

Abstract
The epidermal water barrier resides in the stratum corneum (SC) and is dependent on a highly organized network of multi-lamellar membranes comprised of a critical lipid composition. The SC membranes are formed from precursor membranes packaged in cytoplasmic lamellar bodies in the stratum granulosum and delivered to the SC by exocytosis. An abnormal lipid composition of the SC membranes often results in a disrupted water barrier and the clinical appearance of ichthyosis. This cutaneous feature is characteristic of Sjögren-Larsson syndrome (SLS), an inborn error of lipid metabolism caused by deficiency of fatty aldehyde dehydrogenase (FALDH). The contribution of FALDH to normal epidermal function has become increasingly evident with the recognition that this enzyme has an essential role in metabolism of several lipids, including fatty aldehydes and alcohols, ether glycerolipids, isoprenoid alcohols and certain lipids that undergo ω-oxidation, such as leukotriene B4 and very long-chain fatty acids. In the absence of FALDH, the skin produces lamellar bodies that are empty, lack their surrounding vesicle membranes or contain granular contents rather then the usual cargo membranes. These defective organelles also have impaired exocytosis, which results in structurally abnormal, deficient multi-lamellar membranes in the SC and a leaky water barrier. Although the exact biochemical mechanism for the cutaneous pathology is still unclear, studies in SLS demonstrate the critical importance of FALDH for normal epidermal structure and function.
Key words: ichthyosis, Sjögren-Larsson syndrome, fatty alcohol, stratum corneum, lamellar body, epidermis, membranes
Introduction
The major function of the skin is to prevent loss of water from the body by formation of a permeability barrier. The barrier resides in the stratum corneum (SC) and is dependent on an extensive array of stacked multilamellar membranes that are attached to and intercalate between protein-rich corneocytes.1–3 The SC membranes have a distinctive lipid composition consisting of equimolar ratios of cholesterol, ceramides and free fatty acids, which differs from most other membranes. The multi-lamellar SC membranes are formed from precursor membranes synthesized in the stratum granulosum (SG) and packaged into cytoplasmic lamellar bodies (LBs). These LBs deliver their cargo membranes into the SG-SC boundary through exocytosis, where they are enzymatically modified and assembled into mature SC membranes.
The role of the SC membranes and their lipid components for the epidermal water barrier has been supported by numerous experimental studies indicating that destruction of the SC membranes by topical acetone extraction results in a defective water barrier.4 In an effort to replace the SC membranes and restore the barrier, these experimental manipulations provoke a coordinated biochemical response in the underlying epidermis, with a dramatic increase in epidermal lipid synthesis along with proliferation of LBs carrying their precursor cargo membranes for delivery to the SG-SC interface. This epidermal response to barrier destruction mimics, on an accelerated scale, the normal program of epidermal differentiation that leads to a functional SC. The importance of the lipid composition of the SC membranes is demonstrated by the finding that specific enzymatic inhibitors that block synthesis of each of the three major lipids, when applied topically to the skin, prevent or delay recovery of the barrier.5–7
Inborn errors of lipid metabolism may also affect the SC membranes and cause a chronic barrier defect that is associated with the clinical appearance of ichthyosis.8 These rare genetic diseases have highlighted some seemingly obscure lipid pathways involving phytanic acid, epoxyalcohols, glucosylceramide, triglycerides and fatty alcohols, which would otherwise not have been suspected to be important for epidermal biology. Additional lipid pathways impacting epidermal function will undoubtedly be discovered.
In this review, we illustrate how genetic deficiency of fatty aldehyde dehydrogenase (FALDH) has provided insights into the role of this enzyme in epidermal structure and function.
Fatty Aldehyde Dehydrogenase Deficiency in Sjögren-Larsson Syndrome
Sjögren-Larsson syndrome (SLS) is a rare autosomal recessive disorder of fatty aldehyde and alcohol metabolism that is characterized by the presence of ichthyosis together with neurologic disease.9,10 The cutaneous symptoms are usually apparent at birth and become more pronounced by several months of age. The hyperkeratosis has a generalized distribution but tends to spare the face; over time prominent lichenification is often seen in the flexures of the arms and legs. The hyperkeratotic, scaly skin may vary in appearance from a fine and furfuraceous quality to larger more lamellar-like scales, depending on the body site. The skin often has an erythematous appearance that fades over time. Pruritus is a common complaint of patients. The onset of neurologic symptoms consisting of developmental delay and mental retardation, spastic diplegia or tetraplegia, seizures and photophobia in the first or second year of life usually prompts recognition of SLS rather than another type of ichthyotic disorder.
SLS is caused by mutations in ALDH3A2,11,12 which codes for FALDH, an enzyme that catalyzes the NAD-dependent oxidation of long-chain aliphatic aldehydes to fatty acids.13 FALDH possesses a broad substrate specificity and is capable of oxidizing a variety of aliphatic aldehydes ranging from 6- to 24-carbons long, including saturated, unsaturated and methyl-branched aldehydes.14 The enzyme has a subunit mass of 54 kD and is catalytically active as a homodimer.15 Alternative splicing of the ALDH3A2 gene results in at least two protein isoforms.16 The major isoform is comprised of 485 amino acids and has a carboxy- terminal “tail,” which is necessary for its localization to the endoplasmic reticulum where it encounters a variety of aldehyde substrates.17 A minor protein isoform (FALDHv) is 508 amino acids long and differs from the major isoform by possessing a longer carboxy-terminal region. FALDHv appears to be targeted to the peroxisome, where it probably interacts with a more limited spectrum of aldehyde substrates.18
FALDH is considered a housekeeping enzyme that is expressed in almost all cells and tissues. The enzyme is normally present in keratinocytes located throughout the epidermis, including basal, spinous and granular layers, but is missing from the stratum corneum.19,20
Epidermal Structural Abnormalities Associated with FALDH Deficiency in SLS
FALDH deficiency in SLS leads to distinct structural abnormalities in the skin. With light microscopy, the patients' skin displays pronounced hyperkeratosis, papillomatosis and acanthosis.21 Epidermal hyperplasia is often noted. The granular layer is usually normal or mildly thickened, and a slight mononuclear cell infiltration is occasionally seen in the upper dermis. In the upper spinous layer, keratinocyte nuclei and nucleoli tend to be larger than normal and have more prominent perinuclear halos.22 Image analysis of SLS skin sections using Fast Fourier Transformation reveals periodicities consistent with increased positioning of keratinocyte nuclei in parallel to the basement membrane of the epidermis. This may be related to a hyper-proliferative state in SLS skin as demonstrated by radioactive thymidine incorporation.21
At the ultrastructural level, SLS skin exhibits evidence of a global disruption of LB formation and secretion.19 The LBs in the SG cells are often misshapen, possess granular contents rather than their usual cargo membranes, or are empty (Fig. 1).19,23 Some LBs contain cargo membranes, but have incomplete or missing vesicle membranes surrounding them, suggesting that structural damage has occurred after their formation. Many of the LBs do not undergo exostosis properly and cluster at the apical plasma membrane of the SG cells bordering the SC. They become entombed in the differentiated corneocytes of the SC, where they are seen as discrete cytoplasmic vesicles or lipid inclusions. Membranous lipid inclusions, outside of LBs, can also be seen in the cytoplasm of SG cells.24,25 The extracellular membranes in the SC are reduced in number and the membranes are often disrupted by foci of non-membranous lipid deposits that probably represent lamellar/non-lamellar phase separation (Fig. 2).19 The lipid envelope surrounding corneocytes, however, appears ultrastructurally intact.
Figure 1.

Abnormal LBs in the stratum granulosum of SLS skin. (A–C) Many organelles appear empty (asterisks) or display non-lamellar contents. There is variation in the content and structural appearance of cargo membranes. The limiting vesicle membranes of some LBs appear disrupted or absent (arrows). Reprinted from Rizzo et al. Arch Dermatol Res 2010; 302:443.
