

Abstract
Background
We examined the dose-related influence of alcohol consumption on cerebral ischemia/reperfusion (I/R) injury and the potential mechanism that accounts for the disparate effects of high dose and low dose alcohol consumption on cerebral I/R injury.
Methods
Sprague–Dawley rats were fed a liquid diet without or with 1%, 3%, 5% or 6.4% (v/v) alcohol for 8 weeks, and subjected to a 2-hour middle cerebral artery occlusion (MCAO). We evaluated the brain injury at 24 hours of reperfusion. In addition, we measured protein expression of NMDA receptor and excitatory amino acid transporters (EAATs) in parietal cortex and the effect of NMDA receptor antagonist, memantine, on 2-hour MCAO)/24-hour reperfusion-induced brain injury.
Results
Compared with nonalcohol-fed rats, the total infarct volume was not altered in 3% and 5% alcohol-fed rats, but significantly reduced in 1% alcohol-fed rats and exacerbated in 6.4% alcohol-fed rats. Expression of the NMDA receptor subunit, NR1 was upregulated in 6.4% alcohol-fed rats, whereas expression of EAAT2 was downregulated in 6.4% alcohol-fed rats and upregulated in 1% alcohol-fed rats. Memantine reduced 2-hour MCAO/24-hour reperfusion-induced brain injury in nonalcohol-fed and 6.4% alcohol-fed rats, but not in 1% alcohol-fed rats. The magnitude of reduction in the brain injury was greater in 6.4% alcohol-fed rats compared to nonalcohol-fed rats.
Conclusions
Our findings suggest that chronic consumption of low dose alcohol protects the brain against I/R injury, whereas chronic consumption of high dose alcohol has detrimental effect on cerebral I/R injury. The disparate effects of low dose and high dose alcohol consumption on cerebral I/R may be related to an alteration in NMDA excitotoxicity.
Keywords: Alcohol, brain, ischemia, NMDA receptor, excitatory amino acid transporter
INTRODUCTION
Ischemic stroke is one of the leading causes of death and permanent disability. Alcohol is one of the most commonly used chemical substances, and alcohol may have dose-related influences on the brain. Epidemiological studies suggest that heavy alcohol consumption is a major risk factor for ischemic stroke (Caicoya et al., 1999; Gill et al., 1986; Hillbom and Kaste, 1983), whereas light-moderate consumption of alcohol reduces the morbidity of ischemic stroke (Collins et al., 2009; Gill et al., 1986). In addition, heavy alcohol consumption worsens the outcome of ischemic stroke (Caicoya et al., 1999; Hillbom and Kaste, 1983; Ikehara et al., 2008), whereas light-moderate alcohol consumption reduces mortality (Hansagi et al., 1995; Ikehara et al., 2008) and infarct volume (Caicoya et al., 1999) from ischemic stroke. Recently, a prospective cohort study in men found a beneficial effect of light alcohol consumption on functional outcome from ischemic stroke (Rist et al., 2010). However, there is a paucity of experimental data regarding the influence of alcohol consumption on the consequence of ischemic stroke. Recently, we examined the effect of high dose alcohol consumption on brain injury induced by transient focal cerebral ischemia (Sun et al., 2008). We found that 8-week consumption of 6.4% (v/v) alcohol diet significantly exacerbates cerebral I/R injury in rats. However, the influence of low dose alcohol consumption on cerebral I/R injury has not been experimentally investigated. Thus, our first goal of the present study was to determine the dose-related influence of alcohol consumption on cerebral I/R injury.
