

Abstract
Bacterial endotoxins (lipopolysaccharides (LPS)) are strong elicitors of the human immune system by interacting with serum and membrane proteins such as lipopolysaccharide-binding protein (LBP) and CD14 with high specificity. At LPS concentrations as low as 0.3 ng/ml, such interactions may lead to severe pathophysiological effects, including sepsis and septic shock. One approach to inhibit an uncontrolled inflammatory reaction is the use of appropriate polycationic and amphiphilic antimicrobial peptides, here called synthetic anti-LPS peptides (SALPs). We designed various SALP structures and investigated their ability to inhibit LPS-induced cytokine secretion in vitro, their protective effect in a mouse model of sepsis, and their cytotoxicity in physiological human cells. Using a variety of biophysical techniques, we investigated selected SALPs with considerable differences in their biological responses to characterize and understand the mechanism of LPS inactivation by SALPs. Our investigations show that neutralization of LPS by peptides is associated with a fluidization of the LPS acyl chains, a strong exothermic Coulomb interaction between the two compounds, and a drastic change of the LPS aggregate type from cubic into multilamellar, with an increase in the aggregate sizes, inhibiting the binding of LBP and other mammalian proteins to the endotoxin. At the same time, peptide binding to phospholipids of human origin (e.g., phosphatidylcholine) does not cause essential structural changes, such as changes in membrane fluidity and bilayer structure. The absence of cytotoxicity is explained by the high specificity of the interaction of the peptides with LPS.
Introduction
Bacteria and their pathogenicity factors pose a severe health problem worldwide because of the increasing occurrence of resistance as well as the demographic development of populations, which is leading to higher numbers of immunocompromised people and thus a greater incidence of severe infections, such as septic shock (1). Despite the availability of powerful antibiotics, life-threatening bacterial infections are still a major cause of death due to the inability of the applied therapeutics to not only kill the bacteria but also to neutralize their pathogenicity factors, such as endotoxins (lipopolysaccharides (LPSs)) and lipoproteins (LPs), two of the main toxins of Gram-negative and -positive bacteria, respectively (2). Furthermore, severe septic diseases are a very frequent complication of primary viral infections such the Spanish flu and the recent swine flu of the H1N1 serotype (3). Consequently, most lethal cases result from the bacterial rather than the viral infection.
Clearly, there is a pressing need for new antimicrobial drugs that can neutralize bacterial products such as LPS and LP. A promising strategy is the use of antimicrobial peptides (AMPs), originally based on LPS-binding domains of defense proteins such as lactoferrin (4), NK-lysin (5), and Limulus anti-LPS factor (6). Treatments based on the use of such peptides have been successfully used in animal models of endotoxin-induced septic shock (7). However, no breakthrough regarding the development of a therapeutic commercialized drug has been made, mainly due to the high therapeutic concentrations of AMPs needed to neutralize endotoxins effectively, which inevitably causes severe side effects (8).
To circumvent these restrictions, we developed what we believe is a novel strategy by synthesizing a new class of peptides, called synthetic anti-LPS peptides (SALPs), which are designed to strongly bind to the lipid A part of LPS. The binding constants of these compounds exceed those of mammalian LPS-bindings proteins such as lipopolysaccharide-binding protein (LBP) and CD14. The design of this peptide class was based on previously published details regarding the conformation and physicochemical characteristics of lipid A and LPS (9). In this way, we constructed amphiphilic cationic SALPs with lengths optimized for LPS neutralization, i.e., with a number of amino acid (AA) residues ranging from 19 to 23 (10). In a mouse model of lethal sepsis, these peptides were able to neutralize bacterial endotoxins even at very low concentrations and protect the animals considerably from endotoxic shock. At the same time, their toxicity levels for mammalian cells were far higher than the concentrations used in human therapeutics.
To further investigate these favorable properties, we performed a comprehensive biophysical study of peptide binding to endotoxin to elucidate the mode of action, in particular the extreme selectivity for endotoxin necessary to avoid detrimental side effects in human cells. The results we obtained using a variety of techniques are indicative of unique interaction mechanisms, which are discussed further below in the light of literature data.
Materials and Methods
Lipids
LPS from the rough mutant Ra from Salmonella minnesota (strain R60) was extracted by the phenol/chloroform/petrol ether method from bacteria grown at 37°C, purified, and lyophilized (11). The results obtained by standard assays on the purified LPS (i.e., analyses of the amount of glucosamine, total and organic phosphate, and distribution of the fatty acid residues) were in good agreement with the chemical properties expected for the LPS chemotypes, whose molecular structure has already been solved. S-form LPS from Pseudomonas aeruginosa PAO1 for mouse testing was obtained from the aqueous phase of a water-phenol extract and purified by treatment with chaotropic agents and detergents according to published procedures (12). Cholesterol and the phospholipids phosphatidylcholine (PC) from egg yolk, dioleoylphosphatidylcholine (DOPC), and sphingomyelin (SM, 18:1) were obtained from Avanti Polar Lipids (Alabaster, AL).