Figure 2.
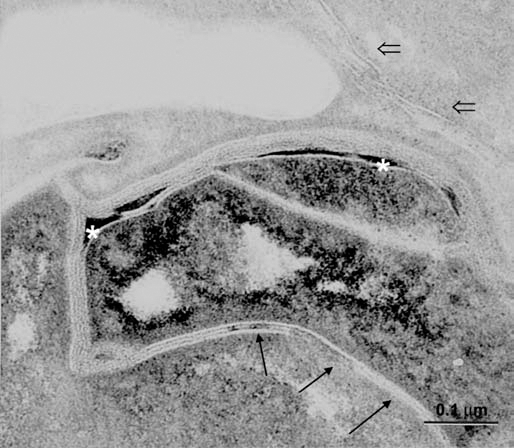
Abnormal SC membranes in SLS skin. Note the paucity of membrane bilayers in some regions (arrows) and lamellar domains that are interspersed with lacunae filled with non-lamellar lipid material (asterisks). Entombed lamellar contents in can be seen in corneocyte cytosol (open arrows). Ruthenium tetroxide post-fixation. Reprinted from Rizzo et al. Arch Dermatol Res 2010; 302:443.
Experimental evidence indicates that the structural abnormalities in SLS skin are associated with a defective epidermal water barrier. In normal skin, the extensive network of intercellular membranes act to prevent entry of water into the SC, a process that is demonstrated by using colloidal lanthanum—an electron-dense, water-soluble marker that can be visualized with electron microscopy.26 When incubated with colloidal lanthanum, this tracer is completely excluded from the normal SC. In SLS skin, however, colloid lanthanum readily permeates into the SC by traversing between corneocytes.19 Lanthanum does not diffuse into the corneocytes, indicating that the cells have a functionally intact cornified lipid envelope.
The Role of FALDH in Lipid Metabolism
Owing to its broad substrate specificity, the role of FALDH in lipid metabolism is not restricted to a single substrate or lipid pathway. Originally thought to primarily oxidize fatty aldehydes derived from straight-chain fatty alcohol metabolism, FALDH is now known to occupy a pivotal place in metabolism of a variety of aliphatic aldehydes generated by several diverse lipid pathways (Fig. 3). Knowledge about the physiologic role of this enzyme has been primarily elucidated by studying cells from SLS patients who lack FALDH activity.
Figure 3.

The role of FALDH in lipid metabolism. Lipid substrates that are inferred to require FALDH for metabolism, but lack experimental confirmation, are indicated with a question mark.
Fatty alcohol metabolism.
Early studies identified FALDH as an enzyme necessary for fatty alcohol oxidation.13,27–29 Fatty alcohols are synthesized from fatty acids via reduction of their acyl-CoA products and used for biosynthesis of wax esters and ether glycerolipids in the skin (Fig. 3). Excess fatty alcohols not used for biosynthesis are recycled back to fatty acid by action of fatty alcohol: NAD oxidoreductase (FAO), a multi-component enzyme complex that consists of fatty alcohol dehydrogenase and FALDH, which act in a sequential fashion to convert fatty alcohol to aldehyde and fatty acid. The pathway for fatty alcohol synthesis and oxidation has been named the fatty alcohol cycle, in part due to evidence that synthesis and oxidation of fatty alcohol occurs simultaneously in cultured skin fibroblasts.30 The initial oxidation of fatty alcohol to fatty aldehyde appears rate-limiting and is followed by a subsequent oxidative step in which FALDH catalyzes the oxidation of fatty aldehyde to fatty acid. In the absence of FAO activity, SLS cells and tissues accumulate long-chain fatty alcohols, primarily hexadecanol (16:0-OH), octadecanol (18:0-OH) and octadecenal (18:1-OH).31–33 Shorter and longer alcohols are also oxidized by FAO, but other isozymes in SLS cells apparently provide enzyme redundancy to prevent their accumulation.
Fatty alcohols are precursors for the synthesis of wax esters and ether glycerolipids, two pathways that are particularly active in skin (Fig. 3). Although wax esters34 and ether glycerolipids35 are thought to be surface lipids that are generated in sebaceous glands, recent studies have demonstrated that both lipids can also be synthesized in cultured keratinocytes.36,37 Fatty alcohols are present in low concentrations in normal cultured keratinocytes, but SLS keratinocytes, which have deficient FAO activity, accumulate up to 25-fold more fatty alcohol than normal.36 Studies using radioactive octadecanol demonstrate that the excess fatty alcohol in SLS keratinocytes is diverted into synthesis of wax esters and neutral ether glycerolipids, resulting in large increases in the cellular content of these lipids.
In contrast to straight-chain alcohols, branched-chain fatty alcohols are often derived from the diet. The most prominent branchedchain alcohol is phytol, a 20-carbon alcohol produced during chlorophyll degradation in the intestine of ruminant animals.38 It is present at high concentrations in dairy products and green leafy vegetables. Phytol is oxidized to phytanic acid in cultured skin fibroblasts by FAO, and this reaction is deficient in SLS cells.39 Subsequent metabolism of phytanic acid is accomplished by the α-oxidation pathway in peroxisomes.40 This pathway generates pristanal, a 1-carbon-shortened, branched-chain aldehyde that is oxidized to pristanic acid. Pristanal is a substrate for FALDH41 and its oxidation to pristanic acid is probably mediated by FALDHv located in peroxisomes.18,42 Thus, FALDH is implicated in two steps in phytol/phytanic acid metabolism. Nevertheless, neither phytol nor phytanic acid have been shown to accumulate in plasma from SLS patients,41 suggesting that alternate enzymes may exist to carry out these reactions in some tissues.
Preliminary studies indicate that other branched-chain alcohols are also oxidized by FAO, including isoprenoid alcohols that are products of the mevalonate pathway.43 Mevalonic acid is a product of hydroxymethylglutaryl-CoA (HMG-CoA) reductase and used to synthesize 5-carbon isoprenoid units that are linked together to generate farnesyl-PP, a key substrate precursor for biosynthesis of cholesterol, dolichols and coenzyme Q. The conversion of farnesyl-PP to geranylgeranyl-PP is a committed step toward cholesterol synthesis. Both farnesyl-PP and geranylgeranyl-PP can be dephosphorylated to farnesol and geranylgeraniol, respectively, and subsequently oxidized to their fatty acids.44,45 This oxidative reaction is impaired in SLS fibroblasts, which implicates FAO/FALDH in this metabolic step.43
Fatty aldehyde metabolism.
Distinct from its role in fatty alcohol metabolism, FALDH is involved in oxidation of fatty aldehydes originating from catabolism of several additional lipids, including ether glycerolipids, and certain fatty acids and eicosanoids that undergo ω-oxidation (Fig. 3).