Glutamate excitotoxicity is a major pathway triggering brain damage following ischemic stroke (McCulloch, 1994). Due to the high Ca2+ permeability, NMDA receptors play a pivotal role in glutamate excitotoxicity following cerebral ischemia (Sattler and Tymianski, 2001). Previous studies including ours have shown that chronic consumption of high dose alcohol upregulates NMDA receptors in the brain (Fadda and Rossetti, 1998; Harper and Matsumoto, 2005; Sun et al., 2002). In addition, chronic consumption of high dose alcohol has been reported to inhibit EAATs (Kim et al., 2005; Park et al., 2008). Thus, it is possible that exacerbated cerebral I/R injury during chronic consumption of high dose alcohol is related to an increased NMDA excitotoxicity. Thus, our second goal of the present study was to determine the role of NMDA exitotoxicity in altered cerebral I/R injury during chronic alcohol consumption. We measured protein expression of the NMDA receptor, EAAT1, EAAT2, and EAAT3 in parietal cortex. In addition, we examined the effect of NMDA antagonist, memantine, on 2-hour MCAO/24-hour reperfusion-induced brain injury.
MATERIALS AND METHODS
Animal models of chronic alcohol consumption
All procedures were in accordance with the “Principle of Laboratory Animal Care” (NIH publication no. 86–23, revised 1985) and were approved by the Institutional Animal Care and Use Committee. At 2 months of age (body weight 200 to 220 g), male Sprague–Dawley rats (n=66) were separated and divided into five groups, nonalcohol-fed (n=18), 1% alcohol-fed (n=18), 3% alcohol-fed (n=6), 5% alcohol-fed (n=6) and 6.4% alcohol-fed (n=18). Rats were fed a liquid diet with or without alcohol (Dyets, Bethlehem, PA) for 8 weeks. Nonalcohol diet and alcohol diets contained same food calories (1.0 kcal/ml). The alcohol was gradually introduced into the diet over a period of 5 days. The total daily volume of diet given to nonalcohol-fed, 1% alcohol-fed, 3% alcohol-fed, 5% alcohol-fed animals was based upon the daily consumption of diet by 6.4% alcohol-fed animals.
Transient focal cerebral ischemia
Transient focal cerebral ischemia will be performed according to our previous study (Sun et al., 2008a). To avoid any acute effect of alcohol and anesthetic accidents during the surgical procedures, rats were fasted for 8 h before the experiments. On the day of the experiment, the rats were anesthetized with ketamine/xylazine (100/15 mg/kg i.p.). A catheter was placed into a femoral artery for measurement of arterial blood pressure and to obtain a blood sample for the measurement of arterial blood gas. A Laser–Doppler flow probe (PeriFlux System 5000, Perimed) was attached to the right side of the dorsal surface of the skull (2 mm caudal and 6 mm lateral to the bregma) to monitor regional cerebral blood flow (rCBF). A 4–0 monofilament nylon suture was prepared by rounding its tip by heating and coating with silicon. The right common and external carotid arteries were exposed and ligated. The middle cerebral artery (MCA) was occluded by inserting the filament from the basal part of the external carotid artery and advancing it cranially into the internal carotid artery to the point where the MCA branched off from the internal carotid artery. Onset of the MCAO was determined by a rapid drop in rCBF to the cerebral hemisphere. In memantine-treated rats, memantine (20 mg/kg) was administrated intraperitoneally in a volume of 15 ml/kg body weight at 15 minutes after the onset of MCAO. In saline-treated rats, saline 15 ml/kg body weight was injected intraperitoneally at 15 minutes after the onset of MCAO. After occluding the right MCA for 2 hours, reperfusion was initiated by removing the suture. Animals were allowed to recover for 24 hours.
Assessment of the brain injury
Neurological evaluation was performed in all animals before MCAO and at 24 hours of reperfusion using a 24-point scoring system. Sensorimotor testing was graded on a scale of 0–3 each on spontaneous activity, symmetry of movement, response to vibrissae touch, floor walking, beam walking, symmetry of forelimbs, climbing wall of wire cage, reaction to touch on either side of trunk. Neurological deficit scores were assigned as follows: 0, complete deficit; 1, definite deficit with some function; 2, decreased response or mild deficit; 3, no evidence of deficit/symmetrical responses. We have used this system previously (Sun et al., 2008). After neurological evaluation, animals were euthanized with Inactin (150 mg/kg body weight). The brains were quickly removed and placed in ice-cold saline for 5 minutes, and cut into six 2 mm-thick coronal sections. Sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Sigma) for 15 minutes at 37 °C. Slice images were digitalized, the infarct lesion was evaluated using Kodak Molecular Imaging Software. Complete lack of staining is defined as infarct lesion. Infarct lesions corrected for cerebral edema were expressed as percentage of the contralateral hemisphere.
