

SUMMARY
Following infection or vaccination, naïve CD8 T cells that receive the appropriate integration of antigenic, co-stimulatory, and inflammatory signals undergo a programmed series of biological changes that ultimately results in the generation of memory cells. Memory CD8 T cells, in contrast to naïve cells, more effectively limit or prevent pathogen re-infection due to both qualitative and quantitative changes that occur following their induction. Unlike vaccination strategies aimed at generating antibody production, the ability to generate protective memory CD8 T cells has proven more complicated and problematic. However, recent experimental results have revealed important principles regarding the molecular and genetic basis for memory CD8 T cell formation, as well as identified ways to manipulate their development through vaccination, resulting in potential new avenues to enhance protective immunity.
INTRODUCTION
Manipulation of the immune system through vaccination to confer protection against human disease is considered one of public health’s greatest accomplishments (Andre, 2003). Although current vaccination methods have been successful at preventing a variety of human diseases, a number of major hurdles remain (Sallusto et al., 2010). Specifically, attempts at vaccinating against malaria, AIDS, and tuberculosis (TB) have been more problematic, largely because the pathogens responsible for causing these diseases are perhaps highly resistant to the humoral arm of the adaptive immune system (Seder et al., 2000). Thus, vaccination strategies/methods that result in host protection against these diseases will likely require the generation of quality memory T cells, including CD8 T cells.
CD8 T cells possess the unique ability to specifically recognize cells infected with intracellular pathogens, ranging from small viruses to large, complex intracellular bacteria and protozoans. In contrast to vaccination strategies that drive the production of high-affinity antibody, the ability to generate large numbers of quality CD8 T cells appears to be much more problematic, especially in humans. However, recent studies have strengthened our understanding of the many factors that contribute to memory CD8 T cell formation. Specifically, this has led to new ideas concerning the generation of memory CD8 T cells through vaccination, including new methods of prime-boost vaccination strategies, how inflammation regulates memory formation, and how subsequent antigenic stimulations impact ensuing memory cell populations.
ACTIVATION OF NAÏVE CD8 T CELLS
Following infection or vaccination, specialized antigen (Ag)-presenting cells known as dendritic cells (DCs) take up foreign antigen, migrate to secondary lymphoid tissues and directly- or cross-present these antigenic peptides on major histocompatibility class I (MHC-I) molecules to naïve CD8 T cells. In addition to their exceptional phagocytic capacity, these cells express a variety of pattern recognitions receptors (PAMPs), such as toll-like receptors (TLRs), which interact with a number of molecules commonly expressed on or within foreign pathogens. Once properly stimulated by PAMPs, DCs undergo maturation that results in enhanced presentation of antigen, expression of co-stimulatory molecules, and production of pro-inflammatory cytokines (Heath et al., 2004). Collectively, these professional Ag-presenting cells provide the necessary signals (commonly referred to as Signal 1, 2, 3) required to efficiently activate a naïve CD8 T cell, resulting in proliferative expansion and generation of effector CD8 T cells, contraction, and formation of long-lived memory cells (Fig 1). Furthermore, the overall balance and integration of these individual signals in vivo influences the CD8 T cell response following activation and impacts both the rate and magnitude of memory CD8 T cell generation (Mescher et al., 2006).
Figure 1. Activation of CD8 T Cells by Dendritic Cells Results in Formation of Long-Lived Memory Cells.

(A) Following maturation and acquisition of antigen, dendritic cells provide the necessary ligands required to activate naïve CD8 T cells. This includes Signal 1 (peptide-MHC Class I complex (p-MHC-I):T Cell Receptor (TCR) ), Signal 2 (co-stimulatory molecules such as CD80/86:CD28), and Signal 3 (Cytokines such as IL-12, IFNα, and IFNβ). (B) Once activated, naïve CD8 T cells undergo rapid and robust proliferation (‘Expansion’) resulting in the generation of effector CD8 T cells, which are able to secrete cytokines and kill pathogen-infected cells. During ‘Contraction’, 90–95% of the newly generated effector CD8 T cells die. However, the remaining 5–10% of CD8 T cells transition into long-lived memory cells and are maintained through cytokines that provide survival signals (such as IL-7) and drive homeostatic proliferation (such as IL-15).