Ether glycerolipids are a source of fatty aldehydes generated through normal catabolism,46 and by non-enzymatic peroxidation (see below). Ether glycerolipids in mammals are characterized by the presence of a long-chain alkyl group attached to the sn-1 carbon of glycerol via an ether bond.47 The alkyl moiety is derived from fatty alcohols that are chiefly 16- to 18-carbons long (Fig. 3).46 Ether glycerolipids include plasmalogen forms of phospholipids (phosphatidyl-choline, -ethanolamine and -serine) in which the sn-1 position is occupied by a 1-O-alkenyl chain with an unsaturated vinyl ether bond. Plasmalogens are present at high concentrations in certain cells (erythrocytes) and tissues (heart, brain, tumors), but not in skin. Instead, neutral ether glycerolipids, such as 1-O-alkyl-2,3-diacyglycerol, are synthesized by sebaceous glands and secreted onto the surface of the skin as a component of sebum.35 1-O-Alkyl-2,3-diacyglycerol is also synthesized in cultured keratinocytes,36,37 but it is not normally present in SC membranes. The catabolism of ether glycerolipids involves enzymatic cleavage of the 1-O-alkyl bond, which releases the alkyl group as fatty aldehyde (Fig. 3).48 Studies on the metabolism of 1-O-alkylglycerol indicate that the fatty aldehyde is normally oxidized to fatty acid by FALDH and this reaction is impaired in SLS fibroblasts and SLS keratinocytes.49
Ceramides comprise one of the three major classes of lipids in the SC membranes. The ceramides are structurally complex and some molecular species, such as acylceramides, are unique to the skin.50 In contrast to its extracellular structural role in SC membranes, intracellular ceramide and its metabolites have a central role in regulating cellular responses to various forms of stress,51 including UV-induced keratinocyte apoptosis.52 Ceramides can be metabolized to generate sphingosine-1-phosphate (S1P), a potent lipid cell-signaling molecule with effects on cell proliferation, calcium homeostasis, cell migration and immune function.53 The degradation of S1P is catalyzed by S1P lyase, an irreversible reaction that yields hexadecenal. This fatty aldehyde is subsequently oxidized to fatty acid by a yet unidentified aldehyde dehydrogenase, possibly FALDH.
Oxidative stress generates several toxic aldehydes, including 4-hydroxynonenal (4-HNE), by peroxidative cleavage of polyunsaturated fatty acids.54 4-HNE is among the most reactive aldehydes and is detoxified via three mechanisms: oxidation to fatty acid, reduction to fatty alcohol and conjugation with glutathione.55 Several aldehyde dehydrogenase isozymes catalyze the oxidation of 4-HNE, including ALDH3A1, which is closely related to FALDH.56,57 Recent studies indicate that overexpression of FALDH protects cultured cells from the cytotoxic effects of 4-HNE, suggesting that FALDH may play a physiologic role in protection of cells from oxidative stress.58,59 The quantitative contribution of FALDH to this process, however, is unclear and may vary among tissues.
With oxidative stress, reactive oxygen species also attack the vinyl ether bond of plasmalogen lipids to release the alkyl chain as fatty aldehyde.60 It is likely, but not proven, that FALDH catalyzes the oxidation of these long-chain aldehydes to fatty acids.
Chlorinated fatty aldehydes (i.e., 2-chloro-hexadecanal) are produced by activated phagocytes as a product of lipid peroxidation.61 Metabolism of 2-chloro-hexadecanal to 2-chloro-hexadecanoic acid has been demonstrated in human endothelial cells,62 and mutant FALDH-deficient Chinese hamster cells have been found to be deficient in this oxidative step.63 It is not known, however, to what extent chlorinated fatty aldehydes are generated in the epidermis.
Fatty acid ω-oxidation.
ω-Oxidation is a pathway for fatty acid degradation in which the ω-terminal end of the aliphatic chain is enzymatically hydroxylated and subsequently oxidized to a carboxyl-group forming a dicarboxylic acid. This alcohol-to-acid conversion proceeds through an aldehyde intermediate (ω-oxo fatty acid) and is analogous to that seen in primary fatty alcohol oxidation (Fig. 3). Evidence is emerging that FAO/FALDH is necessary for ω-oxidation of certain fatty acids, including select eicosanoids. Metabolism of leukotriene B4, a potent eicosanoid inflammatory mediator derived from arachidonic acid, proceeds via P450-mediated hydroxylation of the ω-terminal end of the fatty acid, followed by conversion to a dicarboxylic acid and subsequent degradation in peroxisomes.64 The oxidation of 20-hydroxy-leukotriene B4 to 20-carboxy-leukotriene B4 is catalyzed by FAO/FALDH. This metabolic step is profoundly deficient in SLS patients, and results in tissue accumulation and urinary excretion of leukotriene B4 and 20-hydroxy-leukotriene B4.65 Other eicosanoid lipids also undergo ω-oxidation as the major route of degradation, including the epoxyalcohols66 and certain prostaglandins,67 although FAO/FALDH has not yet been demonstrated to be necessary for their metabolism. In a similar fashion, very long-chain fatty acids can also be ω-oxidized to dicarboxylic acids via a FAO/FALDH-dependent reaction,68 but this represents a minor route for its degradation. In light of these studies, it is possible that FAO/FALDH is involved in metabolism of additional fatty acids that undergo ω-oxidation.
Biochemical Pathogenesis of Epidermal Dysfunction in SLS
The biochemical mechanisms responsible for epidermal dysfunction in SLS are undoubtedly complex. In SLS patients, FALDH activity is deficient in all epidermal cells. Nevertheless, histologic studies of SLS skin point to the SG as the most relevant site of pathogenesis, and abnormalities in LB structure and secretion as the primary target with defective SC membrane arrays as a consequential effect.
Which biochemical abnormalities in SLS might be responsible for the defective LB structure and secretion? Owing to the role of FALDH in several fatty alcohol/aldehyde pathways (Table 1) and very limited biochemical studies of SLS skin, any discussion of pathogenic mechanisms will necessarily appear speculative. It is, however, unlikely that perturbations in all potential lipid pathways contribute equally to the epidermal dysfunction. In most lipid pathways, FALDH acts in a catabolic role to degrade its alcohol/aldehyde substrate, which suggests that accumulation of a toxic lipid is more likely to be pathogenic to the skin than failure to produce a key lipid product. Nevertheless, the epidermal pathogenesis may arise from one or more lipid abnormalities, including (1) accumulation of fatty alcohol or its metabolic products, (2) toxic effects of fatty aldehydes, (3) defective isoprenol oxidation, (4) impaired fatty acid ω-oxidation or (5) altered fatty acid and ceramide metabolism.
Table 1.
Lipid pathways affected by FALDH deficiency in SLS patients or FALDH-deficient cells
| Lipid pathway | Aldehyde precursor lipid | FALDH substrate | Abnormality demonstrated in SLS |
| Fatty alcohol oxidation | Straight-chain alcohols | Hexadecanal, octadecanal, octadecenal | Yes (fibroblasts, keratinocytes, plasma) |
| Branched-chain alcohols (Phytol) | Phytenal | Yes (fibroblasts) | |
| Isoprenoid alcohols (Farnesol, Geranylgeraniol) | Farnesal, Geranylgeranal | Yes (fibroblasts) | |
| Ether glycerolipid catabolism | Ether glycerolipids | Hexadecanal, octadecanal | Yes (fibroblasts, keratinocytes) |
| Fatty acid ω-oxidation | Leukotriene B4 → 20-OH-leukotriene B4 | 20-Oxo-leukotriene B4 | Yes (fibroblasts, urine) |
| Very long-chain fatty acids | ω-Oxo-very long-chain fatty acid | Yes (fibroblasts) | |
| Epoxyalcohols 12R-HXA3 → 12R-TXA3 | 20-OH-TXA3 and 20-oxo-TXA3 | Not determined | |
| Oxidative stress | Polyunsaturated fatty acids | 4-Hydroxynonenal, including other aldehydes | Not determined |
| Ether glycerolipids (plasmalogens) | Hexadecanal, Octadecanal | Not determined | |
| Ether glycerolipids (plasmalogens) | 2-Chloro-hexadecanal | Not determined (demonstrated in FALDH-deficient hamster cells) | |
| Ceramide catabolism | Sphingosine-1-phosphate | Hexadecenal | Not determined |
Accumulation of fatty alcohol or its metabolic products.