Western Blot
Parietal cortex samples obtained from nonalcohol-fed and alcohol-fed rats without MCAO were homogenized in 20% (wt/vol) ice-cold buffer containing 10 mmol/L Tris-HCl, pH 7.4; 1% SDS; 1 mmol/L sodium vanadate; 10 μg/mL aprotinin; 10 μg/mL leupeptin; and 1 mmol/L phenylmethylsulfonyl fluoride. Next, samples were centrifuged at 12,000 g for 20 minutes at 4°C, and protein concentration in supernatant was determined by the Bradford method (Bio-Rad) with BSA as the standard. SDS polyacrylamide gel electrophoresis (SDS-PAGE) was performed on a 10% gel on which 10–20 μg of total protein per well was loaded. After SDS-PAGE, the proteins were transferred onto polyvinylidene difluoride membrane. Immunoblotting was performed with the use of goat anti-NR1, rabbit anti-EAAT1, goat anti-EAAT2 and rabbit anti-EAAT3 (Santa Cruz, CA, USA) as primary and peroxidase conjugated mouse anti-rabbit and mouse anti-goat IgG as the second antibody. The bound antibody was detected by enhanced chemiluminescence (ECL) detection (Pierce Chemical, Rockford, IL, USA) and the bands were analyzed using UVP BioImaging Systems. Protein loading was controlled by probing all Western blots with rabbit anti-beta-actin antibody (Santa Cruz) and normalizing gp91phox protein intensities to that of beta-actin.
Statistical analysis
For evaluation of statistical significance between two groups, the nonparametric Wilcoxon rank-sum test for unpaired samples was used. For comparison of neurological evaluation between the groups, the nonparametric Kruskal Wallis test was performed first, followed by posthoc pairwise comparisons with Bonferroni adjustment. Neurological scores are present as median and the 25th (Q.25) and 75th (Q.75) percentile values. Other values are presented as means ± SEM. A p value of 0.05 or less was considered to be significant.
RESULTS
Control conditions
Alcohol diets did not alter body weight, mean arterial pressure or heart rate. In addition, there were no significant difference in pH, PCO2 and PO2 of arterial blood during MCAO among nonalcohol-fed and alcohol-fed groups. The peak concentration of blood alcohol was reached at 1 hour after feeding the alcohol diets (0.9 ± 0.4 mM in 1% alcohol-fed rats, 5.9 ± 0.7 mM in 3% alcohol-fed rats, 27.4 ± 0.9 mM in 5% alcohol-fed rats and 29.7 ± 3.6 mM in 6.4% alcohol-fed rats). Daily alcohol intake was 1.4 ± 0.3 g/kg in 1% alcohol-fed rats, 4.2 ± 0.9 g/kg in 3% alcohol-fed rats, 7.0 ± 1.5 g/kg in 5% alcohol-fed rats and 9.2 ± 1.7 g/kg in 6.4% alcohol-fed rats.
MCAO/reperfusion-induced brain injury
The total infarct volume induced by 2-hour MCAO/24-hour reperfusion was 32.4 ( 2.0% of controlateral hemisphere in nonalcohol-fed rats (Figure 1). There was a significant decrease in the total infarct volume observed in 1% alcohol-fed rats (20.3 ( 2.3%) compared to nonalcohol-fed rats. In contrast, a significant increase in total infarct volume was found in 6.4% alcohol-fed rats (42.9 (1.9%) compared to nonalcohol-fed rats. Total infarct volume was not significantly changed in 3% alcohol-fed (30.1 (5.6%) and 5% alcohol-fed (35.2 (3.2%) rats when compared to nonalcohol-fed rats (Figure 1).