Presentation of peptide on MHC-I provides ‘Signal 1’ to the naïve CD8 T cell. Appropriate stimulation of the TCR results in a dynamic, rapid, and robust activation of signaling pathways resulting in formation of an immunological synapse between the T cell and DC (Dustin, 2004). In vivo, this interaction is thought to last as long as 18–24 hours (Mempel et al., 2004). However, it is during this time frame that biochemical communication between the two immune cells governs both the magnitude and functional quality of the ensuing CD8 T cell response. Efficient signaling through the TCR is dependent on both the amount of antigen available in vivo and the overall strength (affinity) of the TCR interaction with peptide presented by MHC-I. Indeed, large amounts of high-affinity antigenic peptides drive extensive CD8 T cell proliferation following vaccination (Wherry et al., 1999, Zehn et al., 2009). However, antigen alone in the absence of co-stimulation is thought to drive naïve CD8 T cells towards a state of anergy and/or deletion (Mescher et al., 2006). Thus, appropriate engagement of both the TCR and co-stimulatory molecules (‘Signal 2’) such as CD28, CD27, and OX-40 on CD8 T cells by DCs (via CD80/86, CD70, and OX-40L, respectively) regulate the potential of a naïve CD8 T cell to become either activated or anergic.
Ever increasing amounts of data are being generated demonstrating a critical role for ‘Signal 3’ in shaping the overall magnitude of the primary CD8 T cell response and formation of memory (Harty et al., 2008). Specifically, the cytokines IL-12 and type I Interferons (IFNα and β) appear to be critical regulators of the CD8 T cell response and the absence of these cytokines impacts CD8 T cell activation and expansion (Pearce et al., 2007, Curtsinger et al., 2003, Curtsinger et al., 2005, Aichele et al., 2006, Haring et al., 2006). However, the individual requirement for these cytokines is highly pathogen-dependent (Thompson et al., 2006). IL-2 also plays a role shaping the overall CD8 T cell response and has been shown to promote the generation of effector cells following the initial activation of naïve CD8 T cells (Obar et al., 2010; Kalia et al., 2010, Pipkin et al., 2010). In addition, IL-2 is also essential for the generation of primary memory CD8 T cells capable of re-expansion following secondary challenge (Williams et al., 2006). Other cytokines such as TNFα, IL-21 and IFNγ have also been shown to impact specific facets of the CD8 T cell response. Therefore, the overall cytokine milieu present during naïve CD8 T cell activation could potentially have a profound influence on the rate of memory formation and should be strongly considered when designing vaccination strategies.
CHARACTERISTICS OF MEMORY CD8 T CELLS
Memory CD8 T cells are able to confer protective immunity against a range of pathogens in a number of experimental models. In fact, the overall number of memory CD8 T cells present at the time of infection correlates strongly with host protection from disease (Harty et al., 2008, Kaech et al., 2002a, Schmidt et al., 2008). However, attempts to clearly label specific CD8 T cells as ‘memory’ has remained controversial, other than the overall agreement that a memory CD8 T cell population is quite heterogeneous (Jameson et al., 2009). Thus, memory CD8 T cells may exhibit a variety of characteristics, both functional and phenotypic, that vary dramatically even within a TCR-monoclonal population of cells. Because of this complication, memory CD8 T cell populations are perhaps best characterized by their longevity and functional differences that make them superior to naïve cells in providing host protection.
Probably the most dramatic difference between a population of naïve and memory CD8 T cells is the overall change in absolute number. Representation of naïve CD8 T cells that are able to respond to a specific Ag peptide is an extremely rare event, ranging anywhere from as few as 100 to as many as a few thousand in laboratory mice (Blattman et al., 2002, Obar et al., 2008). However, once activated following either infection or vaccination, these naive CD8 T cells undergo substantial cell division. Indeed, one naïve T cell can give rise to as many as 10,000 daughter cells (Stemberger et al., 2007; Harty et al., 2008, Kaech et al., 2002b). Although the process of contraction will eliminate 90–95% of these newly formed T cells via apoptosis, a substantial number remains, which will result in the formation of an Ag-specific memory CD8 T cell population that is 500–1000 times larger than the naïve precursor pool. This population of memory cells is not only long-lived (Homann et al., 2001), but also can robustly respond to secondary challenge. Therefore, a single vaccination or infection can be sufficient to generate a population of memory CD8 T cells that can confer increased protection against pathogen invasion.
