and remarkably broad and varied physiological responses occur downstream of nucleotide receptor activation [1, 2]. The significance of extracellular nucleotides also is underscored by the ubiquitous distribution of several large classes of ectoenzymes that catalyze nucleotide breakdown and interconversion [3, 4].
Responses to nucleotides, which occur in essentially all tissues, are mediated by two distinct families of cell surface receptors. The P2X receptors are a family of seven different (P2X1 through P2X7) ligand-gated ion channel receptors that conduct extracellular cations in response to ATP [5]. The P2Y receptors (Fig. 1) are a group of eight molecularly defined G protein-coupled receptors (GPCR) that can be divided into two subfamilies based on relative sequence similarity and shared downstream signaling mechanisms [6, 7]. The P2Y1 receptor subfamily consists of the Gq-coupled P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors, and the P2Y12 receptor subfamily consists of the Gi-coupled P2Y12, P2Y13, and P2Y14 receptors. The cognate agonists of the P2Y receptors include at least 5 different nucleotides and nucleotide sugars. The P2Y1, P2Y12, and P2Y13 receptors are selectively activated by ADP, the P2Y2 receptor is activated by both ATP and UTP, the P2Y4 receptor is activated by UTP, the P2Y6 receptor is activated by UDP, the P2Y11 receptor is activated by ATP, and the P2Y14 receptor is activated by UDP-glucose (and other nucleotide sugars). The distribution of these receptors remarkably overlaps in a broad range of cell types and tissues. Pharmacological resolution of the actions of individual P2Y receptors in these tissues is both difficult and complex in part due to the existence of ectoenzymes on the cell surface that, for example, inactivate the cognate agonist for one type of P2Y receptor while producing the cognate agonist for another [3, 8-13].
Fig. (1).

Mammalian P2Y receptors. The eight molecularly and functionally identified mammalian P2Y receptors are listed with their cognate agonists and the heterotrimeric G protein Gα-subunits and effector enzymes most clearly associated with their signaling function. PLC, phospholipase C; AC, adenylyl cyclase. (+), increase in activity; (−) decrease in activity.
Drugs that reliably activate or antagonize a given P2Y receptor subtype would help surmount the difficulties inherent in the study of these receptors with natural nucleotide agonists, but such drugs largely have not been available. Therefore, a major goal of the research of our laboratories has been to generate agonist and antagonist molecules of high affinity and selectivity for individual P2Y receptors. Our focus to date has been on development of drugs for the Gq-coupled members of the P2Y receptor family. Structure-activity studies have led to identification of analogues that exhibit selectivity for the P2Y2, P2Y4, or P2Y6 receptor [14-17], but the most significant progress has been made in development of selective high affinity antagonists, agonists, and radioligands for the P2Y1 receptor. This receptor is broadly distributed in the central nervous system and in peripheral tissues. It plays an indispensable physiological role in ADP-promoted platelet aggregation and is a drug target of considerable therapeutic interest [18-21]. This review focuses on the development, identity, and pharmacological properties of P2Y1 receptor-directed molecules.
STRATEGY
Our progress in drug development has relied heavily on: (i) knowledge and experience in directed medicinal chemistry related to nucleotides, nucleosides, and their analogues; (ii) a robust inositol lipid cell signaling response in a stable cell line expressing the recombinant human P2Y1 receptor; (iii) a radioligand binding assay that was developed from studies applying (i) and (ii) to generate a high affinity radio-labeled antagonist that provides a very sensitive means to quantify affinities of test drugs with membranes from Sf9 insect cells expressing recombinant human P2Y1 receptor at high levels; and (iv) an iterative molecular model of the binding pocket of the P2Y1 receptor developed from the known structure of the seven transmembrane-spanning receptor rhodopsin. Our approach to drug development has applied classical molecular pharmacological approaches that do not rely on high throughput technology. Instead, our philosophy is centered on the view that molecular knowledge of this particular receptor and its associated cell signaling system, together with the attendant biochemical assays we have developed, is well-suited for a focused iterative approach that applies molecular modeling, rational medicinal chemistry, and drug testing with very precise and reproducible model systems in multiple rounds of structure-activity analyses. Optimized research tools for the P2Y1 receptor have evolved from this approach, which is now being optimized and applied for agonist and antagonist development for other P2Y receptor subtypes.
ASSAY SYSTEMS
Phospholipase C Activity
Assays quantifying agonist/antagonist activity of drugs are accomplished by quantification of [3H]inositol phosphate accumulation in 1321N1 human astrocytoma (P2Y1-1321N1) cells stably expressing the human P2Y1 receptor [20]. One advantage of the 1321N1 cells is that they do not express endogenous P2Y receptors. Assays are accomplished in formats using up to a 96-well plate system. We have applied scintillation proximity assay technology for quantification of phospholipase C activity [22], although the traditional Dowex ion exchange chromatography system works equally well for most of our purposes. Briefly, phosphoinositide pools of P2Y1-1321N1 cells (grown on 12- to 96-well plates) are metabolically labeled with [3H]inositol (typically 1 μCi/well) overnight, and drugs are tested the following day during 30 minute incubations in the presence of 10 mM LiCl, which inhibits inositol phosphate phosphatase activity and enhances the signal resulting from P2Y1 receptor-stimulated phospholipase C activity. Full agonists of the P2Y1 receptor promote >10-fold stimulation of [3H]inositol phosphate accumulation over basal levels. Newly synthesized drugs are initially tested at single concentrations (10 μM) alone as well as in the presence of an approximately EC70 concentration (50 nM) of the full agonist 2MeSADP (4); see Fig. 2 for drug structures. If agonist activity is revealed in the test drug, then full concentration-effect curves are generated to obtain an EC50 value relative to that of 2MeSADP (4) and the cognate P2Y1 receptor agonist ADP (1). Representative concentration-effect curves for two agonists are illustrated in Fig. 3. If antagonist activity is revealed, then full Schild analyses are performed to determine a KB value and to establish that antagonist activity is indeed competitive. Briefly, these analyses require the generation of concentration-effect curves for 2MeSADP (4) alone and for 2MeSADP (4) in the presence of increasing concentrations of antagonist (Fig. 4). Typically, 2MeSADP (4) activation curves are generated with 5-6 concentrations of antagonist over at least a 100-fold range of concentrations. These conditions produce a series of increasingly right-shifted 2MeSADP (4) concentration curves that provide dose-ratios defining how far the activation curve is shifted to the right by a given concentration of antagonist. As illustrated in Fig. 4 and amplified in the legend of this figure, a simple plot of log (dose ratio-1) vs antagonist concentration allows regression of a line that provides the pKB on the X-axis. Observation of a slope of unity in these plots also confirms the competitive nature of the interaction of the agonist and antagonist molecules with the receptor [23-25].