Figure 1.
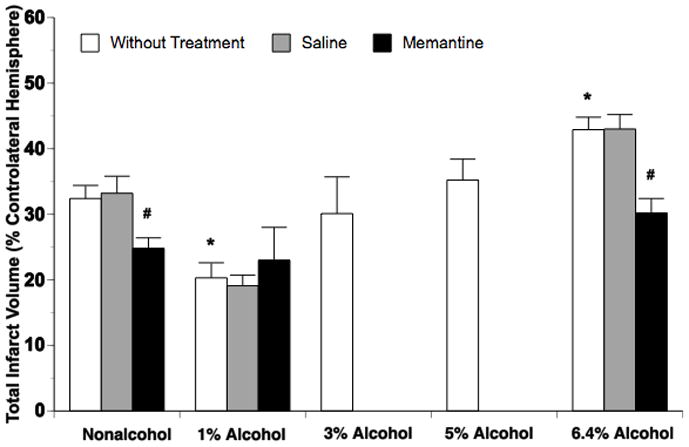
Total infarct volume at 24 hours of reperfusion after a 2-hour MCAO in nonalcohol-fed and 1%, 3%, 5% or 6.4% alcohol-fed rats treated without and with saline or memantine. Values are means ± SE for 6 rats in each group. *P < 0.05 vs. Nonalcohol. #P < 0.05 vs. Without Treatment.
Acute treatment with memantine did not alter the total infarct volume in 1% alcohol-fed rats, but significantly reduced total infarct volume in nonalcohol-fed and 6.4% alcohol-fed rats. The magnitude of reduction in the total infarct volume was greater in 6.4% alcohol-fed rats (12.7%) compared to nonalcohol-fed rats (7.6%) (Figure 1). Consist with the findings regarding total infarct volume, the neurological deficits were significantly improved in 1% alcohol-fed rats and exacerbated in 6.4% alcohol-fed rats. Memantine did not alter 2-hour MCAO/24-hour reperfusion-induced neurological deficits in 1% alcohol-fed rats, but significantly improved the neurological deficits in nonalcohol-fed and 6.4% alcohol-fed rats (Table 1).
Table 1.
Neurological Scores (Median (Q.25/Q.75))
| Without Treatment (n=6) | Saline (n=6) | Memantine (n=6) | |
|---|---|---|---|
| Nonalcohol | 13.5 (12/15) | 13.0 (12/14) | 18.0 (17/20)b |
| 1% Alcohol | 18.0 (17/19)a | 18.0 (16/19) | 20.5 (15/23)b |
| 6.4% Alcohol | 10.5 (8/12)a | 10.5 (10/13) | 16.5 (15/19) |
p < 0.05 vs. Nonalcohol
p < 0.05 vs. Without Treatment
Protein expression of NMDA receptor and EAATs
Protein expression of NMDA receptor subunit, NR1, and EAAT1-3 in parietal cortex was measured in nonalcohol-fed and alcohol-fed rats without MCAO. NR1 expression in parietal cortex was significantly upregulated in 6.4% alcohol-fed rats compared to nonalcohol-fed rats (Figure 2). In contrast, EAAT2 expression in parietal cortex was significantly upregulated in 1% alcohol-fed rats, but downregulated in 6.4% alcohol-fed rats (Figure 2). Expression of EAAT1 and EAAT3 in parietal cortex was not altered in either 1% alcohol-fed or 6.4% alcohol-fed rats.
Figure 2.