The epidermal consequences of fatty alcohol accumulation are poorly understood. Fatty alcohols that accumulate in SLS seem to be restricted to those that are 16- to 18-carbons long.31,33,36 Long-chain alcohols have a high partition coefficient for biological membranes and intercalate into cellular membranes rather than exist free in solution.69,70 Hexadecanol has been reported to inhibit lipase activity in vitro,71 and could potentially cause secondary triglyceride accumulation in the epidermis or prevent production of free fatty acids. On the other hand, feeding studies in rodents have revealed no harmful effects of octadecanol, even at high dietary intake. Moreover, topical lotions containing long-chain alcohols do not induce skin abnormalities in normal humans. It is unclear, however, to what extent these lotion-derived alcohols penetrate the SC and accumulate in the lower layers of the skin.
Cultured skin keratinocytes from SLS patients accumulate large amounts of free fatty alcohols, wax esters and 1-O-alkyl-2,3-diacylglycerol, and it is a reasonable expectation that similar biochemical changes occur in vivo. Wax esters34 and 1-O-alkyl-2,3-diacylglycerol35 are usually present on the surface of the skin, but their abnormal accumulation in cultured SLS keratinocytes suggests a potential role in the pathogenesis of defective LB membranes.
Aside from a direct physical effect on keratinocyte membranes, lipid accumulation in SLS may be detrimental by interfering with normal cell signaling pathways in the skin. Epidermal differentiation is induced by certain physiologic agents, including increased intracellular calcium,72 cholesterol sulfate73,74 and 1,25-dihydroxyvitamin D3,75 that act directly or indirectly to increase 1,2-diacylglycerol and thereby stimulate protein kinase C (PKC) activity.76 Neutral ether glycerolipids can also modulate PKC activity. For example, 1-O-alkylglycerol has been shown to inhibit PKC in rat brain,77,78 whereas 1-O-alkyl-2-acylglycerol has the opposite effect.79 1-O-alkyl-2,3-diacylglycerol accumulates in cultured SLS keratinocytes, but its effect on keratinocyte PKC activity is not known. Furthermore, medium-chain fatty alcohols (C8–10) either enhance or inhibit PKC activity in a complex manner.80 The effects of longer chain alcohols on PKC activity have not been reported.
Unlike straight chain fatty alcohols, mice fed large amounts of phytol develop cutaneous and neurologic symptoms after several days, but these symptoms may be mediated solely or in part by increases in phytanic acid.81 Patients with Refsum disease, who are genetically deficient in α-oxidation and store large amounts of phytanic acid in tissues, exhibit ichthyosis.8 Defective oxidation of phytol to phytanic acid occurs in SLS fibroblasts,39 but neither phytanic acid41 or phytol (Rizzo WB, unpublished data) is elevated in plasma from SLS patients. Furthermore, it is unlikely that phytol/phytanic acid metabolism is responsible for the ichthyosis in SLS, since cutaneous disease is already present at birth before these dietary lipids are consumed.
Toxic effects of fatty aldehydes.
Fatty aldehydes are potentially toxic molecules because of their propensity to form covalent adducts with lipids and proteins.54 Long-chain aldehydes that cannot be metabolized by FALDH can form Schiff base adducts with phosphatidylethanolamine (PE), resulting in accumulation of N-alkyl-PE.41,82,83 Formation of N-alky-PE replaces the positively charged amino group of the ethanolamine moiety of PE with an uncharged hydrophobic alkyl side chain. If sufficient amounts of N-alkyl-PE were to accumulate, LB membrane assembly or stability could be affected. Aldehyde adducts with other lipids containing free amino groups could also contribute to the epidermal pathology in SLS.
In addition to lipid adducts, formation of aldehyde adducts with ε-amino groups of lysine residues in epidermal proteins could have a more widespread effect on epidermal function. To date, however, aldehyde-protein adducts have not been investigated in SLS.
Defective isoprenol oxidation.
Defective farnesol oxidation in SLS may have several biologically important effects on the skin. Farnesol induces differentiation of cultured human keratinocytes via a peroxisome proliferator activated receptor-α (PPARα) dependent mechanism and stimulates transcription of epidermal-specific genes.84 However, activation of PPARα and related PPAR isoforms (PPARβ/δ and PPARγ) tend to stimulate lipid synthesis genes in the epidermis and enhance formation of the permeability barrier.85 It is possible, however, that dysregulation of PPAR-sensitive gene expression by farnesol could occur in the skin and contribute to the pathogenesis of SLS.
Farnesol also has the ability to decrease cholesterol synthesis by promoting the degradation of HMG-CoA reductase86 and inhibiting mevalonate kinase.87 In rodents, topical application of lovastatin, which inhibits HMG-CoA reductase, results in abnormal LB formation, a leaky epidermal water barrier and an ichthyotic appearance.88,89 Although the mechanism for this cutaneous response may be more complex than simply reduction in cholesterol synthesis, farnesol accumulation in SLS skin may mimic the effects of lovastatin on HMG-CoA reductase and cause a similar cutaneous phenotype.
Defective fatty acid ω-oxidation.
It has been speculated that the ichthyosis in SLS arises from defective metabolism of 12R-epoxyalcohol lipids (hepoxilins) that are derived from arachidonic acid.90 Hepoxilins exist as R- or S-isomers, and have potent biological activities that are mediated via an intracellular G-coupled protein receptor.91,92 Genetic defects that cause ichthyosis have been identified in two enzymes, 12R-lipoxygenase and hepoxilin synthase, that sequentially act to synthesize the epoxyalcohol commonly known as (R)-hepoxilin-A3 (abbreviated HXA3).93,94 Subsequent metabolism of HXA3 forms (R)-trioxilin-A3 (abbreviated TXA3) that, in turn, is metabolized by ω-oxidation.66 TXA3 is initially ω-hydroxylated to 20-hydroxy-TXA3 and subsequently oxidized to 20-carboxy-TXA3, analogous to the ω-oxidation of leukotriene B4 that is deficient in SLS. Since genetic defects in synthesis of HXA3 cause ichthyosis, it follows that HXA3 or one of its metabolites is necessary for normal epidermal development. In fact, TXA3 has been found to be a ligand for PPARα95 and may act, like farnesol, in stimulating epidermal gene transcription. If FAO were necessary for 20-hydroxy-TXA3 oxidation, accumulation of TXA3 and its ω-hydroxylated metabolite is expected to induce overexpression of PPARα-sensitive genes, which might disrupt lipid metabolism in keratinocytes. Alternately, if 20-carboxy-TXA3 (or its downstream metabolite) is the key lipid in this pathway required for normal epidermal differentiation, its decreased production in SLS may lead to ichthyosis, similar to the other genetic defects in the HXA3 pathway. The inference that the ichthyosis arises from defective epoxyalcohol metabolism, however, begs experimental support, since other enzymes can catalyze the oxidation of 20-hydroxy-eicosanoids.96
Although not involved in keratinocyte differentiation, accumulation of leukotriene B4 and/or 20-hydroxy-leukotriene B4 in SLS due to FAO/FALDH deficiency is likely responsible for the pruritus seen in SLS patients.97 This conclusion is supported by experimental studies in mice showing that the intradermal injection of leukotriene B4 causes itching.98 In SLS patients, therapeutic reductions in leukotriene B4 levels can be achieved by pharmacologically blocking its synthesis, which leads to clinical improvement in the pruritus of some patients.99
Altered fatty acid and ceramide metabolism.
The free fatty acids in the SC membranes largely originate from hydrolysis of phospholipids during epidermal differentiation.100 It is unlikely that the oxidation of alcohol/aldehyde lipids by FAO/FALDH produces a substantial amount of fatty acids for synthesis of SC membranes, although the relative contribution of this enzyme to the epidermal fatty acid pool has not been measured.
There is evidence, however, for indirect effects of FALDH deficiency on fatty acid metabolism. Certain polyunsaturated fatty acids that arise from δ-6 desaturation of linoleic acid (C18:2) are reduced in SLS serum.101 Linoleic acid itself is an essential fatty acid that is incorporated into acylceramide (ceramide-1).50 This ceramide is important for epidermal barrier function as underscored by a reduced ceramide-1 content and cutaneous desquamation seen in essential fatty acid deficiency.102 However, linoleic acid levels are normal in SLS serum.101 Moreover, SLS fibroblasts have normal δ-6 desaturase activity104 and cultured SLS keratinocytes exhibit no abnormalities in polyunsaturated fatty acids,36 suggesting that the serum fatty acid abnormalities are secondary alterations.