Fig. (2).

Agonists and antagonists of the P2Y1 receptor.
Fig. (3).
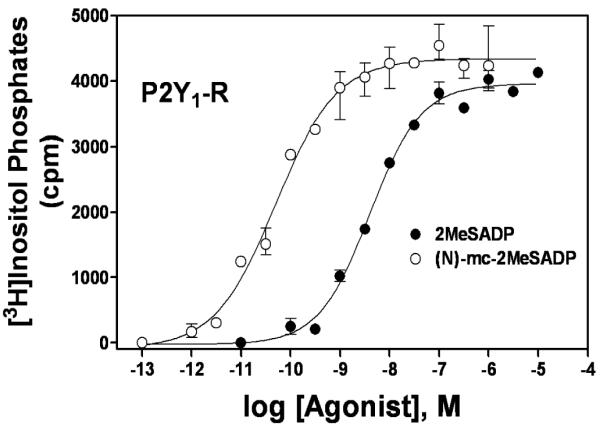
Activation of the P2Y1 receptor by two analogues of ADP. Concentration effect curves for P2Y1 receptor-mediated activation of phospholipase C were determined for 2MeSADP and (N)-methanocarba-2MeSADP ((N)-mc-2MeSADP; MRS2365). This figure is from Ravi et al. [46] and reprinted with permission.
Fig. (4).

Schild analysis of ligand interaction with the P2Y1 receptor. Left Panel, Phospholipase C activity was measured in the indicated concentrations of the agonist 2MeSADP in the absence or presence of increasing concentrations (0 (●), 0.03 (■), 0.1 (▲), 0.3 (◆), 1 (○), 3 (□), 10 (Δ), and 30 (◇) μM) of the antagonist MRS2279. Right Panel, A Schild plot was generated from the data illustrated in A. A dose ratio for each concentration of antagonist was determined by dividing the concentration of agonist necessary to produce 50% of maximal effect in the presence of that concentration of antagonist by the concentration of agonist necessary to produce 50% of maximal effect in the absence of antagonist. Log (dose ratio-1) was plotted vs concentration of antagonist, and the intercept on the X axis is equal to the pKB of the antagonist. This figure is from Boyer et al. [23] and reprinted with permission.
Radioligand Binding
Prior attempts to quantify P2Y1 receptors using a variety of nucleotide radioligands were plagued with inconsistent and often very misleading results. Therefore, progress in the study of this receptor has been impeded by the lack of availability of a generally applicable radioligand. We describe below our success in developing MRS2279 (14) as a high affinity antagonist of the P2Y1 receptor. As we have described in detail [26], this molecule was radiolabeled using [3H]methylamine to high specific radioactivity and used to develop the first generally successful antagonist radioligand binding assay for P2Y1 receptors. Briefly, competition curves for newly synthesized drugs (Fig. 5) are generated by incubating a range of concentrations (10-12 concentrations over a 1000-fold range) of each analogue with a fixed concentration (5 nM) of [3H]MRS2279 (15) near its Kd and a small amount (5 μg) of membranes prepared from Sf9 insect cells expressing recombinant human P2Y1 receptor. Reactions are terminated by filtration over glass fiber filters, and receptor-bound radioactivity determined by scintillation spectrometry. Ki values are calculated from IC50 values of the competing drug according to the equation: Ki = IC50/1 + [[3H]MRS2279]/Kd.
Fig. (5).
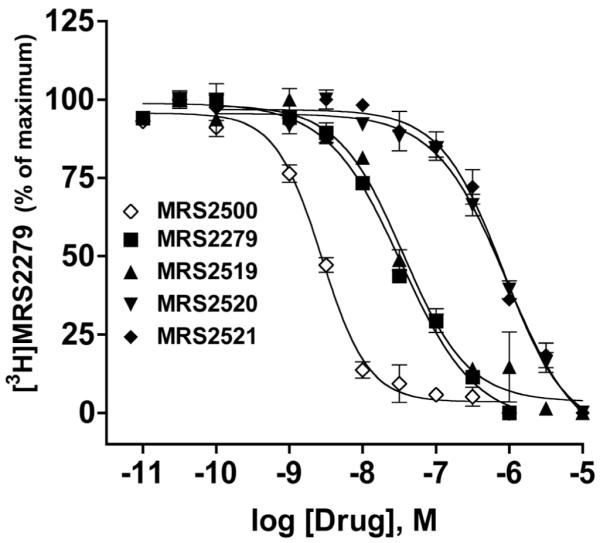
Inhibition of [3H]MRS2279 binding by P2Y1 receptor ligands. A series of 2-substituted analogues of MRS2279 were tested as potential high affinity ligands for the P2Y1 receptor using [3H]MRS2279 as a radioligand and membranes isolated from Sf9 insect cells expressing recombinant human P2Y1 receptor at high levels. The most potent inhibitor in this series was MRS2500, whose structure is illustrated in Fig. 2. Various 2-substituted analogues of MRS2500 are included: 2-fluoro (MRS2520), 2-bromo (MRS2519), and 2-methylthio (MRS2521). This figure is modified from Kim et al. [50] and reprinted with permission.