Preparation of LPS or phospholipid aggregates
The LPS and phospholipids were solubilized in the appropriate buffer (lipid concentrations between 1 nM and 10 mM, depending on the applied technique), extensively vortexed, sonicated for 30 min in a water bath, and subjected to several temperature cycles between 20°C and 60°C. Finally, the lipid suspension was incubated at 4°C for at least 12 h before use.
Fourier-transform infrared spectroscopy and differential scanning calorimetry
The measurements of the gel-to-liquid crystalline phase transitions of the acyl chains of LPS are described in the Supporting Material (13–16).
Isothermal titration calorimetry
Microcalorimetric measurements of peptide binding to endotoxins were performed on a MCS isothermal titration calorimeter (Microcal, Freiburg, Germany)) at 37°C as previously described (17). LPS R60 (0.05 mM, prepared as described above) were dispensed into the microcalorimetric cell (volume: 1.3 ml), and the peptide solutions (2 mM) were loaded into the syringe compartment (volume: 100 μl). After temperature equilibration, the peptides were titrated in 3 μl portions every 5 min into the lipid-containing cell under constant stirring, and the heat of interaction after each injection measured by the isothermal titration calorimetry (ITC) instrument was plotted versus time. The binding constant of the LPS-peptide interaction was determined by fitting the binding curve with a sigmoidal function and applying common thermodynamic analysis, i.e., using ΔG = ΔH − T × ΔS (where ΔG is the Gibbs energy, T is the temperature, ΔS is the entropy change, and ΔH is the measured enthalpy change). Furthermore, the binding constant kb can be taken from ΔG = −R × T × ln kb, where R = gas constant. It should be noted that kb is the reciprocal of the equilibrium dissociation constant kd (kb = 1/kd).
X-ray diffraction
X-ray diffraction measurements of LPS R60 peptide mixtures were performed at the European Molecular Biology Laboratory outstation at the Hamburg synchrotron radiation facility (HASYLAB) using the double-focusing monochromator-mirror camera X33 (18). Diffraction patterns in the range of the scattering vector 0.01 < s < 0.0 nm−1 (s = 2 sin θ/λ, 2θ scattering angle, and wavelength λ = 0.15 nm) were recorded at 20°C, 40°C, and 60°C with exposure times of 1 min using an image plate detector with online readout (MAR345; MarResearch, Norderstedt/Germany) (19). The s axis was calibrated with Ag-Behenate, which has a periodicity of 5.84 nm. We evaluated the diffraction patterns as described previously (20) by assigning the spacing ratios of the main scattering maxima to defined three-dimensional structures. For this study, the lamellar and cubic structures were the most relevant.
Freeze-fracture electron microscopy
For freeze-fracturing, LPS and the LPS/peptide samples, copper sandwich profiles, and instruments for manipulation were incubated at room temperature or at 40°C. A small amount of the sample was sandwiched between two copper profiles as used for the double-replica technique and frozen by plunging the sandwiches immediately into liquified ethane/propane-mixture cooled in liquid nitrogen. Fracturing and replication were performed at –150°C in a BAF 400T freeze-fracture device (BAL-TEC, Liechtenstein) equipped with electron guns and a film sheet thickness monitor. In a first step 2 nm of Pt(C) were evaporated under an angle of 35°, followed by perpendicular evaporation of C for a second replica layer of 20 nm thickness. The replicas were placed on copper grids, cleaned with a chloroform-methanol mixture, and examined in an EM 901 electron microscope (Zeiss, Oberkochen, Germany).
Fluorescence resonance energy transfer spectroscopy
The ability of the peptides to intercalate into phospholipid liposomes or LPS R60 aggregates, as investigated by fluorescence resonance energy transfer (FRET), is described in the Supporting Material.
Determination of the ζ-potential
We used a Zetasizer Nano (Malvern Instruments, Herrsching, Germany) to determine the ζ-potentials of endotoxin aggregates from the electrophoretic mobility by laser-Doppler anemometry at a scattering angle of 90° as described previously (21). The ζ-potential was calculated according to the Helmholtz-Smoluchowski equation from the mobility of the aggregates in a driving electric field of 19.2 Vcm−1. LPS aggregates (0.05 mM) and peptide stock solutions (1 mM) were prepared in 20 mM Hepes, which yielded the most reproducible results.
Stimulation of human mononuclear cells by LPS
Mononuclear cells (MNCs) were isolated from heparinized blood of healthy donors as described previously (10). The cells were resuspended in medium (RPMI 1640) and their number was equilibrated at 5 × 106 cells/ml. For stimulation, 200 μl MNC (1 × 106 cells) were transferred into each well of a 96-well culture plate. LPS Ra and LPS/peptide mixtures were preincubated for 30 min at 37°C, and then added to the cultures at 20 μl per well. The cultures were incubated for 4 h at 37°C under 5% CO2. Supernatants were collected after centrifugation of the culture plates for 10 min at 400 × g and stored at −20°C until immunological determination of tumor necrosis factor α (TNFα), carried out in a Sandwich enzyme-linked immunosorbent assay using a monoclonal antibody against TNF (clone 6b; Intex AG, Switzerland).
Animal model of endotoxicity
LPSs from P. aeruginosa PA01 or S. minnesota R60 (see above for details of preparation) were used to induce endotoxic shock.