Scales from SLS patients have mildly reduced levels of ceramide-1 and ceramide-6,103 but a direct metabolic connection between FALDH deficiency and ceramide synthesis in skin is not known. Nor is it known whether the fatty aldehyde produced from ceramide degradation influences S1P or intracellular ceramide levels in keratinocytes. The reduced ceramide content in scales of SLS patients may nevertheless contribute to the water barrier defect, especially in combination with other lipid changes.
Conclusion
In summary, the available evidence suggests that the cutaneous pathogenesis of SLS originates from one or more lipid abnormalities that disrupts the formation and secretion of LBs, and leads to defective SC membranes and a leaky water barrier. It is likely that the skin responds by a process of reactive hyperproliferation in an attempt to restore the water barrier by making more defective SC, which results in hyperkeratosis and overt ichthyosis. FALDH deficiency causes several potentially deleterious lipid abnormalities. To date, however, no single lipid pathway has emerged as primarily responsible for the cutaneous disease and it is possible that multiple lipid abnormalities are involved. For understanding the epidermal pathogenesis, it will be critically important to better define the lipid composition of the skin of SLS patients and to develop an animal model for investigation. The potential contribution of the lipid abnormalities to signaling pathways and epidermal gene expression are intriguing possibilities that remain to be explored. Nevertheless, elucidation of the biochemical mechanisms resulting in abnormal epidermal structure and function should lead to the development of rational, targeted therapeutic approaches for the cutaneous symptoms of SLS.
Acknowldgements
The author gratefully acknowledges support from grant AR044552 from the National Institute of Arthritis, Musculoskeletal and Skin Diseases of the NIH; the Sjögren-Larsson Syndrome Research Fund of the University of Nebraska Foundation; and the Foundation for Ichthyosis and Related Skin Types.
Abbreviations
- FALDH
fatty aldehyde dehydrogenase
- FAO
fatty alcohol:NAD oxidoreductase
- HMG-CoA
hydroxymethylglutaryl-CoA
- 4-HNE
4-hydroxynonenal
- HXA3
(R)-hepoxilin-A3
- LB
lamellar body
- PE
phosphatidylethanolamine
- PKC
protein kinase C
- PPARα
peroxisome proliferator activated receptor-α
- SC
stratum corneum
- SG
stratum granulosum
- SLS
Sjögren-Larsson syndrome
- S1P
sphingosine-1-phosphate
- TXA3
(R)-trioxilin-A3
References
- 1.Elias PM, Menon GK. Structural and lipid biochemical correlates of the epidermal permeability barrier. Adv Lipid Res. 1991;24:1–26. doi: 10.1016/b978-0-12-024924-4.50005-5. [DOI] [PubMed] [Google Scholar]
- 2.Madison KC. Barrier function of the skin: “la raison d'être” of the epidermis. J Invest Dermatol. 2003;121:231–241. doi: 10.1046/j.1523-1747.2003.12359.x. [DOI] [PubMed] [Google Scholar]
- 3.Feingold KR. Thematic review series: skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J Lipid Res. 2007;48:2531–2546. doi: 10.1194/jlr.R700013-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Feingold KR. The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res. 2009;50:417–422. doi: 10.1194/jlr.R800039-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao-Qiang M, Elias PM, Feingold KR. Fatty acids are required for epidermal permeability barrier function. J Clin Invest. 1993;92:791–798. doi: 10.1172/JCI116652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feingold KR, Man MQ, Menon GK, Cho SS, Brown BE, Elias PM. Cholesterol synthesis is required for cutaneous barrier function in mice. J Clin Invest. 1990;86:1738–1745. doi: 10.1172/JCI114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holleran WM, Man MQ, Gao WN, Menon GK, Elias PM, Feingold KR. Sphingolipids are required for mammalian epidermal barrier function. Inhibition of sphingolipid synthesis delays barrier recovery after acute perturbation. J Clin Invest. 1991;88:1338–1345. doi: 10.1172/JCI115439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizzo WB. The metabolic & molecular bases of inherited disease. New York, NY: McGraw-Hill; 2001. Sjogren-Larsson syndrome: fatty aldehyde dehydrogenase deficiency; pp. 2239–2258. [Google Scholar]
- 10.Sjögre T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorders. Acta Psychiatr Neurol Scand. 1957;32:1–113. [PubMed] [Google Scholar]
- 11.De Laurenzi V, Rogers GR, Hamrock DJ, Marekov LN, Steinert PM, Compton JG, et al. Sjögren-Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52–57. doi: 10.1038/ng0196-52. [DOI] [PubMed] [Google Scholar]
- 12.Rizzo WB, Carney G. Sjögren-Larsson syndrome: diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2) Hum Mutat. 2005;26:1–10. doi: 10.1002/humu.20181. [DOI] [PubMed] [Google Scholar]
- 13.Rizzo WB, Craft DA. Sjögren-Larsson syndrome. Deficient activity of the fatty aldehyde dehydrogenase component of fatty alcohol:NAD+ oxidoreductase in cultured fibroblasts. J Clin Invest. 1991;88:1643–1648. doi: 10.1172/JCI115478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelson TL, Secor McVoy JR, Rizzo WB. Human liver fatty aldehyde dehydrogenase: microsomal localization, purification and biochemical characterization. Biochim Biophys Acta. 1997;1335:99–110. doi: 10.1016/s0304-4165(96)00126-2. [DOI] [PubMed] [Google Scholar]
- 15.Keller MA, Watschinger K, Golderer G, Maglione M, Sarg B, Lindner HH, et al. Monitoring of fatty aldehyde dehydrogenase by formation of pyrenedecanoic acid from pyrenedecanal. J Lipid Res. 2010;51:1554–1559. doi: 10.1194/jlr.D002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers GR, Markova NG, De Laurenzi V, Rizzo WB, Compton JG. Genomic organization and expression of the human fatty aldehyde dehydrogenase gene (FALDH) Genomics. 1997;39:127–135. doi: 10.1006/geno.1996.4501. [DOI] [PubMed] [Google Scholar]
- 17.Masaki R, Yamamoto A, Tashiro Y. Microsomal aldehyde dehydrogenase is localized to the endoplasmic reticulum via its carboxyl-terminal 35 amino acids. J Cell Biol. 1994;126:1407–1420. doi: 10.1083/jcb.126.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashibe B, Hirai T, Higashi K, Sekimizu K, Motojima K. Dual subcellular localization in the endoplasmic reticulum and peroxisomes and a vital role in protecting against oxidative stress of fatty aldehyde dehydrogenase are achieved by alternative splicing. J Biol Chem. 2007;282:20763–20773. doi: 10.1074/jbc.M611853200. [DOI] [PubMed] [Google Scholar]