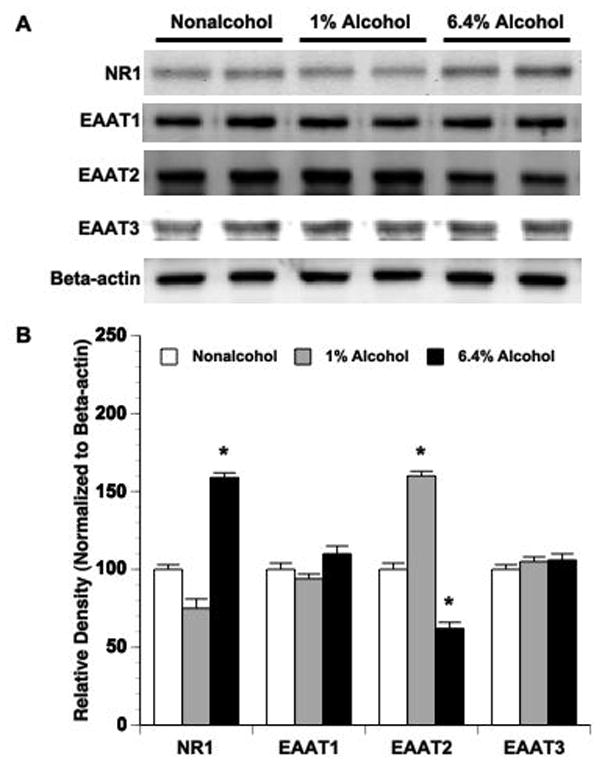
Expression of NR1, EAAT1 and EAAT2 in parietal cortex of nonalcohol-fed, 1% alcohol-fed and 6.4% alcohol-fed rats. (A) Representative Western blots. (B) Values are means ± SE for 6 rats in each group. *P < 0.05 vs. Nonalcohol.
DISCUSSION
There are several new findings from this study. First, in contrast to chronic consumption of high dose alcohol, chronic consumption of low dose alcohol reduces cerebral I/R injury. Second, chronic consumption of high dose alcohol downregulates EAAT2, whereas chronic consumption of low dose alcohol upregulates EAAT2. Third, treatment with an NMDA receptor antagonist can restore exacerbated cerebral I/R injury during chronic consumption of high dose alcohol. We suggest that the disparate effects of low dose and high dose alcohol consumption on cerebral I/R injury may be related to an altered NMDA excitotoxicity.
Although some epidemiological studies found that chronic alcohol consumption may have dose-related influences on the consequence of ischemic stroke (Caicoya et al., 1999; Hansagi et al., 1995; Ikehara et al., 2008), these studies failed to provide detailed information regarding ischemic stroke subtype, ischemic duration and ischemic region. In addition, mechanisms underlying disparate influences of high and low dose alcohol consumption have not been examined. Due to the advancesin intravascular techniques and thrombolytic agents, transient focal cerebral ischemia has become one of the most common types of stroke. Thus, we determined the dose-related influences of chronic alcohol consumption on cerebral I/R injury. Interestingly, 8-week consumption of 6.4% alcohol worsens cerebral I/R injury, whereas 8-week consumption of 1% alcohol protected the brain against I/R injury in rats. Thus, our findings from the present study are consistent with the findings from epidemiological studies. In the present study, the daily intake of alcohol was 1.4 ± 0.3 g/kg and 9.2 ± 1.7 g/kg in 1% alcohol-fed and 6.4% alcohol-fed rats, respectively. In the epidemiological studies, the alcohol quantity required to reduce infarct volume and mortality from ischemic stroke in humans was up to 30 g/day (Caicoya et al., 1999), and the lowest alcohol quantity to increase infarct volume and mortality from ischemic stroke in humans was 69 g/day (Ikehara et al., 2008). However, if both numbers are presented according to the body weight, they will be much smaller than those found in rats. Although ischemic stroke subtype might be a contributing factor, the difference in alcohol metabolism between humans and rodents may be a major reason (von Wartburg, 1976).