Along with this overall change in absolute number, memory CD8 T cells exhibit several other functional qualities that distinguish them from their naïve counterparts. Upon secondary TCR stimulation, memory CD8 T cells can quickly produce a variety of cytokines including IFNγ, TNFα and, to a lesser extent, IL-2. In addition, memory CD8 T cells have the capacity to immediately kill target cells in a TCR-dependent fashion, which does not require de novo protein synthesis (Cho et al., 1999, Barber et al., 2003). Thus, preformed mediators of cytolysis (such as perforin and granzyme) are retained in populations of memory CD8 T cells. Also, in contrast to naïve CD8 T cells, memory CD8 T cells have the ability to localize into peripheral tissues such as the liver, skin, and lung, and this localization becomes enhanced during episodes of inflammation (Woodland et al., 2009, Masopust et al., 2001). Furthermore, memory CD8 T cells that localize to areas of inflammation exhibit more robust killing capacities and increased expression of granzyme B (Kohlmeier et al., 2010, Marzo et al., 2007). More recent studies have also suggested that there may be populations of memory CD8 T cells that are retained long-term in tissues such as the skin and gut (Masopust et al., 2010, Gebhardt et al., 2009). Thus, memory CD8 T cells can be differentiated from naïve cells based on (but not limited to) rapid cytokine production following TCR stimulation, cytolytic activity, and peripheral tissue localization.
In the most general (although most widely accepted) classification, memory CD8 T cell subsets can be differentiated based on the expression of L-selectin (CD62L) and the chemokine receptor CCR7 (Sallusto et al., 1999, Wherry et al., 2003). Memory T cells that express these receptors are termed ‘central memory’ (Tcm) and are able to traffic into lymph nodes directly from the blood through high endothelial venules. In contrast, ‘effector memory’ cells (Tem), by virtue of lacking these receptors, are excluded from lymph nodes, localize more efficiently to peripheral tissues and are thought to be the first line of defense against invading pathogens (Masopust et al., 2001). In addition to these different trafficking capabilities, Tcm also exhibit a more robust proliferative response to antigen than Tem (Wherry et al., 2003). Because of this, Tcm are thought to provide a sustained CD8 T cell secondary response and probably are important in controlling prolonged and/or systemic infections.
Long-term survival and maintenance of memory CD8 T cells does not require TCR signaling or MHC-I (Leignadier et al., 2008, Murali-Krishna et al., 1999), but is dependent upon the pro-survival cytokines IL-7 and IL-15 (Schluns et al., 2003). IL-7 signaling is important for both naïve and memory CD8 T cell survival. In fact, effector CD8 T cells that express the IL-7 receptor α-chain (IL-7Rα; CD127) shortly after the peak of expansion progress into long-lived memory cells (Kaech et al., 2003). In addition, IL-7Rα−/− CD8 T cells expand normally following viral infection, but do not generate memory cells (Schluns et al., 2000). However, it has also been shown that enforced expression of IL-7Rα alone is not sufficient to generate memory cells during a normal CD8 T cell response (Haring et al., 2008, Hand et al., 2007). Thus, IL-7 signals are required, but not sufficient to generate memory CD8 T cells.
At late time-points following infection or vaccination, memory CD8 T cell populations undergo substantial homeostatic proliferation in vivo. In contrast to the pro-survival characteristics of IL-7 signaling, the cytokine IL-15 appears to be required for the long-term ‘maintenance’ of memory CD8 T cell populations (Schluns et al., 2003). Although in the absence of IL-15 signaling, the initial generation of memory cells still occurs (early memory), these memory cells are not maintained over time (Becker et al., 2002). Since IL-15 is a potent inducer of memory CD8 T cell proliferation (in the absence of TCR stimulation), this suggests that IL-7-mediated survival signals alone are not sufficient to maintain memory CD8 T cell populations at late time points. In addition, since homeostatic proliferation occurs and is thought to be required for the long-term maintenance of memory CD8 T cell populations, this proliferation must be accompanied by a ‘nearly equal’ death rate of memory CD8 T cells, since the overall number of memory CD8 T cells doesn’t change over time. However, it is currently unknown how this decision process (life vs. death of a memory CD8 T cell) occurs in vivo.
Collectively, these findings highlight the characteristics of memory CD8 T cells that underlie their enhanced effector function and longevity, which ultimately result in their ability to mediate enhanced protection. However, most of the results discussed here involved the use of laboratory mice and pathogens, which generate an immense CD8 T cell response leading to a large number of memory CD8 T cells. In all practicality, generating large numbers of memory CD8 T cells with a single vaccination or infection in humans is highly unlikely. Thus, vaccination strategies in humans will likely involve multiple rounds of Ag-stimulation in order to appropriately boost memory CD8 T cells numbers to a level that are able to provide a sufficient level of host protection. Indeed, recent studies have begun to uncover important clues regarding CD8 T cell biology that impact memory formation, which has direct implications regarding the ‘boostability’ of a memory CD8 T cell population.