Female C57/BL6 mice (6 weeks old, 14–16 g) were purchased from Harlan Spain (Harlan Interfauna Iberica S.A., Barcelona, Spain) and randomly distributed in experimental groups (n = 8). Endotoxic shock was induced in the animals by co-inoculation of LPS and galactosamine following the method of Galanos et al. (22).
Specifically, each animal received an intraperitoneal injection containing a mixture of 25 ng of LPS of S. minnesota R60 (150 ng in the case of the less-endotoxic LPS of P. aeruginosa PA01) and 18 mg of galactosamine resuspended in 200 μl of endotoxin-free saline. Immediately after LPS administration, the animals were inoculated at a different site of the peritoneum, with the test peptide resuspended in 150 μl of pyrogen-free saline at a LPS/peptide ratio of 1:500. To facilitate the therapeutic action of the peptide, the mice were gently massaged at the site of inoculation for a few seconds. Animal mortality was monitored at least every 24 h for 7 days.
In each independent experiment, a group of animals received a dose of polymyxin B (PMB) identical to that used for the peptide, whereas another group was left untreated.
Results regarding animal mortality at all experimental time points were globally analyzed by means of a Kaplan-Meier survival analysis (SPSS 15.0). When the survival plots were parallel, the data were compared by the log-rank test, whereas the Breslow-Gehan-Wilcoxon test was applied when the plots intersected. The p-values were obtained by comparing data from the same experiment (i.e., mortality in the treated versus the untreated group). All of the animal protocols used in this study were approved by the Animal Research Committee of the University of Navarra (protocol No. 069-09).
Cytotoxicity
The techniques used to detect cytotoxic effects are described in the Supporting Material.
Results
In a first step, the effectivity of selected peptides (amino acid sequence; see Table 1) to neutralize endotoxin in vitro and in a mouse model of sepsis is described.
Table 1.
Sequences and molecular weights of the investigated peptides
| Pep19-2.5 | GCKKYRRFRWKFKGKFWFWG | 2711 |
| Pep19-2.5KO | KfGKwRfGKYRFCwKfRGwK | 2711 |
| Pep19-4 | GKKYRRfRwKFKGK wfwfG | 2750 |
| Pep19-8 | GRRYKKfRwKFKGRwfwfG | 2636 |
AA identification: Bold corresponds to basic, lower case to hydrophobic, and italic to polar character.
Inhibition of the LPS-induced stimulation of cytokines in MNCs
We investigated the ability of LPS to induce TNFα secretion in human MNCs at 100, 10, and 1 ng/ml LPS concentrations, and the results show a high cytokine production with a slight decrease at the lowest LPS concentration (Fig. 1 A). In all cases, the addition of the peptides Pep19-2.5, Pep19-2.5KO, Pep19-4, and Pep19-8 leads to a decreased secretion of TNFα, but at completely different concentrations. Pep19-2.5 inhibits cytokine production at an extremely low concentration, whereas at the lowest LPS concentration (1 ng/ml), there is almost no measurable TNFα secretion. Of interest, for Pep19-2.5KO, which has the same amino acids as Pep19-2.5 but in a random sequence, there is still effective inhibition at a [Pep]/[LPS] 100:1 excess concentration ratio. The other two compounds also differ in their ability to neutralize LPS; in particular, compound Pep19-8 has only a weak cytokine inhibitory ability, and a strong excess on a weight scale (100:1) is necessary to induce some inhibition (Fig. 1 B, right-hand bars).
Figure 1.
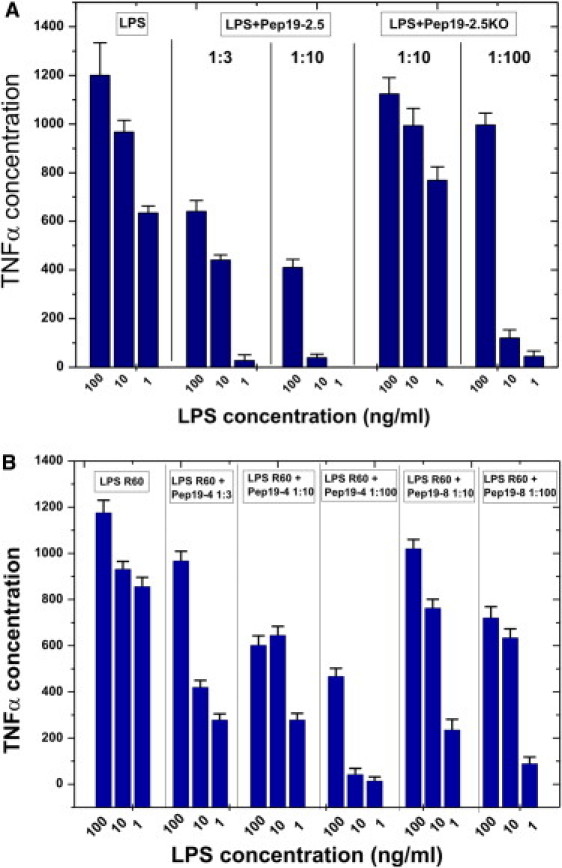
Inhibition of the LPS-induced inflammation reaction in vitro. LPS R60-induced production of TNFα of human MNCs and inhibition by Pep19-2.5 and Pep19-2.5KO (A), and Pep19-4 and Pep19-8 (B) at different weight ratios.