Molecular Modeling
A molecular model of the P2Y1 receptor (Fig. 6) was constructed using the electron density map of bovine rhodopsin as a template [27]. Using molecular dynamics simulations, an energetically sound conformational hypothesis was generated identifying transmembrane domains (TMs), extracellular loops (ELs), and a short amino terminus. Various ligands were docked into the model to establish a putative binding site, and these studies were complemented with site-directed mutagenesis to confirm the requirement of specific residues for the coordination of agonists and antagonists in the binding site. A principal binding domain was established identifying residues necessary for coordinating various atoms of the nucleotide (Fig. 7). Gln307 and Ser314 of TM7 are in contact with the adenine ring while the phosphate groups of both polyphosphate agonists and bisphosphate antagonists reside in a positively charged pocket formed by Arg128 (TM3), Lys280 (TM6), and Arg310 (TM7). Based on site-directed mutagenesis studies, the physical properties of these residues appeared to be more important than their absolute conservation, since agonist activation was restored in S314T and R310K mutant receptors [28]. Supporting the identity of the proposed binding pocket and the accuracy of the models, a ligand and structure-based consensus model (LIST-CM) proved capable of predicting quantitatively the potencies of a novel set of bisphosphate antagonists [29].
Fig. (6).

Molecular model of the human P2Y1 receptor complexed with the antagonist MRS2179 in space filling representation. Nucleotide agonists and antagonists bind within the upper portion of the helical bundle, with the phosphate group coordinated by three cationic residues and the base in contact with residues from TM7. The ribbon representation of the receptor backbone is depicted with the colors of the rainbow going from the N-terminus to the C-terminus (TM1: orange; TM2: yellow; TM3: green; TM4: green/blue; TM5: cyan; TM6: blue; TM7: violet).
Fig. (7).

Schematic representation of the putative interaction of MRS2179 with the P2Y1 receptor. The phosphate moieties are coordinated by a number of basic residues located in TM3, TM6, and TM7. The nucleobase establishes contacts with residues located in TM1 (Tyr58), TM3 (Phe131), and TM7 (Gln307 and Ser314). The picture has been generated with the ligand interaction application of MOE and uses the following annotations: hydrogen bonds are represented with green arrows; residues highly exposed to the ligand are denoted with a blue half moon; ligand atoms highly exposed to the receptor are represented denoted with a blue shadow; polar residues are colored in pink; greasy residues are colored in green; basic residues are marked with a blue circle.
In addition to the principal binding domain, two additional binding sites comprised by EL2 and EL3, termed meta-binding sites, were identified and a three-step model for ligand binding proposed, with association at the meta-binding sites necessarily preceding binding at the principal site (Fig. 7). In meta-binding site I, Glu209 and Arg287 help coordinate the ribose ring and α and β phosphates of ATP, respectively. Meta-binding site II exists just beneath EL2 and shares amino acids Arg128, Lys280, and Gln307 with the principal binding site. Residues determined to be essential for ligand recognition are likely necessary for the formation of stable intermediates during the multistep ligand binding process [27].
DEVELOPMENT OF COMPETITIVE ANTAGONISTS OF THE P2Y1 RECEPTOR
Discovery of Adenosine Bisphosphates as Competitive Antagonists of the P2Y1 Receptor
Molecules such as reactive blue-2, suramin, and PPADS were first used as potential antagonists of P2 receptors. Although small degrees of selectivity were observed with certain receptors including the P2Y1 receptor [30-32], these drugs largely proved to be at best only slightly selective antagonists that exhibited only low to moderate potency. The original breakthrough in identification of P2Y1 receptor antagonists came with our discovery that adenosine-3′-phosphate-5′-phosphosulfate (A3P5PS, 8) is a partial agonist at the avian P2Y1 receptor; follow-up Schild analyses illustrated that this molecule behaved as a true competitive antagonist [25]. We subsequently discovered that the sulfate substitution was dispensable and that adenosine-3′-phosphate-5′-phosphate, (A3P5P, 7), also was a competitive antagonist. Moreover, studies with the human P2Y1 receptor stably expressed in 1321N1 human astrocytoma cells also revealed that both A3P5PS (8) and A3P5P (7) exhibited competitive antagonist activity at the mammalian P2Y1 receptor ortholog [25]. The pKB value for A3P5P (7) at the human P2Y1 receptor was 6.05. The selectivity of these bisphosphate molecules as P2Y1 receptor antagonists was revealed by demonstration of lack of antagonist activity at the Gq-linked P2Y2, P2Y4, or P2Y6 receptors or at a Gi-linked P2Y receptor of C6 glioma cells now known to be the P2Y12 receptor. The availability of these P2Y1 receptor selective antagonists played a key role in pharmacological delineation of the existence of two distinct receptors (the P2Y1 and the P2Y12 receptors) on platelets that mediate the aggregation response to ADP [33, 34]. Moreover, the discovery of these bisphosphate analogues was logically followed by synthetic efforts designed to increase the affinity for P2Y1 receptors, the level of receptor selectivity, and the resistance to degradation in multiple series of synthetic bisphosphate analogues.