PRIME-BOOST VACCINATION FOR THE GENERATION OF MEMORY CD8 T CELL POPULATIONS
In general, the effective protective capacity of a population of memory CD8 T cells is largely a function of their absolute numbers within the host (Harty et al., 2008, Sallusto et al., 2010). As discussed previously, to become efficiently activated, expand greatly in number and survive to establish a high frequency Ag-specific memory population, naïve CD8 T cells must receive appropriate stimuli via signals 1, 2 and 3. Although effective integration of these signals results in maximal numerical induction of primary CD8 T cell responses, an alternative way to substantially increase the absolute numbers of pathogen-specific memory CD8 T cells is through prime-boost vaccination (Woodland, 2004). In this scenario, ‘booster’ immunization (i.e. re-stimulation of Ag-specific immune cells) additionally expands the preexisting memory CD8 T cell pool to even larger numbers, as well as additionally enriches high affinity Ag-specific CD8 T cells.
Classically, prime-boost vaccination has entailed repeated immunization with the same (homologous) vector. However, homologous prime-boost vaccination, particularly for viral vectors that encode target immunogens, invariably leads to humoral and cellular immune responses that effectively neutralize the vector and accelerate its clearance from the host upon secondary exposure. This rapid clearance of the vector subsequently dampens secondary CD8 T cell responses to the encoded immunogen. In more recent years, an accumulation of experimental evidence has clearly shown that the most effective prime-boost immunizations are structured such that the second dose of antigen (i.e. the boost) is delivered via ‘heterologous’ formulations (Sallusto et al., 2010, Woodland, 2004). For example, the use of heterologous recombinant viral vectors for priming and boosting, such as different serotypes of Adenovirus or different poxvirus vectors, effectively circumvents rapid clearance of the boost vector, thereby allowing sufficient time, recruitment and re-stimulation of preexisting memory CD8 T cells (Woodland, 2004). Such heterologous prime-boost vaccination strategies are now currently under clinical evaluation for diseases such as malaria, TB and HIV (reviewed in (Good et al., 2010, Kaufmann, 2010, McElrath et al., 2010), and are mainly focused on the use of viral vectors to deliver the pathogen-specific immunogens.
While in the simplest terms, prime-boost vaccination appears to be straightforward approach, a great deal of recent experimental evidence suggests that several factors must be taken into consideration when designing prime-boost vaccination strategies. For example, in scenarios of high inflammation during priming, memory CD8 T cells with high proliferative capacity do not form for several weeks to months after the first immunization (Harty et al., 2008, Kaech et al., 2002a). Moreover, repeated boosting of Ag-specific CD8 T cells has been shown to drive memory CD8 T cells towards terminal differentiation, effectively rendering them unresponsive to subsequent re-stimulation (Masopust et al., 2006, Wirth et al. 2010). Thus, three of the most critical issues associated with prime-boost vaccination strategies for the generation of memory CD8 T cells involve the relationships between the inflammation associated with the primary immunization, the timing between the priming and booster immunizations, and the net effect of multiple antigen encounters on the quality and function of the memory CD8 T cells.
THE IMPACT OF INFLAMMATION DURING THE GENERATION OF MEMORY CD8 T CELL POPULATIONS
As described earlier, under most circumstances, the absolute number of Ag-specific memory CD8 T cells that persist after initial vaccination constitutes ~5–10% of the size of the effector pool that arises during the clonal expansion phase. However, inflammatory cytokines have been shown to directly modulate the various phases of the CD8 T cell response. Specifically, pro-inflammatory cytokines have been shown to shape both primary CD8 T cell expansion and contraction, through enhanced survival of effector cells, and the conversion rate of surviving CD8 T cells from early- to late-memory (Butler et al., 2010, Haring et al., 2006). Because each phase of the CD8 T cell response dramatically influences the relative size, function and stability of the resultant Ag-specific memory CD8 T cell pool, the effect of adjuvants and inflammation, or the relative virulence of the vaccine vectors, are important considerations when developing strategies to elicit protective CD8 T cells through vaccination. Although it is clear that inflammatory cytokines enhance the numerical accumulation of both effector and memory cells during a primary CD8 T cell response, inflammation is also known to negatively impact the rate of memory CD8 T cell development. Specifically, CD8 T cell priming coupled with measurable systemic inflammation markedly prolongs the time interval required for the generation of memory cells with late-memory characteristics capable of undergoing robust secondary expansion following booster immunization (Fig. 2A). Thus, although exposure to inflammation (e.g. virulent vectors or adjuvants) during CD8 T cell priming results in larger overall responses, inflammation negatively impacts late-memory generation, thereby potentially diminishing the efficacy of booster immunization.