Animal model of endotoxicity
To ascertain whether three selected peptides (Pep19-2.5, Pep19-4, and Pep19-8) could neutralize LPS in vivo, we used a mouse model of acute septic shock and measured the ability of the compounds to increase animal survival (Fig. 2, A and B). To induce septic shock, we first used LPS from P. aeruginosa, whose endotoxic activity is known to be lower than that of Salmonella (see Materials and Methods). As shown in Fig. 2 A, whereas Pep19-2.5 protected the animals very efficiently, Pep19-8 showed no antiendotoxic activity in this model. Of interest, Pep19-4, which displayed an intermediate level of inhibitory activity in the cytokine assay (Fig. 1 A), showed an antiendotoxic activity halfway between those of Pep19-2.5 and Pep19-8.
Figure 2.
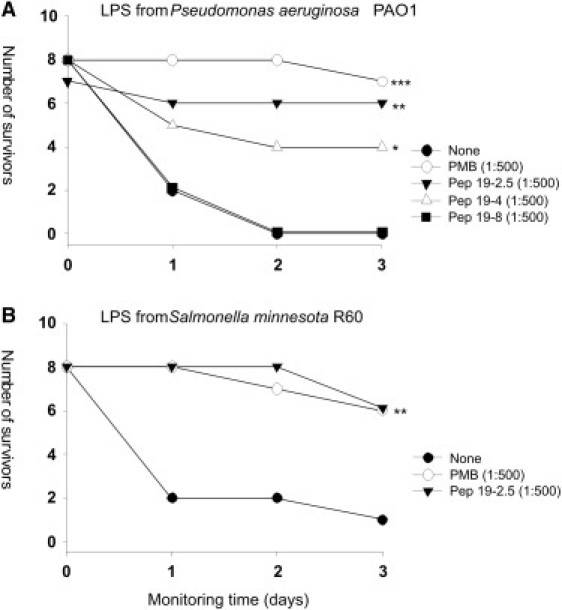
Protection against lethal septic shock caused by injection of LPS. On day 0, a group of C57/BL6 mice (n = ≤7) were inoculated intraperitoneally with a mixture of 18 mg of galactosamine and either (A) 150 ng of LPS from P. aeruginosa PAO1 or (B) 25 ng of LPS from S. minnesota R60. Immediately afterward, the animals received at a different site of the peritoneum either 150 μg (A) or 12.5 μg (B) of the test peptide. Duplicate groups of LPS-challenged animals received an amount of PMB identical to that of the peptide or were left untreated. Animal mortality was monitored at daily intervals for 3 days. Statistical differences were analyzed by means of the Kaplan-Meier survival test (∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05).
To determine whether the peptides could neutralize an LPS of enhanced endotoxicity, we used LPS from S. enterica serovar Minnesota R60 and tested the most potent compound, Pep19-2.5, in the same animal model. As shown in Fig. 2 B, administration of Pep19-2.5 conferred a high level of protection, which was indistinguishable from that of PMB.
Below, we characterize the physical parameters that are prerequisites for effective LPS neutralization by the peptides.
Aggregate structure of LPS
The biologically active aggregate structure of LPS was previously shown to have three-dimensional cubic symmetry (23). To determine the aggregate structure of LPS R60 in the presence of the peptides Pep19-2.5, Pep19-2.5KO, and Pep19-8, we applied synchrotron radiation small-angle x-ray scattering (SAXS). The scattering patterns presented in Fig. 3 for [LPS]/[peptide] 3:1 molar ratios for Pep19-2.5 show (A) two strong reflections corresponding to the bilayer repeat and the second order, according to previous data for LPS R60 (18). These are even more intensely expressed at 60°C with the appearance of the third and fourth order (Fig. 3, bottom). In contrast, the data for Pep19-2.5 KO (Fig. 3 B, middle) and Pep19-8 (Fig. 3 B, bottom) are not indicative of a multilamellar structure; rather, they show that a different cubic or mixed aggregate structure is adopted. (For example, the scattering maxima shown in Fig. 5 B, bottom, could be assigned to 8.85 nm = aQ /√2, 5.52 nm = aQ /√5, and 4.54 nm = aQ /√8 of a cubic periodicity aQ = 12.5 ± 0.3 nm.) However, the number of reflections is too low to obtain an unequivocal assignment. For example, we completely exclude the possibility that the reflections in the diagram in Fig. 3 B (bottom) correspond to the two orders of two lamellar spacings. In each case, the most important information is that the ability of the peptide to inhibit the cytokine protection in both in vitro and in vivo assays (Figs. 1 and 2) directly corresponds to its ability to convert LPS into a multilamellar aggregate. Thus, further measurements show that the conversion of the basic cubic aggregate structure of LPS R60 into a multilamellar form takes place for Pep19-2.5KO and Pep19-8 at a much higher peptide concentration, i.e., at a molar excess of the peptide (data not shown).
Figure 3.