Synthetic Nucleotide Analogues as Competitive Antagonists
A3P5P (7) was utilized as the core molecule for initial synthetic efforts directed toward obtaining antagonist molecules that exhibited increased affinity for the P2Y1 receptor. Initial studies utilized turkey erythrocyte membranes as a model system for rapid assessment of molecules at a Gq/phospholipase C-linked P2Y1 receptor. This system was eventually replaced with the engineered cell line, i.e. hP2Y1-1321N1 cells, discussed above. Substitutions on various positions of the adenine ring of adenosine bisphosphates were explored, followed by modifications and replacement of the ribose ring with various cyclic and acyclic structures and substitutions of the 3′ and 5′ phosphates with uncharged functional groups. These studies were complemented with molecular modeling of the receptor to predict the energy and space determinants governing recognition of the various nucleotide analogues in the putative receptor binding site.
Substitutions at the 2-position of the adenine ring were well-tolerated at the avian P2Y1 receptor with preference for chloro and methylthio substitutions, which conferred up to 3-fold increases in antagonist potency while retaining partial agonist activity [35, 36]. Substitutions at the 6- and 8-positions of adenine were largely disadvantageous, resulting in molecules that interacted poorly with the receptor.
Of particular importance was the finding that addition of a methyl group at the N6 position eliminated partial agonist activity at the turkey P2Y1 receptor and increased the potency of adenosine bisphosphates as antagonists [35]. The N6-methyl substitution was sufficient to eliminate the partial agonist activity of a broad range of modified adenine nucleotide derivatives, generating a template on which to design purely competitive antagonists. Other substitutions at this position, including alkyl groups of longer chain length, a benzoyl group, or chloro or hydroxyl groups resulted in either decreased potency or complete loss of activity. Modification of the ribose ring indicated that the unphosphorylated hydroxyl group of A3P5P (7) was not required for recognition at the turkey P2Y1 receptor. The 2′-deoxy modification also tended to remove any residual agonism of the nucleotide derivatives at the P2Y1 receptor and eliminated activity at adenosine receptors, thus favoring a selective, pure antagonist.
MRS2179
The series of structure-activity studies described in the previous section resulted in synthesis of N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate (MRS2179, 9). This molecule was the first highly selective, competitive antagonist of mid-nanomolar affinity for a P2Y receptor, and has been widely applied in pharmacological studies of the P2Y1 receptor in a variety of tissues. Schild analysis of the capacity of MRS2179 (9) to inhibit 2MeSATP-promoted inositol phosphate accumulation in turkey erythrocyte membranes revealed pure competitive antagonist activity [24]. A pKB of 6.99 was determined. Similar results were obtained with the corresponding 2′,5′ bisphosphate analogue. The activity of MRS2179 (9) also was examined at the P2Y1 receptor of human and rat platelets. MRS2179 (9) inhibited ADP-promoted shape change, aggregation and Ca2+ release with a pKB of 6.55 [38]. Moreover, MRS2179 (9) exhibited no effect on the ADP-promoted inhibition of adenylyl cyclase. Thus, MRS2179 (9) does not interact with the ADP-activated P2Y12 receptor of platelets, which has led to its wide use as a selective and potent inhibitor of ADP-induced platelet aggregation occurring via activation of the P2Y1 receptor.
The identification of MRS2179 (9) as a highly selective competitive antagonist and its subsequent commercial availability made possible the use of this molecule to identify or exclude a role for P2Y1 receptor function in other tissues and physiological systems. For example, roles for P2Y1 receptor activity were revealed in astrocyte survival and in inhibitory neurotransmission in the central and peripheral nervous systems [39, 40].
Acyclic Derivatives and Other Non-Nucleotide P2Y1 Receptor Antagonists
A major goal of our drug development has been to produce high affinity P2Y1 receptor ligands that do not contain a core nucleotide structure. Such molecules predictably should not interact with the thousands of nucleotide binding proteins encoded by the mammalian genome, and also should exhibit reduced susceptibility to metabolism by ectonucleotidases, which very specifically recognize nucleotides as substrates. Toward this end, several replacements of the ribose entity were explored. A series of acyclic nucleotide analogues bearing symmetrically branched phosphorylated aliphatic chains or carbocyclic rings was produced.
Phosphorylated and symmetrically branched isopentyl, isobutyl, and isopropyl groups attached at the 9-position of the adenine ring all retained competitive antagonist activity at the P2Y1 receptor and exhibited no agonist activity [40]. Indeed, the analogue containing an isobutyl group, MRS2298 (10), exhibited antagonist potency at the turkey P2Y1 receptor (IC50 = 0.48 μM) similar to that of its N6-methyl bisphosphate precursor, MRS2179 (9). Replacement of one of the phosphate groups with an uncharged ester greatly decreased potency at the turkey erythrocyte P2Y1 receptor and in washed human platelets while replacement of both phosphate groups with uncharged esters greatly decreased potency at the avian P2Y1 receptor while retaining inhibitory activity in washed human platelets [41, 42]. Interestingly, the uncharged diesters, such as the dipivaloyl derivative MRS2395 (11), were capable of blocking ADP-promoted inhibition of cAMP accumulation in platelets but not the Ca2+ increase, suggesting that these compounds were recognized by the P2Y12 but not the P2Y1 receptor [42]. However, replacement of the bisphosphate groups of MRS2298 (10) with the more stable phosphonates, in MRS2496 (12), resulted in retention of P2Y1 receptor affinity (Ki = 76 nM, in P2Y1 receptor binding assays). Additionally, an analogue containing a bisphosphorylated alkyl group with a cyclopropyl core attached at the 9-position of the N6-methlyadenine ring, MRS2290 (13), was recognized by the turkey P2Y1 receptor albeit with lower potency (IC50 = 1.74 μM) than the riboside [43].