To determine possible mechanisms that underlie altered cerebral I/R injury during chronic alcohol consumption, we elected to measure expression of the NMDA receptor and EAATs in the parietal cortex. Overactivation of NMDA receptors induced by glutamate is known to play a predominant role in the pathogenesis of ischemic brain damage (McCulloch, 1994; Meldrum, 2000; Sattler and Tymianski, 2001). Previous studies including ours have found that chronic consumption of high dose alcohol up-regulates NMDA receptor (Fadda and Rossetti, 1998; Harper and Matsumoto, 2005; Sun et al., 2002). In the present study, we found that chronic consumption of high dose alcohol significantly upregulates NR1, an obligatory subunit of NMDA receptor (Gascon et al., 2005), in the parietal cortex. Thus, it is possible that chronic alcohol consumption influences the consequence of transient focal cerebral ischemia via altering NMDA receptor expression. This possibility is further supported by experiments using the NMDA receptor antagonist. A number of NMDA receptor antagonists including MK-801 and memantine were developed and provided substantial protection against cerebral I/R injury (Gorgulu et al., 2000; Simon et al., 1984; Yang et al., 1994; Zhao et al., 2008b). However, MK-801 causes schizophrenic symptoms (Olney et al., 1991). Memantine does not have those toxicities and does not stimulate acetylcholine release in the cerebral cortex. We found that acute treatment with memantine significantly reduced cerebral I/R injury in nonalcohol-fed and high dose alcohol-fed rats, but not in low dose alcohol-fed rats. In addition, the magnitude of reduction in cerebral I/R injury was greater in high dose alcohol-fed rats compared nonalcohol-fed rats. Thus, the NMDA receptor may play an important role in the disparate influences of low dose and high dose alcohol consumption on cerebral I/R injury. In addition to the inhibitory effect on NMDA receptors, however, memantine has been reported to block alpha 7 nicotinic acetylcholine receptors (α7nAChRs) in rat hippocampal neurons (Aracava et al., 2005). Neuronal α7nAChRs has been reported to be involved in neuroprotection against ischemia (Hejmadi et al., 2003). Thus, the inhibitory effect of memantine on neuronal α7nAChRs may suppress its neuroprotective effect against I/R injury.
EAATs control the extracellular glutamate concentration (Takahashi et al., 1997). To date, five subtypes of EAATs, EAAT1-5, have been identified. EAAT1 is expressed in astrocytes throughout the brain. EAAT2 and EAAT3 are mainly expressed in astrocytes and neurons, respectively. EAAT2 is the main regulator of extracellular glutamate levels, contributing to >90% of transport activity in most brain regions (Danbolt, 2001). EAAT4 and EAAT5 are restricted to the cerebellum and the retina, respectively (Beschorner et al., 2007). During ischemia, impaired energy interferes with the re-uptake of glutamate by EAATs, and thus enhances the abnormal accumulation of synaptic glutamate. Chronic exposure of high dose alcohol has been reported to reduce the activity of EAAT3 and EAAT4 in Xenopus oocytes (Kim et al., 2005; Park et al., 2008). In the present study, we measured expression of EAAT1, EAAT2 and EAAT3 in the parietal cortex. Although the expression of EAAT1 and EAAT3 was not altered by chronic alcohol consumption, the expression of EAAT2 was upregulated in low dose alcohol-fed rats and downregulated in high dose alcohol-fed rats. Thus, altered EAAT2 expression may also contribute to the disparate influences of low dose and high dose alcohol consumption on cerebral I/R injury by influencing the re-uptake of glutamate and eventually activation of the NMDA receptors.
The mechanism underlying the alteration in expression of the NMDA receptor and EAAT2 during chronic alcohol consumption remains unknown. It is well known that alcohol, when administrated acutely in a pharmacologically relevant dose, selectively and potently inhibits the function of NMDA receptors. In addition, acute exposure of high dose alcohol may increase EAAT2 expression and activity (Wu et al., 2010). Thus, upregulation of NMDA receptor and downregulation of EAAT2 during chronic consumption of high dose alcohol may be a major neuroadaptive response to the chronic blockade by high dose alcohol (Fadda and Rossetti, 1998; Harper and Matsumoto, 2005; Sun et al., 2002). This sensitization of NMDA receptors is one of the most important factors in mechanisms underlying high dose alcohol-induced brain damage (Harper and Matsumoto, 2005). However, cellular pathways underlying altered expression of the NMDA receptor and EAAT2 during chronic consumption of high dose alcohol remain unknown. In addition, we are not aware of studies that have examined the chronic effect of low dose alcohol on EAAT2 expression/activity.