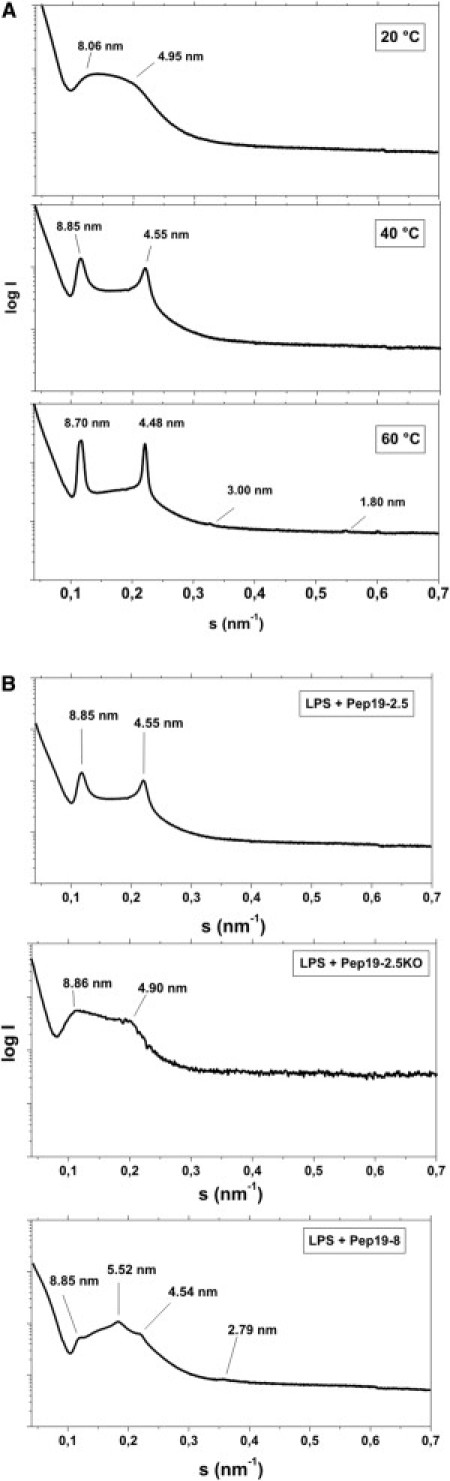
Supramolecular aggregate structure of LPS. SAXS was performed using synchrotron radiation of LPS R60 in the presence of Pep19-2.5 at (A) three temperatures and (B) at 40°C in the presence of Pep19-2.5 (top), Pep19-2.5KO (middle), or Pep19-8 (bottom). All samples were prepared at [LPS]/[Pep] 3:1 weight %. The logarithm of the scattering intensity logI is plotted versus the scattering vector s (= 1/d, d = lattice spacings).
Figure 5.
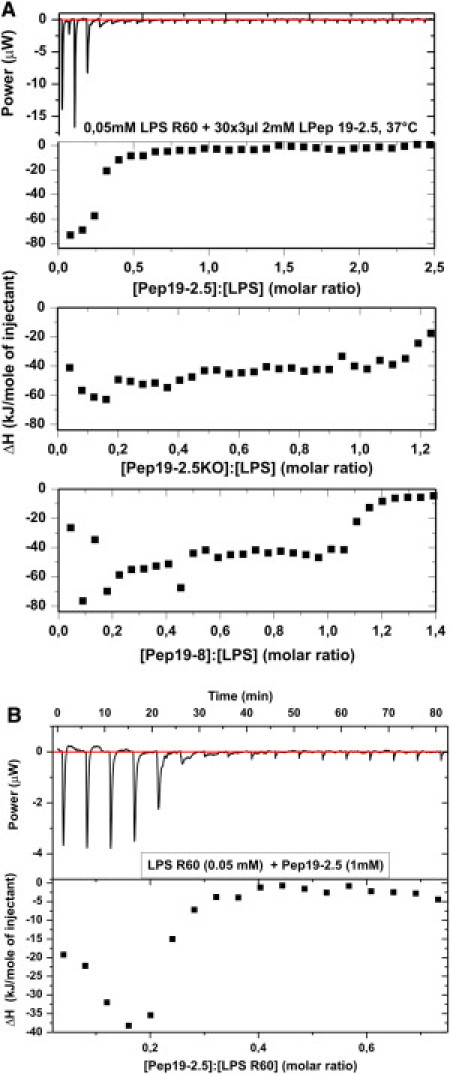
(A) Binding saturation of peptides. ITC of mixtures of LPS R60 with Pep19-2.5 (top), Pep19-2.5KO (middle), and Pep19-8 (bottom) at 37°C. For the LPS dispersion (0.05 mM), 5 μl of the peptide solution (2 mM) was titrated every 5 min, and the calorimetric signal was recorded. (B) Binding saturation of peptides. ITC of mixtures of LPS R60 with Pep19-2.5 (LPS concentration: 0.05 mM; Pep19-2.5 concentration: 2 mM). The measurements were done at 37°C.
The morphologies of the pure LPS samples and the mixtures with peptide Pep19-2.5 investigated with freeze-fracture electron microscopy are shown in Fig. 4. LPS alone exhibits ribbon-like structures with lengths up to a few hundred μm (see inset), thicknesses of 14–20 nm, and variable widths (A). In the presence of LPS (B), these separated structures change completely into large and densely packed multilamellar aggregates, with many multilamellar stackings corresponding to the multiple lamellae found in the x-ray experiment.