Analogues containing several different types of ring structures retained activity at the P2Y1 receptor. For example, six-membered ring anhydrohexitol bisphosphate analogues displayed micromolar potency. These molecules exhibited agonist action when containing a 6-NH2 substitution while N6-methyl substitution resulted in competitive antagonist action [44]. In contrast, another set of morpholine ring-substituted analogues were essentially inactive at the P2Y1 receptor [45]. Collectively, these studies revealed that the ribose ring is not necessary for recognition by the P2Y1 receptor binding site, and opened up several new avenues for development of non-nucleotide P2Y1 receptor ligands [43, 45-47].
Conformationally Constrained Nucleotide Analogues as Competitive P2Y1 Receptor Antagonists
The importance of sugar puckering in adenine nucleotides has been assessed previously, and for example, conformational preference was illustrated in a series of nucleotide analogues acting as antiviral agents [48]. Thus, the possibility was considered that phosphorylated carbocyclic rings constrained in either the Northern or the Southern conformation by a fused cyclopropane ring would prove advantageous in development of selective ligands of the P2Y1 receptor. In the case of the weak agonist ATP (2), the Northern-constrained derivative was 200-fold more potent as a P2Y1 receptor agonist than the unconstrained molecule and 250-fold more potent than the Southern-constrained derivative, indicating a substantial role for sugar puckering in recognition at the P2Y1 receptor [46]. Using the molecular homology model of the P2Y1 receptor based on the structure of bovine rhodopsin (Figs. 6,7), ATP (2) and MRS2179 (9) were docked in the putative binding site and found to reside in the binding pocket preferably in the Northern (N) conformation, which was energetically favored by 20 kcal/mol over the Southern conformation [45]. These results translated well to several nucleotide derivatives and enhanced potency was observed for (N)-constrained agonists and antagonists at the P2Y1 receptor. This conformational preference applied to some, but not all of the Gq-coupled P2Y receptor subtypes [46], indicating similarities in the binding pockets of nucleotide receptors, which were later further analyzed through a series of molecular modeling studies [14, 15, 17, 28].
MRS2279
Our studies with conformationally constrained pseudosugar-containing analogues led to identification of a non-nucleotide antagonist for the P2Y1 receptor, 2-chloro-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate (MRS2279, 14) that has proved to be a widely useful pharmacological agent [23]. MRS2279 (14) exhibited a pKB of 8.10 for the human P2Y1 receptor. Moroever, it blocked the human platelet P2Y1 receptor and platelet aggregation with a pKB of 8.05, while exhibiting no effect on ADP-dependent activation of the platelet P2Y12 receptor. MRS2279 (14) also had no effect on activation of the P2Y2, P2Y4, P2Y6, or P2Y11 receptors by their cognate agonists.
MRS2500
Substitutions at the 2-position of adenine nucleotides increase ligand affinity for the P2Y1 receptor and P2Y12 receptors [30, 36, 49]. Thus, we determined whether halogen substitutions of the 2-position in various methanocarba analogues would result in molecules with increased affinity for the P2Y1 receptor. Indeed, as is illustrated in Fig. 5, a 2-iodo derivative (MRS2500, 16) exhibited a 10-fold higher affinity than MRS2279 (14) for the P2Y1 receptor [50]. The sub-nanomolar affinity and non-nucleotide structure of this molecule provides an important new reagent for the study of P2Y1 receptors both for pharmacological studies in tissues and animals and for quantification of receptors with a very high affinity radioligand (see below).
Although MRS2179 (9) and MRS2279 (14) effectively inhibited platelet aggregation through blockade of P2Y1 receptors in vivo, MRS2500 (16) proved more stable in vivo, and its much higher affinity make it an attractive candidate for antiplatelet testing. MRS2500 (16) potently inhibited collagen and epinephrine-induced thrombus formation in mice, as well as thrombus formation in response to laser-induced vascular injury [51]. Additionally, the combination of MRS2500 (16) and the irreversible P2Y12 antagonist clopidogrel was more effective than either drug alone in a mouse model of arterial thrombosis, suggesting benefits of combination antiplatelet therapy targeting P2Y1 and P2Y12 receptors. MRS2500 (16) alone also caused only a modest increase in bleeding time, suggesting advantages of reversible, competitive P2Y1 receptor antagonists with regard to safety.