In summary, the present study further defines the influence of chronic alcohol consumption on transient focal ischemia-induced brain injury. We suggest that altered NMDA excitotoxicity may be involved in the disparate influences of high dose and low dose alcohol consumption on cerebral I/R injury.
Acknowledgments
This study was supported by National Institutes of Health Grants DA 14258, HL79587, HL090657, AA 11288, a Scientist Development Grant from the American Heart Association (0635052N), and funds from the University of Nebraska Medical Center.
References
- Aracava Y, Pereira EF, Maelicke A, Albuquerque EX. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 2005;312(3):1195–205. doi: 10.1124/jpet.104.077172. [DOI] [PubMed] [Google Scholar]
- Beschorner R, Simon P, Schauer N, Mittelbronn M, Schluesener HJ, Trautmann K, Dietz K, Meyermann R. Reactive astrocytes and activated microglial cells express EAAT1, but not EAAT2, reflecting a neuroprotective potential following ischaemia. Histopathology. 2007;50(7):897–910. doi: 10.1111/j.1365-2559.2007.02703.x. [DOI] [PubMed] [Google Scholar]
- Caicoya M, Rodriguez T, Corrales C, Cuello R, Lasheras C. Alcohol and stroke: a community case-control study in Asturias, Spain. Journal of Clinical Epidemiology. 1999;52:677–684. doi: 10.1016/s0895-4356(98)00074-2. [DOI] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, Korthuis RJ. Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcohol Clin Exp Res. 2009;33(2):206–19. doi: 10.1111/j.1530-0277.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;56(4):385–431. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Gascon S, Deogracias R, Sobrado M, Roda JM, Renart J, Rodriguez-Pena A, Diaz-Guerra M. Transcription of the NR1 subunit of the N-methyl-D-aspartate receptor is down-regulated by excitotoxic stimulation and cerebral ischemia. J Biol Chem. 2005;280(41):35018–27. doi: 10.1074/jbc.M504108200. [DOI] [PubMed] [Google Scholar]
- Gill JS, Zezulka AV, Shipley MJ, Gill SK, Beevers DG. Stroke and alcohol consumption. New England Journal of Medicine. 1986;315:1041–1046. doi: 10.1056/NEJM198610233151701. [DOI] [PubMed] [Google Scholar]
- Gorgulu A, Kins T, Cobanoglu S, Unal F, Izgi NI, Yanik B, Kucuk M. Reduction of edema and infarction by Memantine and MK-801 after focal cerebral ischaemia and reperfusion in rat. Acta Neurochir (Wien) 2000;142(11):1287–92. doi: 10.1007/s007010070027. [DOI] [PubMed] [Google Scholar]
- Hansagi H, Romelsjo A, de Verdier MG, Andreasson S, Leifman A. Alcohol consumption and stroke mortality. Stroke. 1995;26:1768–1773. doi: 10.1161/01.str.26.10.1768. [DOI] [PubMed] [Google Scholar]
- Harper C, Matsumoto I. Ethanol and brain damage. Curr Opin Pharmacol. 2005;5(1):73–8. doi: 10.1016/j.coph.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Hejmadi MV, Dajas-Bailador F, Barns SM, Jones B, Wonnacott S. Neuroprotection by nicotine against hypoxia-induced apoptosis in cortical cultures involves activation of multiple nicotinic acetylcholine receptor subtypes. Mol Cell Neurosci. 2003;24(3):779–86. doi: 10.1016/s1044-7431(03)00244-6. [DOI] [PubMed] [Google Scholar]
- Hillbom M, Kaste M. Ethanol intoxication: a risk factor for ischemic brain infarction. Stroke. 1983;14:694–699. doi: 10.1161/01.str.14.5.694. [DOI] [PubMed] [Google Scholar]