Figure 4.

Morphology of LPS aggregates. Freeze-fracture micrographs of LPS R60 and LPS/peptide mixtures. (A) LPS alone shows aggregates of ribbon-like structures appearing more or less cross fractured (area marked by star) or tangentially fractured (area marked by triangle). The extension of the ribbons is more clearly visible in the case of adsorbed ribbons (inset). (B) After addition of Pep19-2.5, multilamellar aggregates are visible.
Neutralization of the LPS headgroup charges
We next sought to investigate the ability of a given peptide to neutralize the negative surface charges of an LPS sample. To that end, we applied isothermal titration calorimetry (ITC) and electrophoretic mobility determination to determine the ζ-potential. ITC has been shown to be a sensitive and direct means of determining the strength of a binding complex (13). The ITC data for the three selected peptides are given in Fig. 5 A. All single titrations (for example, representation of the power versus time in the diagram at the top of the figure) indicate negative values corresponding to a strong exotherm of the binding of the peptides to LPS, which disappear at different concentrations for the three peptides. The presentation of the integrals of these exotherms (diagrams at the bottom of the figure) shows a transition into binding saturation at different peptide/LPS ratios. Whereas the Pep19-2.5/LPS system is already in saturation at molar ratios < 0.4, for the other peptides this value is ≥1.1. Therefore, the binding affinity of Pep19-2.5 is much higher than those for the other peptides. A more precise ITC experiment was performed with the latter peptide (Fig. 5 B). The evaluation of such curves from five independent experiments, using a sigmoidal saturation curve as shown in Fig. 5 B, gave a binding constant of k = (2.8 ± 3.0) × 10−8 /Mol.
We determined the ζ-potential of the LPS/peptide mixtures by measuring their electrophoretic mobilities. A comparison of Pep19-2.5 with Pep19-8 (Fig. 6) shows that the ζ-potential, starting from negative values due to the negative lipid A backbone charges, does not end at zero potential. Instead, it adopts clearly positive values, i.e., additional binding occurs beyond pure charge compensation. Of interest, the less-active Pep19-8 reaches a positive ζ-potential at significantly lower LPS/peptide ratios than the highly active Pep19-2.5. Thus, in addition to purely electrostatic interactions, hydrophobic interactions between the lipid and the peptides have to be considered.
Figure 6.
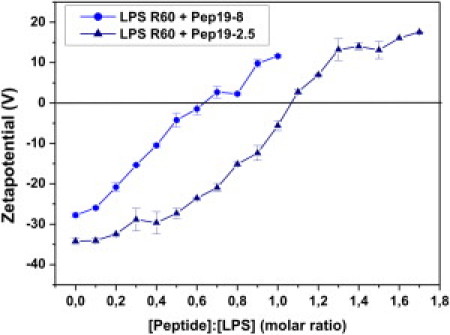
Compensation of LPS backbone charges. The ζ-potential of LPS R60 aggregates depends on different concentrations of Pep19-8 and Pep19-2.5, as determined by their electrophoretic mobility with Laser-Doppler anemometry.
Peptide incorporation into phospholipid and LPS membranes
We investigated the ability of the peptides Pep19-2.5, Pep19-2.5KO, and Pep19-8 to intercalate into phospholipid membranes from PC, phosphatidylserine (PS), and LPS by applying FRET (Fig. S3, A–C).
LPS inactivation in the Limulus test
In the Limulus amoebocyte lysate test, which is a very sensitive test for endotoxin contamination, the recognition structure for LPS is the diglucosamine 4′-phosphate backbone region (24). The data in Fig. 7 show a complete inhibition of endotoxic activity (in endotoxin units (EUs)) at all concentrations of LPS (10, 1, and 0.1 ng/ml) and an [LPS]/[Pep19-2.5] weight ratio of 1:1. Thus, binding of the peptide to the 4′-phosphate should take place very effectively.
Figure 7.
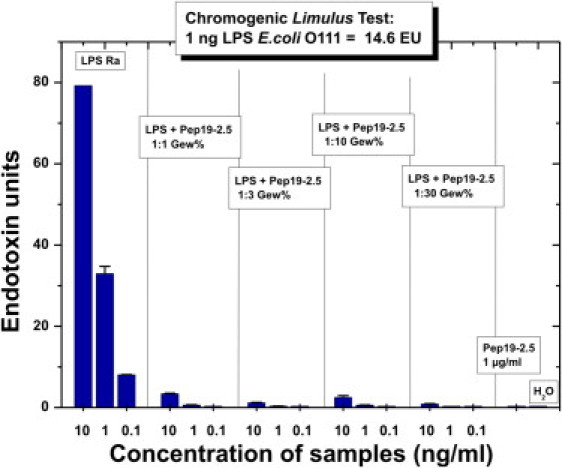
Biological activity in the Limulus test system. The activities (in EUs) of LPS R60 and LPS/Pep19-2.5 mixtures in the Limulus amebocyte lysate test at three different LPS concentrations and LPS/Pep ratios of 1:1–1:30 weight % are shown. As the standard in this test, the LPS S-form from E. coli O111 is used; 1 ng of LPS corresponds to 14.6 EUs.