DEVELOPMENT OF RADIOLIGAND BINDING ASSAYS FOR THE P2Y1 RECEPTOR
[3H]MRS2279
The notable advantages contributed by generation of a non-nucleotide high affinity competitive antagonist of the P2Y1 receptor placed us in position to develop the first generally applicable antagonist radioligand for this receptor. The presence of a methyl-substitution at the N6 position in bisphosphate analogues increases P2Y1 receptor affinity and selectivity while decreasing the propensity of some of these bisphosphate analogs to exhibit partial agonist activity. We took advantage of this substitution by synthesizing a precursor molecule that, when reacted with [3H]methylamine, provided [3H]MRS2279 (15) harboring three tritium atoms in the N6-methyl group and a radioligand with a specific radioactivity of 89 Ci/mmole. [3H]MRS2279 (15) bound to the human P2Y1 receptor overexpressed in Sf9 insect cell membranes with a KD of 7.05 nM, and various nucleotide antagonists inhibited [3H]MRS2279 (15) binding with pharmacological selectivity predicted from previous studies [26]. [3H]MRS2279 (15) also specifically bound to the endogenous P2Y1 receptor of rat brain and human platelets with affinity and selectivity similar to that observed for the recombinant human receptor. Due to extensive metabolism of nucleotide agonists, agonist affinities were not determined in these membrane preparations. However, [3H]MRS2279 (15) was utilized to quantify active receptor during purification to homogeneity of the human P2Y1 receptor after its overexpression from a recombinant baculovirus in Sf9 insect cells [52]. Moreover, the availability of purified P2Y1 receptor allowed us to determine for the first time the affinities of agonists for the P2Y1 receptor. These include 2MeSADP (4), 2MeSATP (5), ADP (1), ATP (2), and ADPβS (3), which exhibited Ki values of 0.1 μM, 1.9 μM, 0.9 μM, 18 μM, and 2.4 μM, respectively. The availability of [3H]MRS2279 (15) also provided us with the first useful radioligand binding assay suitable for screening P2Y1 receptor ligands (Fig. 5). This binding assay complements previous and subsequent SAR and molecular modeling studies and has provided us with a very reliable and rapidly accomplished means of assessing newly synthesized nucleotide analogues.
[32P]MRS2500
Our synthesis of MRS2500 (16) provided a non-nucleotide antagonist with an affinity of 1 nM or less. Moreover, the presence of an iodine in the 2-position of MRS2500 (16) suggested the possibility of generation of a high specific radioactivity radioligand for the P2Y1 receptor. Introduction of trimethyltin at the 2-position allowed introduction of 125I through a reaction catalyzed by chloramine T. Unfortunately, the procedure to obtain [125I]MRS2500 requires multiple steps, and reliable production of high quantities of radiolabeled antagonist proved impractical. As such, we sought other means to label MRS2500 (16) to high specific radioactivity and discovered that a precursor 3′-monophosphate molecule was a substrate for phosphorylation catalyzed by polynucleotide kinase. Thus, 32P could be introduced into the 5′-position of the precursor to yield 32P-labeled MRS2500 in a single step from a reaction that included [γ32P]ATP, the precursor, and polynucleotide kinase [53]. Thus, we have produced a means for the routine, facile production of high μCi amounts of [32P]MRS2500 (17) with a theoretical specific activity of approximately 9120 Ci/mmol. The ease of synthesis of this radioligand obviates potential problems introduced by rapid radioactive decay of the short half life radioisotope, 32P. [32P]MRS2500 (17) binds to the recombinant human P2Y1 receptor with a KD of approximately 1 nM [52]. Moreover, the extremely low concentration of protein (< 1 μg) needed in typical binding assays with membranes from P2Y1 receptor-expressing Sf9 cells has allowed us to determine agonist affinities for the first time for the P2Y1 receptor in its membrane environment. These values are very close to those listed above for the purified P2Y1 receptor determined in binding assays with [3H]MRS2279 (15). [32P]MRS2500 (17) has proved very useful for quantification of P2Y1 receptors in a variety of native tissues. A binding assay was established measuring relative distribution of P2Y1 receptors in adult rat organs and brain regions, representing the first determination of the distribution of functional binding sites. Interestingly, P2Y1 receptors were most densely expressed in the lung followed by near equal expression in the brain and the liver. Among brain regions, functional receptor binding sites varied by approximately 6-fold with the highest expression observed in cerebellum and the lowest in cortex [53]. Because P2Y1 receptors are expressed on astrocytes, glia and neurons, cellular localization by receptor autoradiography is an important future application of [32P]MRS2500 (17). Additionally, a binding assay for [32P]MRS2500 (17) has been established in human platelets. Current work is aimed at using [32P]MRS2500 (17) to study agonist-promoted regulation of P2Y1 receptor surface expression in native tissues including platelets and cell lines expressing native and recombinant receptors.
Development of a P2Y1 Receptor-Selective Agonist
Our observation that the affinity of P2Y1 receptor selective antagonists was enhanced by replacing the ribose moiety with a constrained carbocyclic ring [45] suggested that such modifications might enhance affinity and selectivity in agonist molecules. Therefore, various methanocarba analogues were synthesized, and N-methanocarbo-ATP was found to be greater than 100-fold more potent than ATP (2) or S-methanocarba-ATP as an agonist at the P2Y1 receptor [46]. Moreover, combination of the ring-constrained (N)-methanocarba modification with other functionalities resulted in identification of a remarkably potent P2Y1 receptor agonist [47]. Thus, (N)-methanocarba-2-methylthio-ADP (MRS2365, 6) displayed a potency (EC50 = 0.4 nM) at the human P2Y1 receptor that was 20-fold greater than the corresponding riboside 5′-diphosphate (Fig. 3). The fact that ADP (1) is the cognate agonist for three different P2Y receptors, the P2Y1, P2Y12, and P2Y13 receptors, led us to determine whether MRS2365 (6) exhibited selectivity among these receptors. Indeed, the (N)-methanocarba moiety is specifically favored at the P2Y1 receptor, since MRS2365 (6) proved to be neither an agonist nor antagonist at the P2Y12 or P2Y13 receptors [54]. This development of a high affinity, highly selective P2Y1 receptor agonist has provided an important advance for studying platelet function. Thus, the platelet P2Y1 receptor can be selectively activated without necessitating concomitant blockade of the P2Y12 receptor with an antagonist of that receptor [54], and therefore, studies of the function and regulation of the platelet P2Y1 receptor can be accomplished under conditions in which no P2Y12 receptor-directed drugs are present [55]. These studies are ongoing and should lead to better understanding of the interplay of the P2Y1 receptor and the P2Y12 receptor in platelet function.