- 19.Rizzo WB, S'Aulis D, Jennings MA, Crumrine DA, Williams ML, Elias PM. Ichthyosis in Sjögren-Larsson syndrome reflects defective barrier function due to abnormal lamellar body structure and secretion. Arch Dermatol Res. 2010;302:443–451. doi: 10.1007/s00403-009-1022-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Judge MR, Lake BD, Smith VV, Besley GT, Harper JI. Depletion of alcohol (hexanol) dehydrogenase activity in the epidermis and jejunal mucosa in Sjögren-Larsson syndrome. J Invest Dermatol. 1990;95:632–634. doi: 10.1111/1523-1747.ep12514294. [DOI] [PubMed] [Google Scholar]
- 21.Jagell S, Lidén S. Ichthyosis in the Sjögren-Larsson syndrome. Clin Genet. 1982;21:243–252. doi: 10.1111/j.1399-0004.1982.tb00758.x. [DOI] [PubMed] [Google Scholar]
- 22.Auada MP, Adam RL, Leite NJ, Puzzi MB, Cintra ML, Rizzo WB, Metze K. Texture analysis of the epidermis based on fast Fourier transformation in Sjögren-Larsson syndrome. Anal Quant Cytol Histol. 2006;28:219–227. [PMC free article] [PubMed] [Google Scholar]
- 23.Shibaki A, Akiyama M, Shimizu H. Novel ALDH3A2 heterozygous mutations are associated with defective lamellar granule formation in a Japanese family of Sjögren-Larsson syndrome. J Invest Dermatol. 2004;123:1197–1199. doi: 10.1111/j.0022-202X.2004.23505.x. [DOI] [PubMed] [Google Scholar]
- 24.Matsuoka LY, Kousseff BG, Hashimoto K. Studies of the skin in Sjögren-Larsson syndrome by electron microscopy. Am J Dermatopathol. 1982;4:295–301. doi: 10.1097/00000372-198208000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Ito M, Oguro K, Sato Y. Ultrastructural study of the skin in Sjögren-Larsson syndrome. Arch Dermatol Res. 1991;283:141–148. doi: 10.1007/BF00372053. [DOI] [PubMed] [Google Scholar]
- 26.Schmuth M, Yosipovitch G, Williams ML, Weber F, Hintner H, Ortiz-Urda S, et al. Pathogenesis of the permeability barrier abnormality in epidermolytic hyperkeratosis. J Invest Dermatol. 2001;117:837–847. doi: 10.1046/j.0022-202x.2001.01471.x. [DOI] [PubMed] [Google Scholar]
- 27.Ichihara K, Kusunose E, Noda Y, Kusunose M. Some properties of the fatty alcohol oxidation system and reconstitution of microsomal oxidation activity in intestinal mucosa. Biochim Biophys Acta. 1986;878:412–418. doi: 10.1016/0005-2760(86)90250-x. [DOI] [PubMed] [Google Scholar]
- 28.Ichihara K, Noda Y, Tanaka C, Kusunose M. Purification of aldehyde dehydrogenase reconstitutively active in fatty alcohol oxidation from rabbit intestinal microsomes. Biochim Biophys Acta. 1986;878:419–425. [PubMed] [Google Scholar]
- 29.Lee T. Characterization of fatty alcohol:NAD+ oxidoreductase from rat liver. J Biol Chem. 1979;254:2892–2896. [PubMed] [Google Scholar]
- 30.Rizzo WB, Craft DA, Dammann AL, Phillips MW. Fatty alcohol metabolism in cultured human fibroblasts. Evidence for a fatty alcohol cycle. J Biol Chem. 1987;262:17412–17419. [PubMed] [Google Scholar]
- 31.Rizzo WB, Craft DA. Sjögren-Larsson syndrome: accumulation of free fatty alcohols in cultured fibroblasts and plasma. J Lipid Res. 2000;41:1077–1081. [PubMed] [Google Scholar]
- 32.Rizzo WB, Dammann AL, Craft DA, Black SH, Tilton AH, Africk D, et al. Sjögren-Larsson syndrome: inherited defect in the fatty alcohol cycle. J Pediatr. 1989;115:228–234. doi: 10.1016/s0022-3476(89)80070-8. [DOI] [PubMed] [Google Scholar]
- 33.Rizzo WB, Dammann AL, Craft DA. Sjögren-Larsson syndrome. Impaired fatty alcohol oxidation in cultured fibroblasts due to deficient fatty alcohol:nicotinamide adenine dinucleotide oxidoreductase activity. J Clin Invest. 1988;81:738–744. doi: 10.1172/JCI113379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolaides N. The monoene and other Wax alcohols of human skin surface lipid and their relation to the fatty acids of this lipid. Lipids. 1967;2:266–275. doi: 10.1007/BF02532567. [DOI] [PubMed] [Google Scholar]
- 35.Oku H, Shudo J, Mimura K, Haratake A, Nagata J, Chinen I. 1-O-alkyl-2,3-diacylglycerols in the skin surface lipids of the hairless mouse. Lipids. 1995;30:169–172. doi: 10.1007/BF02538271. [DOI] [PubMed] [Google Scholar]
- 36.Rizzo WB, Craft DA, Somer T, Carney G, Trafrova J, Simon M. Abnormal fatty alcohol metabolism in cultured keratinocytes from patients with Sjögren-Larsson syndrome. J Lipid Res. 2008;49:410–419. doi: 10.1194/jlr.M700469-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oku H, Shudo J, Nagata J, Chinen I. Accumulation of 1-o-alkyl-2,3-diacylglycerols in cultured rat keratinocytes. Biochim Biophys Acta. 1996;1300:35–41. doi: 10.1016/0005-2760(95)00244-8. [DOI] [PubMed] [Google Scholar]
- 38.van den Brink DM, Wanders RJ. Phytanic acid: production from phytol, its breakdown and role in human disease. Cell Mol Life Sci. 2006;63:1752–1765. doi: 10.1007/s00018-005-5463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Brink DM, van Miert JN, Dacremont G, Rontani JF, Jansen GA, Wanders RJ. Identification of fatty aldehyde dehydrogenase in the breakdown of phytol to phytanic acid. Mol Genet Metab. 2004;82:33–37. doi: 10.1016/j.ymgme.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Jansen GA, Wanders RJ. Alpha-oxidation. Biochim Biophys Acta. 2006;1763:1403–1412. doi: 10.1016/j.bbamcr.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Verhoeven NM, Jakobs C, Carney G, Somers MP, Wanders RJ, Rizzo WB. Involvement of microsomal fatty aldehyde dehydrogenase in the alpha-oxidation of phytanic acid. FEBS Lett. 1998;429:225–228. doi: 10.1016/s0014-5793(98)00574-2. [DOI] [PubMed] [Google Scholar]
- 42.Jansen GA, van den Brink DM, Ofman R, Draghici O, Dacremont G, Wanders RJ. Identification of pristanal dehydrogenase activity in peroxisomes: conclusive evidence that the complete phytanic acid alpha-oxidation pathway is localized in peroxisomes. Biochem Biophys Res Commun. 2001;283:674–679. doi: 10.1006/bbrc.2001.4835. [DOI] [PubMed] [Google Scholar]
- 43.Roullet J-B, Steiner R, Rizzo W. Impaired isoprenoid metabolism in Sjögren-Larsson syndrome. J Inherit Metab Dis. 2006;29:>49. [Google Scholar]
- 44.Crick DC, Andres DA, Waechter CJ. Novel salvage pathway utilizing farnesol and geranylgeraniol for protein isoprenylation. Biochem Biophys Res Commun. 1997;237:483–487. doi: 10.1006/bbrc.1997.7145. [DOI] [PubMed] [Google Scholar]
- 45.Joo JH, Jetten AM. Molecular mechanisms involved in farnesol-induced apoptosis. Cancer Lett. 2010;287:123–135. doi: 10.1016/j.canlet.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagan N, Zoeller RA. Plasmalogens: biosynthesis and functions. Prog Lipid Res. 2001;40:199–229. doi: 10.1016/s0163-7827(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 47.Horrocks LA. Plasmalogens and O-alkyl glycerophospholipids. In: Hawthorne JN, editor. Phospholipids. New York: Elsevier Biomedical Press; 1982. pp. 51–93. [Google Scholar]