- Ikehara S, Iso H, Toyoshima H, Date C, Yamamoto A, Kikuchi S, Kondo T, Watanabe Y, Koizumi A, Wada Y, Inaba Y, Tamakoshi A. Alcohol consumption and mortality from stroke and coronary heart disease among Japanese men and women: the Japan collaborative cohort study. Stroke. 2008;39(11):2936–42. doi: 10.1161/STROKEAHA.108.520288. [DOI] [PubMed] [Google Scholar]
- Kim JH, Do SH, Kim YL, Zuo Z. Effects of chronic exposure to ethanol on glutamate transporter EAAT3 expressed in Xenopus oocytes: evidence for protein kinase C involvement. Alcohol Clin Exp Res. 2005;29(11):2046–52. doi: 10.1097/01.alc.0000187594.92476.07. [DOI] [PubMed] [Google Scholar]
- McCulloch J. Glutamate receptor antagonists in cerebral ischaemia. J Neural Transm Suppl. 1994;43:71–9. [PubMed] [Google Scholar]
- Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(4S Suppl):1007S–15S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA. NMDA antagonist neurotoxicity: mechanism and prevention. Science. 1991;254(5037):1515–8. doi: 10.1126/science.1835799. [DOI] [PubMed] [Google Scholar]
- Park HY, Kim JH, Zuo Z, Do SH. Ethanol increases the activity of rat excitatory amino acid transporter type 4 expressed in Xenopus oocytes: role of protein kinase C and phosphatidylinositol 3-kinase. Alcohol Clin Exp Res. 2008;32(2):348–54. doi: 10.1111/j.1530-0277.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- Rist PM, Berger K, Buring JE, Kase CS, Gaziano JM, Kurth T. Alcohol consumption and functional outcome after stroke in men. Stroke. 2010;41(1):141–6. doi: 10.1161/STROKEAHA.109.562173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24(1–3):107–29. doi: 10.1385/MN:24:1-3:107. [DOI] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226(4676):850–2. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- Sun H, Patel KP, Mayhan WG. Impairment of neuronal nitric oxide synthase-dependent dilatation of cerebral arterioles during chronic alcohol consumption. Alcoholism: Clinical and Experimental Research. 2002;26:663–670. [PubMed] [Google Scholar]
- Sun H, Zhao H, Sharpe GM, Arrick DM, Mayhan WG. Effect of chronic alcohol consumption on brain damage following transient focal ischemia. Brain Res. 2008;1194:73–80. doi: 10.1016/j.brainres.2007.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. J Exp Biol. 1997;200(Pt 2):401–9. doi: 10.1242/jeb.200.2.401. [DOI] [PubMed] [Google Scholar]
- von Wartburg JP. Biologic aspects of alcohol: An introduction. Annals of the New York Academy of Sciences. 1976;173:146–150. doi: 10.1111/j.1749-6632.1976.tb52876.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Lee MR, Choi S, Kim T, Choi DS. ENT1 regulates ethanol-sensitive EAAT2 expression and function in astrocytes. Alcoholism: Clinical and Experimental Research. 2010;34(6):1110–1117. doi: 10.1111/j.1530-0277.2010.01187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Chan PH, Chen SF, Babuna OA, Simon RP, Weinstein PR. Reduction of vasogenic edema and infarction by MK-801 in rats after temporal focal cerebral ischemia. Neurosurgery. 1994;34:339–345. doi: 10.1227/00006123-199402000-00018. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li W, Chow PC, Lau DT, Lee NT, Pang Y, Zhang X, Wang X, Han Y. Bis(7)-tacrine, a promising anti-Alzheimer’s dimer, affords dose- and time-dependent neuroprotection against transient focal cerebral ischemia. Neurosci Lett. 2008;439(2):160–4. doi: 10.1016/j.neulet.2008.05.007. [DOI] [PubMed] [Google Scholar]