CONCLUDING REMARKS
We have designed a set of selective and versatile ligand tools for studying the P2Y1 receptor. Thus, both high affinity agonists and antagonists have been crafted using a rational approach to ligand design that derived from the native ligands of the P2Y1 receptor. An iterative approach led to enhancement of affinity and selectivity and optionally to the engineered loss of efficacy (while retaining affinity) to arrive at ligand structures having targeted pharmacological properties. Receptor modeling and computational ligand docking have supported this effort and provided a putative structural framework for the drug-receptor interactions. By substituting the ribose moiety with either less rigid or more sterically-constrained bicyclic ring systems we have optimized the pharmacological profiles. The achievement of nanomolar affinity at the P2Y1 receptor has enabled the preparation of radioligands that can provide broad library screening to discover more novel and chemically diverse ligand structures. This approach is now being applied to the rational design of drug probes for other subtypes of P2Y receptors.
ACKNOWLEDGEMENTS
This work was supported in part by a grant from the National Institute of General Medical Sciences (GM38213) and by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. Dayle Houston was supported by a predoctoral fellowship from the Howard Hughes Medical Institute.
ABBREVIATIONS
- A3P5P
Adenosine 3′, 5′-bisphosphate
- MRS2179
N6-methyl-2′-deoxyadenosine-3′,5′-bisphosphate
- MRS2279
((1′R,2′S,4′S,5′S)-4-(2-chloro-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane)
- MRS2365
(1′S,2′R,3′S,4′R,5′S)-4-[(6-amino-2-methylthio-9H-purin-9-yl)-1-diphosphoryloxymethyl]bicyclo[3.1.0] hexane-2,3-diol
- MRS2500
((1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane)
REFERENCES
- [1].Burnstock G. Pharmacol. Rev. 2006;58:58. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- [2].Burnstock G. Physiol. Rev. 2007;87:659. doi: 10.1152/physrev.00043.2006. [DOI] [PubMed] [Google Scholar]
- [3].Harden TK, Lazarowski ER, Boucher RC. Trends Pharmacol. Sci. 1997;18:43. [PubMed] [Google Scholar]
- [4].Zimmermann H, Braun N. Prog. Brain Res. 1999;120:371. [PubMed] [Google Scholar]
- [5].Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Seguela P, Voigt M, Humphrey PP. Pharmacol. Rev. 2001;53:107. [PubMed] [Google Scholar]
- [6].Harden TK, Barnard EA, Boeynaems J-M, Burnstock G, Dubyak G, Hourani SMO, Insel PA. In: The IUPHAR Compendium of Receptor Characterization and Classification. Girdlestone D, editor. IUPHAR Media Ltd.; Cambridge: 1998. p. 209. [Google Scholar]
- [7].Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA. Pharmacol. Rev. 2006;58:281. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. J. Biol. Chem. 1996;271:33116. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- [9].Kegel B, Braun N, Heine P, Maliszewski CR, Zimmermann H. Neuropharmacology. 1997;36:1189. doi: 10.1016/s0028-3908(97)00115-9. [DOI] [PubMed] [Google Scholar]
- [10].Kirley TL. J. Biol. Chem. 1997;272:1076. doi: 10.1074/jbc.272.2.1076. [DOI] [PubMed] [Google Scholar]
- [11].Mateo J, Harden TK, Boyer JL. Br. J. Pharmacol. 1999;128:396. doi: 10.1038/sj.bjp.0702805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zimmermann H. Naunyn Schmiedebergs Arch. Pharmacol. 2000;362:299. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- [13].Zimmermann H. Drug Develop. Res. 2001;52:44. [Google Scholar]
- [14].Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. J. Med. Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jacobson KA, Costanzi S, Ivanov AA, Tchilibon S, Besada P, Gao ZG, Maddileti S, Harden TK. Biochem. Pharmacol. 2006;71:540. doi: 10.1016/j.bcp.2005.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Besada P, Shin DH, Costanzi S, Ko H, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. J. Med. Chem. 2006;49:5532. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ivanov AA, Fricks I, Harden TK, Jacobson KA. Bioorg. Med. Chem. Lett. 2007;17:761. doi: 10.1016/j.bmcl.2006.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Janssens R, Communi D, Pirotton S, Samson M, Parmentier M, Boeynaems JM. Biochem. Biophys. Res. Commun. 1996;221:588. doi: 10.1006/bbrc.1996.0640. [DOI] [PubMed] [Google Scholar]
- [19].Leon C, Vial C, Cazenave JP, Gachet C. Gene. 1996;171:295. doi: 10.1016/0378-1119(96)00027-3. [DOI] [PubMed] [Google Scholar]
- [20].Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Br. J. Pharmacol. 1996;118:167. doi: 10.1111/j.1476-5381.1996.tb15381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moore D, Chambers J, Waldvogel H, Faull R, Emson P. J. Comp. Neurol. 2000;421:374. doi: 10.1002/(sici)1096-9861(20000605)421:3<374::aid-cne6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- [22].Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Methods Enzymol. 2006;406:489. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- [23].Boyer JL, Adams M, Ravi RG, Jacobson KA, Harden TK. Br. J. Pharmacol. 2002;135:2004. doi: 10.1038/sj.bjp.0704673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK. Br. J. Pharmacol. 1998;124:1. doi: 10.1038/sj.bjp.0701837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Boyer JL, Romero-Avila T, Schachter JB, Harden TK. Mol. Pharmacol. 1996;50:1323. [PubMed] [Google Scholar]