- 48.Taguchi H, Armarego WL. Glyceryl-ether monooxy-genase [EC 1.14.16.5]. A microsomal enzyme of ether lipid metabolism. Med Res Rev. 1998;18:43–89. doi: 10.1002/(sici)1098-1128(199801)18:1<43::aid-med3>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 49.Rizzo WB, Heinz E, Simon M, Craft DA. Microsomal fatty aldehyde dehydrogenase catalyzes the oxidation of aliphatic aldehyde derived from ether glycerolipid catabolism: implications for Sjögren-Larsson syndrome. Biochim Biophys Acta. 2000;1535:1–9. doi: 10.1016/s0925-4439(00)00077-6. [DOI] [PubMed] [Google Scholar]
- 50.Uchida Y, Holleran WM. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci. 2008;51:77–87. doi: 10.1016/j.jdermsci.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–25850. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 52.Uchida Y, Houben E, Park K, Douangpanya S, Lee YM, Wu BX, et al. Hydrolytic pathway protects against ceramide-induced apoptosis in keratinocytes exposed to UVB. J Invest Dermatol. 2010;130:2472–2480. doi: 10.1038/jid.2010.153. [DOI] [PubMed] [Google Scholar]
- 53.Serra M, Saba JD. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv Enzyme Regul. 2010;50:349–362. doi: 10.1016/j.advenzreg.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 55.Alary J, Guéraud F, Cravedi JP. Fate of 4-hydroxynonenal in vivo: disposition and metabolic pathways. Mol Aspects Med. 2003;24:177–187. doi: 10.1016/s0098-2997(03)00012-8. [DOI] [PubMed] [Google Scholar]
- 56.Pappa A, Estey T, Manzer R, Brown D, Vasiliou V. Human aldehyde dehydrogenase 3A1 (ALDH3A1): biochemical characterization and immunohistochemical localization in the cornea. Biochem J. 2003;376:615–623. doi: 10.1042/BJ20030810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demozay D, Rocchi S, Mas JC, Grillo S, Pirola L, Chavey C, Van Obberghen E. Fatty aldehyde dehydrogenase: potential role in oxidative stress protection and regulation of its gene expression by insulin. J Biol Chem. 2004;279:6261–6270. doi: 10.1074/jbc.M312062200. [DOI] [PubMed] [Google Scholar]
- 59.Demozay D, Mas JC, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes. 2008;57:1216–1226. doi: 10.2337/db07-0389. [DOI] [PubMed] [Google Scholar]
- 60.Stadelmann-Ingrand S, Favreliere S, Fauconneau B, Mauco G, Tallineau C. Plasmalogen degradation by oxidative stress: production and disappearance of specific fatty aldehydes and fatty alpha-hydroxyaldehydes. Free Radic Biol Med. 2001;31:1263–1271. doi: 10.1016/s0891-5849(01)00720-1. [DOI] [PubMed] [Google Scholar]
- 61.Albert CJ, Crowley JR, Hsu FF, Thukkani AK, Ford DA. Reactive chlorinating species produced by myeloperoxidase target the vinyl ether bond of plasmalogens: identification of 2-chlorohexadecanal. J Biol Chem. 2001;276:23733–23741. doi: 10.1074/jbc.M101447200. [DOI] [PubMed] [Google Scholar]
- 62.Wildsmith KR, Albert CJ, Anbukumar DS, Ford DA. Metabolism of myeloperoxidase-derived 2-chlorohexadecanal. J Biol Chem. 2006;281:16849–16860. doi: 10.1074/jbc.M602505200. [DOI] [PubMed] [Google Scholar]
- 63.Anbukumar DS, Shornick LP, Albert CJ, Steward MM, Zoeller RA, Neumann WL, Ford DA. Chlorinated lipid species in activated human neutrophils: lipid metabolites of 2-chlorohexadecanal. J Lipid Res. 2010;51:1085–1092. doi: 10.1194/jlr.M003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murphy RC, Gijón MA. Biosynthesis and metabolism of leukotrienes. Biochem J. 2007;405:379–395. doi: 10.1042/BJ20070289. [DOI] [PubMed] [Google Scholar]
- 65.Willemsen MA, Rotteveel JJ, de Jong JG, Wanders RJ, IJlst L, Hoffmann GF, Mayatepek E. Defective metabolism of leukotriene B4 in the Sjögren-Larsson syndrome. J Neurol Sci. 2001;183:61–67. doi: 10.1016/s0022-510x(00)00474-3. [DOI] [PubMed] [Google Scholar]
- 66.Reynaud D, Rounova O, Demin PM, Pivnitsky KK, Pace-Asciak CR. Hepoxilin A3 is oxidized by human neutrophils into its omega-hydroxy metabolite by an activity independent of LTB4 omega-hydroxylase. Biochim Biophys Acta. 1997;1348:287–298. doi: 10.1016/s0005-2760(97)00064-7. [DOI] [PubMed] [Google Scholar]
- 67.Kikuta Y, Kusunose E, Kusunose M. Prostaglandin and leukotriene omega-hydroxylases. Prostaglandins Other Lipid Mediat. 2002;68:345–362. doi: 10.1016/s0090-6980(02)00039-4. [DOI] [PubMed] [Google Scholar]
- 68.Sanders RJ, Ofman R, Dacremont G, Wanders RJ, Kemp S. Characterization of the human omega-oxidation pathway for omega-hydroxy-very-long-chain fatty acids. FASEB J. 2008;22:2064–2071. doi: 10.1096/fj.07-099150. [DOI] [PubMed] [Google Scholar]
- 69.Franks NP, Lieb WR. Partitioning of long-chain alcohols into lipid bilayers: implications for mechanisms of general anesthesia. Proc Natl Acad Sci USA. 1986;83:5116–5120. doi: 10.1073/pnas.83.14.5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller KW, Firestone LL, Alifimoff JK, Streicher P. Nonanesthetic alcohols dissolve in synaptic membranes without perturbing their lipids. Proc Natl Acad Sci USA. 1989;86:1084–1087. doi: 10.1073/pnas.86.3.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferreira GC, Patton JS. Inhibition of lipolysis by hydrocarbons and fatty alcohols. J Lipid Res. 1990;31:889–897. [PubMed] [Google Scholar]
- 72.Bikle DD, Ng D, Tu CL, Oda Y, Xie Z. Calcium- and vitamin D-regulated keratinocyte differentiation. Mol Cell Endocrinol. 2001;177:161–171. doi: 10.1016/s0303-7207(01)00452-x. [DOI] [PubMed] [Google Scholar]
- 73.Denning MF, Kazanietz MG, Blumberg PM, Yuspa SH. Cholesterol sulfate activates multiple protein kinase C isoenzymes and induces granular cell differentiation in cultured murine keratinocytes. Cell Growth Differ. 1995;6:1619–1626. [PubMed] [Google Scholar]
- 74.Ikuta T, Chida K, Tajima O, Matsuura Y, Iwamori M, Ueda Y, et al. Cholesterol sulfate, a novel activator for the eta isoform of protein kinase C. Cell Growth Differ. 1994;5:943–947. [PubMed] [Google Scholar]
- 75.Bollag WB. Differentiation of human keratinocytes requires the vitamin d receptor and its coactivators. J Invest Dermatol. 2007;127:748–750. doi: 10.1038/sj.jid.5700692. [DOI] [PubMed] [Google Scholar]
- 76.Denning MF. Epidermal keratinocytes: regulation of multiple cell phenotypes by multiple protein kinase C isoforms. Int J Biochem Cell Biol. 2004;36:1141–1146. doi: 10.1016/j.biocel.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 77.Warne TR, Buchanan FG, Robinson M. Growth-dependent accumulation of monoalkylglycerol in Madin-Darby canine kidney cells. Evidence for a role in the regulation of protein kinase C. J Biol Chem. 1995;270:11147–11154. doi: 10.1074/jbc.270.19.11147. [DOI] [PubMed] [Google Scholar]