Figure 2. Inflammation during CD8 T cell activation regulates the rate at which vaccine-induced CD8 T cells acquire boosting potential.
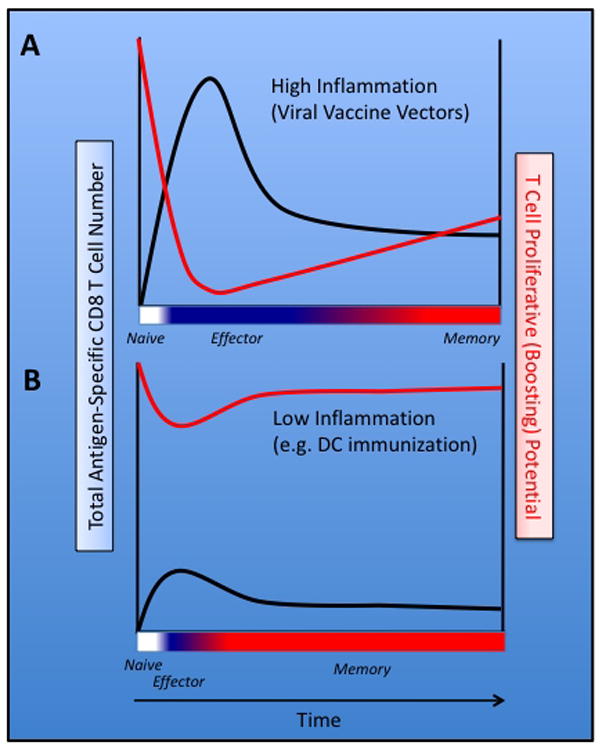
(A) Following vaccination with a virulent vector, high systemic inflammation increases the survival of Ag-specific effector T cells resulting in a protracted effector phase (blue) and relatively enhanced accumulation of memory CD8 T cells. In this scenario, the majority of vaccine-induced Ag-specific CD8 T cells are refractory to additional antigenic stimulation (boosting) for weeks to months. (B) Vaccination with particulate immunogens, such as those encapsulated in PLGA microspheres, get cross-presented to CD8 T cells in an environment of low inflammation. The resulting CD8 T cells, albeit low in number, proceed through a condensed effector phase and rapidly develop characteristics of late-memory CD8 T cells. In this scenario, the CD8 T cells are capable of responding to booster immunization in as few as 4–5 days following priming.
Clearly, the rate at which CD8 T cells acquire characteristics of functional memory cells following immunization is not ‘hardwired’ and manipulating this facet of CD8 T cell priming could potentially maximize the efficacy of prime-boost vaccination. Indeed, work from our lab has demonstrated that delivery of an immunogen in absence of measurable systemic inflammation significantly accelerates the rate at which CD8 T cells acquire late-memory characteristics (Badovinac et al., 2005, Pham et al., 2009). For example, vaccination of mice with peptide-coated, mature DC primes Ag-specific CD8 T cells that exhibit characteristics of late-memory T cells (e.g. CD62Lhi, CD127hi) as early as 6–7 days following priming (Badovinac et al., 2005). Strikingly, these memory-like CD8 T cells are able to robustly proliferate in response to booster immunization as soon as 4 days following DC-priming (Fig. 2B). In this scenario, CD8 T cells appropriately receive and integrate signals 1, 2 and 3, however signal 3 consists of localized cytokine production by the DC. Thus, the CD8 T cells are not exposed to overt systemic inflammation, which occurs following virulent acute infection or vaccination in combination with adjuvants. In support of this, co-administration of peptide-coated DC and a TLR9 agonist (CpG DNA) prevents the accelerated memory CD8 T cell transition after peptide-DC vaccination, while only minimally changing the magnitude of Ag-specific CD8 T cell expansion (Badovinac et al., 2005, Pham et al., 2009).