- [26].Waldo GL, Corbitt J, Boyer JL, Ravi G, Kim HS, Ji XD, Lacy J, Jacobson KA, Harden TK. Mol. Pharmacol. 2002;62:1249. doi: 10.1124/mol.62.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moro S, Guo D, Camaioni E, Boyer JL, Harden TK, Jacobson KA. J. Med. Chem. 1998;41:1456. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Costanzi S, Mamedova L, Gao ZG, Jacobson KA. J. Med. Chem. 2004;47:5393. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Costanzi S, Tikhonova IG, Ohno M, Roh EJ, Joshi BV, Colson AO, Houston D, Maddileti S, Harden TK, Jacobson KA. J. Med. Chem. 2007;50:3229. doi: 10.1021/jm0700971. [DOI] [PubMed] [Google Scholar]
- [30].Boyer JL, Zohn IE, Jacobson KA, Harden TK. Br. J. Pharmacol. 1994;113:614. doi: 10.1111/j.1476-5381.1994.tb17034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Charlton SJ, Brown CA, Weisman GA, Turner JT, Erb L, Boarder MR. Br. J. Pharmacol. 1996;118:704. doi: 10.1111/j.1476-5381.1996.tb15457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lambrecht G, Braun K, Damer M, Ganso M, Hildebrandt C, Ullmann H, Kassack MU, Nickel P. Curr. Pharm. Des. 2002;8:2371. doi: 10.2174/1381612023392973. [DOI] [PubMed] [Google Scholar]
- [33].Jin J, Daniel JL, Kunapuli SP. J. Biol. Chem. 1998;273:2030. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- [34].Hechler B, Leon C, Vial C, Vigne P, Frelin C, Cazenave JP, Gachet C. Blood. 1998;92:152. [PubMed] [Google Scholar]
- [35].Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. J. Med. Chem. 1998;41:183. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fischer B, Boyer JL, Hoyle CH, Ziganshin AU, Brizzolara AL, Knight GE, Zimmet J, Burnstock G, Harden TK, Jacobson KA. J. Med. Chem. 1993;36:3937. doi: 10.1021/jm00076a023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baurand A, Raboisson P, Freund M, Leon C, Cazenave JP, Bourguignon JJ, Gachet C. Eur. J. Pharmacol. 2001;412:213. doi: 10.1016/s0014-2999(01)00733-6. [DOI] [PubMed] [Google Scholar]
- [38].Rodrigues RJ, Almeida T, Richardson PJ, Oliveira CR, Cunha RA. J. Neurosci. 2005;25:6286. doi: 10.1523/JNEUROSCI.0628-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mamedova LK, Gao ZG, Jacobson KA. Biochem. Pharmacol. 2006;72:1031. doi: 10.1016/j.bcp.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim HS, Barak D, Harden TK, Boyer JL, Jacobson KA. J. Med. Chem. 2001;44:3092. doi: 10.1021/jm010082h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cattaneo M, Lecchi A, Ohno M, Joshi BV, Besada P, Tchilibon S, Lombardi R, Bischofberger N, Harden TK, Jacobson KA. Biochem. Pharmacol. 2004;68:1995. doi: 10.1016/j.bcp.2004.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu B, Stephens A, Kirschenheuter G, Greslin AF, Cheng X, Sennelo J, Cattaneo M, Zighetti ML, Chen A, Kim SA, Kim HS, Bischofberger N, Cook G, Jacobson KA. J. Med. Chem. 2002;45:5694. doi: 10.1021/jm020173u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim YC, Gallo-Rodriguez C, Jang SY, Nandanan E, Adams M, Harden TK, Boyer JL, Jacobson KA. J. Med. Chem. 2000;43:746. doi: 10.1021/jm9905211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nandanan E, Camaioni E, Jang SY, Kim YC, Cristalli G, Herdewijn P, Secrist JA, 3rd, Tiwari KN, Mohanram A, Harden TK, Boyer JL, Jacobson KA. J. Med. Chem. 1999;42:1625. doi: 10.1021/jm980657j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Nandanan E, Jang SY, Moro S, Kim HO, Siddiqui MA, Russ P, Marquez VE, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. J. Med. Chem. 2000;43:829. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjo M, Boyer JL, Harden TK, Jacobson KA. J. Med. Chem. 2002;45:208. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. J. Med. Chem. 2002;45:2090. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. J. Med. Chem. 1996;39:3739. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- [49].Gough G, Maguire MH, Penglis F. Mol. Pharmacol. 1972;8:170. [PubMed] [Google Scholar]
- [50].Kim HS, Ohno M, Xu B, Kim HO, Choi Y, Ji XD, Maddileti S, Marquez VE, Harden TK, Jacobson KA. J. Med. Chem. 2003;46:4974. doi: 10.1021/jm030127+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hechler B, Nonne C, Roh EJ, Cattaneo M, Cazenave JP, Lanza F, Jacobson KA, Gachet C. J. Pharmacol. Exp. Ther. 2006;316:556. doi: 10.1124/jpet.105.094037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Waldo GL, Harden TK. Mol. Pharmacol. 2004;65:426. doi: 10.1124/mol.65.2.426. [DOI] [PubMed] [Google Scholar]
- [53].Houston D, Ohno M, Nicholas RA, Jacobson KA, Harden TK. Br. J. Pharmacol. 2006;147:459. doi: 10.1038/sj.bjp.0706453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chhatriwala M, Ravi RG, Patel RI, Boyer JL, Jacobson KA, Harden TK. J. Pharmacol. Exp. Ther. 2004;311:1038. doi: 10.1124/jpet.104.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bourdon DM, Mahanty SK, Jacobson KA, Boyer JL, Harden TK. J. Thromb. Haemost. 2006;4:861. doi: 10.1111/j.1538-7836.2006.01866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]