Influence of peptides on a membrane mimetic bilayer
We investigated the influence of the peptide 19-2.5 on the mimetic of the immune cell membrane as described above by applying SAXS (Fig. 8). As can be seen in the figure, this mixed lipid system exhibits a bilayer periodicity of 5.05 nm with a shoulder at 4.65 nm and an additional reflection at 7.46 nm (top). The latter reflection may reflect three-dimensional phase separation of SM (see Discussion). The first two values may correspond to different periodicities due to the domain structures of this lipid mixture. Upon addition of Pep19-2.5 (middle, bottom), the main periodicity hardly changes, whereas the values corresponding to the domains change, i.e., the domain structure changes.
Figure 8.
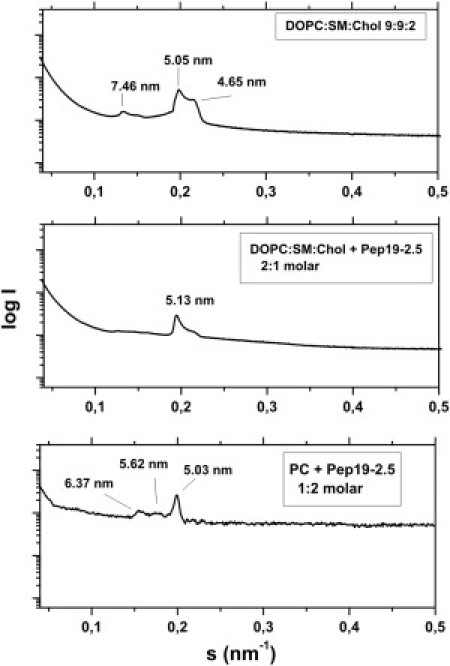
Aggregate structure of a mimetic of the mammalian phospholipids membrane. SAXS was performed using synchrotron radiation of DOPC/SM/Cho mixtures 9:9:2 molar in the presence of Pep19-2.5 at three molar ratios 1:0 (top), 2:1 (middle), and 1:2 (bottom) at 40°C. The logarithm of the scattering intensity logI is plotted versus the scattering vector s (= 1/d, d = lattice spacings).
Cytotoxicity of the peptides
The cytotoxic effects of the peptides are described in the Supporting Material.
Discussion
Our aim in this systematic study of the mechanisms of interaction of selected SALPs with LPS (endotoxin) and with eukaryotic membrane systems was to characterize both the neutralization mechanisms of LPS and the selectivity of these processes in different membrane systems. The data clearly show that the selected SALPs differ strongly in their ability to suppress the cytokine response in human MNCs (Fig. 1), and to confer protection in the animal model of sepsis (Fig. 2). We analyzed various physical parameters to find out whether they are determinants of the inactivation process of LPS.
A comparison of the data for the gel-to-liquid crystalline phase behavior reveals that the Pep19-2.5 peptide with the highest inhibitory activity results in only a slight fluidization of the acyl chains (Fig. S1 A), whereas the Pep19-8 peptide with much lower inhibitory activity shows a stronger fluidization (Fig. S1 B). It was previously shown that the gold standard of AMPs, PMB, leads to a strong acyl chain fluidization of LPS (25). However, its nonapeptide, PMBN, behaved nearly identically to PMB, although this compound exhibits no or only low antimicrobial activity (26). Therefore, the parameter fluidization of the acyl chains of the lipid A moiety of LPS by itself does not correlate with the capacity of a peptide to block the stimulation capacity of LPS. This is also in accord with a previous study of the inhibiting effect of human lactoferrin on LPS-induced cell stimulation, which showed that the binding of LF to LPS leads to acyl-chain rigidification (4).
Various studies have emphasized the importance of the aggregate structure of endotoxins for the expression of biological activity. In one study, cubic aggregate structures were assigned to their bioactive structure, whereas the inhibition induced by AMPs converted these into a multilamellar assembly (23). The diffraction patterns seen here do not give strong evidence for cubic phases, which may reflect the fact that it was our aim to realize near-physiological conditions, i.e., a water content >95%, whereas cubic phases for LPS are well expressed at 70–85% water content (18). However, the most important finding is that the extent of multilamellarization directly correlates with the biological efficiency of the single peptides (Figs. 1, 2, and 3 B), as evidenced by the fact that compound Pep19-8, which has the lowest inhibition capacity, maintained the original aggregate structure more strongly than the more-active compounds.
This behavior is directly connected with the drastic increase in the size of the aggregate structures (Fig. 4), with small ribbon-like structures changing into large, agglomerated, three-dimensional patches in which expanded multilamellar stackings are found in correspondence to the SAXS data.