Although peptide-DC vaccination has been shown to induce the rapid acquisition of late-memory characteristics by responding CD8 T cells, human immunogenetic complexity and logistical hurdles preclude translation of DC-based vaccines into widespread use. However, we have also recently identified that pathways of cross-presentation and cross-priming by DC can be exploited to rapidly generate CD8 T cell immunity (Pham et al., 2010). Cross-presentation involves DC-mediated acquisition, display and induction of CD8 T cell responses against exogenous particulate or cell-associated antigens (Kurts et al., 2010), and recently our lab showed that vaccination of laboratory mice with autologous, apoptotic cells artificially coated with whole immunizing protein cross-primes a small but detectable CD8 T cell population (Pham et al., 2010). Importantly, these CD8 T cells also exhibit characteristics of late-memory CD8 T cells, including the ability to be boosted as early as 4–5 days following priming. Most strikingly, when this population of cells is boosted using conventional vectors (high virulence and measurable systemic inflammation) the CD8 T cell response undergoes massive secondary expansion resulting in long-lived, protective secondary memory CD8 T cell populations. Of note, these results could be recapitulated when using a particulate immunogen; whole protein antigen coated onto biodegradable poly(lactic-co-glycolic) acid (PLGA) microspheres (Pham et al., 2010). Most importantly, since the PLGA microspheres are coated with whole proteins, this approach could be used in humans as an “off the shelf” vaccine regimen to rapidly generate highly ‘boostable’ populations of memory CD8 T cells without special consideration for host immunogenetics. Thus, although CD8 T cell priming in environments of low inflammation leads to relatively reduced expansion and accumulation of effector cells, it markedly reduces the time interval required before cells become responsive to subsequent booster immunization (Fig. 2B).
Collectively, the results described above raise important questions about the utility of adjuvants during prime-boost vaccine strategies designed to generate high magnitude memory CD8 T cell responses. Indeed, new information has revealed that priming CD8 T cell responses in the absence of overt inflammation (‘Signal 3’) results in a population of CD8 T cells, albeit small, that rapidly exhibit characteristics of boostable, long-lived memory CD8 T cells. Therefore, prime-boost vaccines designed to minimize or compress the effector phase of the CD8 T cell response following initial priming, coupled with a strong, heterologous booster vaccination may represent the most efficient way to rapidly generate very large numbers of protective memory CD8 T cells.
THE IMPACT OF REPEATED STIMULATION ON THE FUNCTIONAL CHARACTERISTICS OF MEMORY CD8 T CELLS
Much of what we currently understand about the formation, function and protective capacity of memory CD8 T cells has been evaluated following a single infection or immunization. Thus, a great deal is known about the characteristics of primary memory cells. However, outside of the experimental laboratory rodent, a population of memory CD8 T cells that has been exposed to only a single stimulation is likely very rare. Moreover, as outlined above, the most common strategy for increasing the absolute number of vaccine-induced memory CD8 T cells is prime-boost vaccination, which results in a pool of secondary memory CD8 T cells that have encountered antigen twice. Indeed, many of the most successful human vaccine regimens employ one or more rounds of booster immunization in order to elicit robust protective immunity (Sallusto et al., 2010, Woodland, 2004). Since, by design, prime-boost vaccination leads to a population of memory CD8 T cells that has encountered antigen twice, it is of critical importance that we understand both short- and long-term effects of repeated stimulation on the biology, behavior and function of memory CD8 T cells.
Several studies have begun to shed light on the immunologic consequences of multiple antigen encounters by memory CD8 T cells (Badovinac et al., 2003, Jabbari et al., 2006, Masopust et al., 2006, Wirth et al., 2010). Perhaps one of the most striking differences between primary and secondary memory CD8 T cells is the rate at which each population acquires (or re-acquires) a late, central memory (Tcm) phenotype. For example, upon antigen stimulation, secondary memory CD8 T cells take significantly longer to re-express molecules important for lymph node trafficking, such as CD62L and CCR7, and on the population level secondary memory CD8 T cells appear to undergo reduced contraction (Jabbari et al., 2006, Masopust et al., 2006). Thus, relative to primary memory CD8 T cells, secondary memory CD8 T cells exhibit a more effector-like phenotype.