- 78.McNeely TB, Rosen G, Londner MV, Turco SJ. Inhibitory effects on protein kinase C activity by lipophosphoglycan fragments and glycosylphosphatidylinositol antigens of the protozoan parasite Leishmania. Biochem J. 1989;259:601–604. doi: 10.1042/bj2590601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ford DA, Miyake R, Glaser PE, Gross RW. Activation of protein kinase C by naturally occurring ether-linked diglycerides. J Biol Chem. 1989;264:13818–13824. [PubMed] [Google Scholar]
- 80.Slater SJ, Kelly MB, Larkin JD, Ho C, Mazurek A, Taddeo FJ, et al. Interaction of alcohols and anesthetics with protein kinase Calpha. J Biol Chem. 1997;272:6167–6173. doi: 10.1074/jbc.272.10.6167. [DOI] [PubMed] [Google Scholar]
- 81.Van den Branden C, Vamecq J, Wybo I, Roels F. Phytol and peroxisome proliferation. Pediatr Res. 1986;20:411–415. doi: 10.1203/00006450-198605000-00007. [DOI] [PubMed] [Google Scholar]
- 82.James PF, Zoeller RA. Isolation of animal cell mutants defective in long-chain fatty aldehyde dehydrogenase. Sensitivity to fatty aldehydes and Schiff 's base modification of phospholipids: implications for Sjögren-Larsson syndrome. J Biol Chem. 1997;272:23532–23539. doi: 10.1074/jbc.272.38.23532. [DOI] [PubMed] [Google Scholar]
- 83.Stadelmann-Ingrand S, Pontcharraud R, Fauconneau B. Evidence for the reactivity of fatty aldehydes released from oxidized plasmalogens with phosphatidylethanolamine to form Schiff base adducts in rat brain homogenates. Chem Phys Lipids. 2004;131:93–105. doi: 10.1016/j.chemphyslip.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 84.Hanley K, Kömüves LG, Ng DC, Schoonjans K, He SS, Lau P, et al. Farnesol stimulates differentiation in epidermal keratinocytes via PPARalpha. J Biol Chem. 2000;275:11484–11491. doi: 10.1074/jbc.275.15.11484. [DOI] [PubMed] [Google Scholar]
- 85.Schmuth M, Jiang YJ, Dubrac S, Elias PM, Feingold KR. Thematic review series: skin lipids. Peroxisome proliferator-activated receptors and liver X receptors in epidermal biology. J Lipid Res. 2008;49:499–509. doi: 10.1194/jlr.R800001-JLR200. [DOI] [PubMed] [Google Scholar]
- 86.Meigs TE, Roseman DS, Simoni RD. Regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation by the nonsterol mevalonate metabolite farnesol in vivo. J Biol Chem. 1996;271:7916–7922. doi: 10.1074/jbc.271.14.7916. [DOI] [PubMed] [Google Scholar]
- 87.Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res. 1997;38:2216–2223. [PubMed] [Google Scholar]
- 88.Menon GK, Feingold KR, Mao-Qiang M, Schaude M, Elias PM. Structural basis for the barrier abnormality following inhibition of HMG CoA reductase in murine epidermis. J Invest Dermatol. 1992;98:209–219. doi: 10.1111/1523-1747.ep12555880. [DOI] [PubMed] [Google Scholar]
- 89.Feingold KR, Man MQ, Proksch E, Menon GK, Brown BE, Elias PM. The lovastatin-treated rodent: a new model of barrier disruption and epidermal hyperplasia. J Invest Dermatol. 1991;96:201–209. doi: 10.1111/1523-1747.ep12461153. [DOI] [PubMed] [Google Scholar]
- 90.Lefèvre C, Bouadjar B, Ferrand V, Tadini G, Mégarbané A, Lathrop M, et al. Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet. 2006;15:767–776. doi: 10.1093/hmg/ddi491. [DOI] [PubMed] [Google Scholar]
- 91.Nigam S, Zafiriou MP, Deva R, Ciccoli R, Roux-Van der Merwe R. Structure, biochemistry and biology of hepoxilins: an update. FEBS J. 2007;274:3503–3512. doi: 10.1111/j.1742-4658.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- 92.Pace-Asciak CR, Reynaud D, Demin P, Nigam S. The hepoxilins. A review. Adv Exp Med Biol. 1999;447:123–132. [PubMed] [Google Scholar]
- 93.Yu Z, Schneider C, Boeglin WE, Brash AR. Mutations associated with a congenital form of ichthyosis (NCIE) inactivate the epidermal lipoxygenases 12RLOX and eLOX3. Biochim Biophys Acta. 2005;1686:238–247. doi: 10.1016/j.bbalip.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 94.Jobard F, Lefèvre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, et al. Lipoxygenase-3 (ALOXE3) and 12®-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11:107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- 95.Yu Z, Schneider C, Boeglin WE, Brash AR. Epidermal lipoxygenase products of the hepoxilin pathway selectively activate the nuclear receptor PPARalpha. Lipids. 2007;42:491–497. doi: 10.1007/s11745-007-3054-4. [DOI] [PubMed] [Google Scholar]
- 96.Collins XH, Harmon SD, Kaduce TL, Berst KB, Fang X, Moore SA, et al. Omega-oxidation of 20-hydroxyeicosatetraenoic acid (20-HETE) in cerebral microvascular smooth muscle and endothelium by alcohol dehydrogenase 4. J Biol Chem. 2005;280:33157–33164. doi: 10.1074/jbc.M504055200. [DOI] [PubMed] [Google Scholar]
- 97.Willemsen MA, de Jong JG, van Domburg PH, Rotteveel JJ, Wanders RJ, Mayatepek E. Defective inactivation of leukotriene B4 in patients with Sjögren-Larsson syndrome. J Pediatr. 2000;136:258–260. doi: 10.1016/s0022-3476(00)70113-2. [DOI] [PubMed] [Google Scholar]
- 98.Ando T, Katsube N, Maruyama M, Kuraishi Y. Involvement of leukotriene B(4) in substance P-induced itch-associated response in mice. J Invest Dermatol. 2001;117:1621–1626. doi: 10.1046/j.0022-202x.2001.01585.x. [DOI] [PubMed] [Google Scholar]
- 99.Willemsen MA, Lutt MA, Steijlen PM, Cruysberg JR, van der Graaf M, Nijhuis-van der Sanden MW, et al. Clinical and biochemical effects of zileuton in patients with the Sjögren-Larsson syndrome. Eur J Pediatr. 2001;160:711–717. doi: 10.1007/s004310100838. [DOI] [PubMed] [Google Scholar]
- 100.Mao-Qiang M, Feingold KR, Jain M, Elias PM. Extracellular processing of phospholipids is required for permeability barrier homeostasis. J Lipid Res. 1995;36:1925–1935. [PubMed] [Google Scholar]
- 101.Hernell O, Holmgren G, Jagell SF, Johnson SB, Holman RT. Suspected faulty essential fatty acid metabolism in Sjögren-Larsson syndrome. Pediatr Res. 1982;16:45–49. doi: 10.1203/00006450-198201001-00009. [DOI] [PubMed] [Google Scholar]
- 102.Wertz PW, Swartzendruber DC, Abraham W, Madison KC, Downing DT. Essential fatty acids and epidermal integrity. Arch Dermatol. 1987;123:1381–1384. [PubMed] [Google Scholar]
- 103.Paige DG, Morse-Fisher N, Harper JI. Quantification of stratum corneum ceramides and lipid envelope ceramides in the hereditary ichthyoses. Br J Dermatol. 1994;131:23–27. doi: 10.1111/j.1365-2133.1994.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 104.Avigan J, Campbell BD, Yost DA, Hernell O, Holmgren G, Jagell SF. Sjögren-Larsson syndrome: delta 5-and delta 6-fatty acid desaturases in skin fibroblasts. Neurology. 1985;35:401–403. doi: 10.1212/wnl.35.3.401. [DOI] [PubMed] [Google Scholar]