Consistent with this effector-like phenotype, secondary memory CD8 T cells also appear to exhibit higher cytolytic potential, as evidence by relatively higher expression of Granzyme B, and increased in vivo cell killing (Wirth et al., 2010, Jabbari et al., 2006). Interestingly, since primary memory CD8 T cells more efficiently traffic to lymph nodes, while secondary memory are more commonly associated with peripheral tissues, this suggests that primary and secondary memory CD8 T cells likely differ in their ability to protect against certain acute or chronic infections. For example, although repeated stimulation appears to enforce an effector-like phenotype, thereby decreasing lymph node localization and allowing for priming of new naïve CD8 T cells, in certain scenarios of infection it is possible that a very large population of lymph node homing memory CD8 T cells is optimal for host protection. Indeed, viruses that establish chronic/latent infection in humans, including Epstein-Barr virus (EBV) and human immunodeficiency virus (HIV), undergo significant replication and reactivation in lymph nodes. It is interesting to speculate that EBV and HIV have coevolved with the host immune system to drive the differentiation of virus-specific effector-like memory CD8 T cells, effectively evading a potent arm of the adaptive immune response. In line with this hypothesis, a large fraction of CD8 T cells found within HIV-infected individuals exhibit a CD62LloCCR7lo Tem phenotype (Chen et al., 2001), and therefore would not be expected to localize in appreciable numbers within lymph nodes that harbor virus.
In addition to prime-boost approaches, some current vaccination strategies rely on multiple booster immunizations. While it is well established that repeated immunization works well for augmenting humoral responses, recent work has revealed that repeated antigen stimulations effectively drives memory CD8 T cells to a terminally differentiated state (Wirth et al., 2010). These studies revealed that repeated boosting markedly decreases the representation of Tcm cells within a population of memory CD8 T cells, while at the same time significantly expands the number of differentially expressed genes. Thus, repeated stimulation appears to increase functional diversity of memory CD8 T cells at the cost of proliferative potential (Sallusto et al., 2010, Wirth et al., 2010). Indeed, the study by Wirth et al. highlights both the complex regulation of memory CD8 T cell differentiation and identifies potential new targets for manipulating the function of multiply stimulated memory CD8 T cells.
Lastly, despite the effects of repeated stimulations on CD8 T cells, recent studies reveal the potential for manipulating both numbers and quality (i.e function) of vaccine-induced memory CD8 T populations through the use of pharmacologic agents. Specifically, rapamycin-mediated inhibition of a key regulator of cellular metabolism, mTOR (mammalian target of rapamycin), has been shown to both decrease CD8 T cell contraction and increase the rate of conversion of Tem to Tcm (Araki et al., 2009, Pearce et al., 2009, Prlic et al., 2009). Thus, despite the potential negative impact of prime-boost (or prime-boost-boost) vaccination on phenotypic, functional or proliferative characteristics of memory CD8 T cells, in the future it is possible that co-delivery of booster immunizations with novel drugs could serve as a strategy to specifically modulate memory CD8 T cell function.
CONCLUDING REMARKS
For some major human pathogens such as HIV, M. tuberculosis and P. falciparum, efficacious vaccines have yet to be developed, although it is generally agreed that success against these pathogens will likely require the induction of very large memory CD8 T cell responses. Indeed, some of the most promising human vaccines under development are targeted at augmenting both the number and function of memory CD8 T cells through prime-boost vaccination (Good and Doolan, 2010). However, in recent years a large number of studies have revealed how repeated antigen stimulation significantly impacts both the quality and the absolute number of memory CD8 T cells. Developing approaches to more effectively manipulate both the number and function of vaccine-induced memory CD8 T cells will required further understanding of the molecular and genetic basis for their differentiation and survival. Further examination of the biology and behavior of vaccination-induced memory CD8 T cells in rodent models will continue to inform efforts to develop vaccines against major human diseases, including AIDS, malaria and TB. Such studies in rodents have thus far revealed the profound relationship between low-inflammation scenarios of CD8 T cell priming coupled to the ability to massively expand CD8 T cell numbers within days of the initial immunization. Collectively, these data have the potential to provide a critical framework for more efficient development or improvement of human vaccines.
Acknowledgments
The authors would like to acknowledge former and current members of the Harty laboratory for their contributions to this work. We also offer apologies to the many investigators whose contributions we were unable to discuss owing to space limitations. Work in the Harty Lab is supported by grants from the NIH and a Career Development Award from the Leukemia and Lymphoma Society to JCN.
References
- Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- Andre FE. Vaccinology: past achievements, present roadblocks and future promises. Vaccine. 2003;21:593–595. doi: 10.1016/s0264-410x(02)00702-8. [DOI] [PubMed] [Google Scholar]
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Messingham KA, Hamilton SE, Harty JT. Regulation of CD8+ T cells undergoing primary and secondary responses to infection in the same host. J Immunol. 2003;170:4933–4942. doi: 10.4049/jimmunol.170.10.4933. [DOI] [PubMed] [Google Scholar]
- Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. 2005;11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171:27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, Ahmed R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195:1541–1548. doi: 10.1084/jem.20020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler NS, Harty JT. The role of inflammation in the generation and maintenance of memory T cells. Adv Exp Med Biol. 2010;684:42–56. doi: 10.1007/978-1-4419-6451-9_4. [DOI] [PubMed] [Google Scholar]
- Chen G, Shankar P, Lange C, Valdez H, Skolnik PR, Wu L, et al. CD8 T cells specific for human immunodeficiency virus, Epstein-Barr virus, and cytomegalovirus lack molecules for homing to lymphoid sites of infection. Blood. 2001;98:156–164. doi: 10.1182/blood.v98.1.156. [DOI] [PubMed] [Google Scholar]
- Cho BK, Wang C, Sugawa S, Eisen HN, Chen J. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci U S A. 1999;96:2976–2981. doi: 10.1073/pnas.96.6.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- Dustin ML. Stop and go traffic to tune T cell responses. Immunity. 2004;21:305–314. doi: 10.1016/j.immuni.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- Good MF, Doolan DL. Malaria vaccine design: immunological considerations. Immunity. 2010;33:555–566. doi: 10.1016/j.immuni.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci U S A. 2007;104:11730–11735. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Haring JS, Jing X, Bollenbacher-Reilley J, Xue HH, Leonard WJ, Harty JT. Constitutive expression of IL-7 receptor alpha does not support increased expansion or prevent contraction of antigen-specific CD4 or CD8 T cells following Listeria monocytogenes infection. J Immunol. 2008;180:2855–2862. doi: 10.4049/jimmunol.180.5.2855. [DOI] [PubMed] [Google Scholar]
- Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- Heath WR, Belz GT, Behrens GM, Smith CM, Forehan SP, Parish IA, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev. 2004;199:9–26. doi: 10.1111/j.0105-2896.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7:913–919. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- Jabbari A, Harty JT. Secondary memory CD8+ T cells are more protective but slower to acquire a central-memory phenotype. J Exp Med. 2006;203:919–932. doi: 10.1084/jem.20052237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002a;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002b;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH. Future vaccination strategies against tuberculosis: thinking outside the box. Immunity. 2010;33:567–577. doi: 10.1016/j.immuni.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- Leignadier J, Hardy MP, Cloutier M, Rooney J, Labrecque N. Memory T-lymphocyte survival does not require T-cell receptor expression. Proc Natl Acad Sci U S A. 2008;105:20440–20445. doi: 10.1073/pnas.0806289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo AL, Yagita H, Lefrancois L. Cutting edge: migration to nonlymphoid tissues results in functional conversion of central to effector memory CD8 T cells. J Immunol. 2007;179:36–40. doi: 10.4049/jimmunol.179.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D, Ha SJ, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol. 2006;177:831–839. doi: 10.4049/jimmunol.177.2.831. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- McElrath MJ, Haynes BF. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity. 2010;33:542–554. doi: 10.1016/j.immuni.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–1381. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28:859–869. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrancois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci U S A. 2010;107:193–198. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. 2007;179:2074–2081. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NL, Badovinac VP, Harty JT. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol. 2009;183:2337–2348. doi: 10.4049/jimmunol.0901203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NL, Pewe LL, Fleenor CJ, Langlois RA, Legge KL, Badovinac VP, Harty JT. Exploiting cross-priming to generate protective CD8 T-cell immunity rapidly. Proc Natl Acad Sci U S A. 2010;107:12198–12203. doi: 10.1073/pnas.1004661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic M, Bevan MJ. Immunology: A metabolic switch to memory. Nature. 2009;460:41–42. doi: 10.1038/460041a. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- Schluns KS, Lefrancois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol. 2003;3:269–279. doi: 10.1038/nri1052. [DOI] [PubMed] [Google Scholar]
- Schmidt NW, Podyminogin RL, Butler NS, Badovinac VP, Tucker BJ, Bahjat KS, et al. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc Natl Acad Sci U S A. 2008;105:14017–14022. doi: 10.1073/pnas.0805452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Hill AV. Vaccines against intracellular infections requiring cellular immunity. Nature. 2000;406:793–798. doi: 10.1038/35021239. [DOI] [PubMed] [Google Scholar]
- Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, Busch DH. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007;27:985–997. doi: 10.1016/j.immuni.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Puorro KA, Porgador A, Eisenlohr LC. The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J Immunol. 1999;163:3735–3745. [PubMed] [Google Scholar]
- Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth TC, Xue HH, Rai D, Sabel JT, Bair T, Harty JT, Badovinac VP. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]