The literature contains only limited data regarding changes in LPS aggregate structures and sizes due to the action of peptides. Rosenfeld et al. (27,28) studied the inhibiting effects of different peptides on the stimulating capacity of S-form LPS and concluded that peptide binding to LPS is accompanied by a dissociation of the LPS aggregates. They based their conclusions on the results of FITC-labeled LPS and fluorescence dequenching experiments, in which an increase of the fluorescence intensity was interpreted as resulting from a disaggregation process. It is known, however, that other effects, such as an increase of the LPS acyl-chain fluidity, may also be connected to an increase in fluorescence intensity (K. Brandenburg, unpublished results), and that in both cases, the binding of LPS to LBP, as well as to BPI, lead to a fluorescence increase due to dequenching, although the former protein should result in a disaggregation, and the latter in an complexation, i.e., an increase in aggregation (29). Furthermore, using negative-staining electron microscopy, Rosenfeld et al. found a decrease in aggregate size. This can be understood in the light of the fact that the aggregational behavior of LPS is extremely dependent on the water content, leading to completely different phases when water is removed from the dispersions, i.e., the inherent cubic aggregates of enterobacterial lipid A and LPS would convert into multilamellar complexes at ∼≤50% water content (30,31). Another possibility would be a different mechanism as described by Rosenfeld et al., because these authors used S-form LPS, which has a rather high heterogeneity. The aggregate structure of this complex mixture is not known in detail; therefore, other mechanisms may be involved. To resolve these differences, we plan to conduct experiments with LPS S-form from Escherichia coli, which is similar to the S-form LPS used by Rosenfeld et al. We will perform electron microscopic analyses using special cryo-techniques, including cryo-transmission electron microscopy and freeze-fracturing (32).
The ITC data are important for an interpretation of the peptide-induced inhibition of the cell stimulation by LPS. The strong exothermic reaction of the LPS peptide interaction, found for all peptides due to Coulomb interaction between the positive charges of the peptides and the negative backbone charges of LPS, is already in saturation at [Pep19-2.5]/[LPS] = 0.3, i.e., three peptide molecules per 10 LPS molecules already lead to charge saturation. In contrast, for the much less effective compounds Pep19-2.5KO and Pep19-8, saturation is reached at [Pep]/[LPS] molar ratios of >1 (Fig. 5). The strong inhibitory capacity of Pep19-2.5 can be explained by the high binding constant (estimated as k = 2.8 × 10−8 /Mol from Fig. 5) of the peptide to LPS putatively exceeding that for the human LPS-binding proteins LBP and CD14, which are responsible for the initial steps of cell activation. It should be mentioned that such binding constants of LPS with LBP and CD14 have been reported to lie in the range of 10−8 to 10−9/Mol (33,34). These data were obtained by using FITC-labeled or 3H-metabolically labeled LPS from S. minnesota R595. Since binding constants measured with other techniques, which afford the use of labels, frequently differ strongly from values reported with ITC, comparative measurements should be done. We will address this question in a separate investigation.
An important inactivation step is the competitive interaction of the peptide with human binding proteins such as LBP and CD14. However, this may not be the only step that is important for understanding LPS neutralization. The FRET data indicate an intercalation of the peptides not only into LPS aggregates (Fig. S3 C), which is in accord with the above-discussed data, but also into normal PC (or DOPC/SM/Chol mixtures) and PS liposomal membranes (Fig. S3, A and B). Together with the observation that the neutralization ability of Pep19-2.5 is still present when it is administered up to 3 h after LPS addition (data not shown), and the reported data that LPS by itself is transported into target membranes by the action of LBP (35), a second step in LPS neutralization is the interaction of LPS and peptides within the membranes of immune cells. The details and mechanisms of this interaction will be presented in a separate study.
It was previously shown that the potential binding sites in LPS, the phosphates, differ by their conformation, with the 1-phosphate looking into the aqueous environment of the LPS layer, and the 4′-phosphate sticking into the hydrophobic/interface region (36). In similarity to what was recently reported for cyclic peptides based on the Limulus-anti LPS factor (20), we found that the 1-phosphate is the first point of binding of the peptide (IR fingerprint region; Supporting Material). This is in excellent agreement with the data from the Limulus assay (Fig. 7), which unequivocally indicate a complete blocking of the reaction at LPS/Pep19-2.5 1:1 weight %. It was previously reported that the presence of an acylated diglucosamine 4′-phosphate headgroup of LPS/lipid A is a prerequisite for Limulus activity (24).
For therapeutic use, the question of the specificity of the SALP, or, in pharmacological terms, the therapeutic index, is of utmost importance. This corresponds to the ratio of the dose at which side effects occur to the therapeutic dose. Here, the in vitro data of the cytotoxicity and hemolysis (Fig. S4, A and B) are indicative of the beginning of adverse effects at concentrations of 30–50 μg/ml, whereas the therapeutic dose (Fig. 2) is ∼1 μg/ml. In support of these findings are the SAXS experiments of the interaction of Pep19-2.5 with a mimetic of an immune cell membrane (DOPC/SM/Chol 9:9:2 molar ratio). Various authors have investigated such membrane models with variations in the single components (37–39). In the light of their data, it can be concluded that the SM exhibits a three-dimensional phase separation, which can be deduced from the extra reflection at 7.46 nm in Fig. 8 (top). This reflection disappears in the presence of the peptide, which apparently is a result of the membrane incorporation as described above. Of most importance, however, is the observation that the basic structure and hence its integrity are maintained, in accordance with the determination of cytotoxic effects.
A previous study in mice showed that a considerable degree of protection occurs at [Pep19-2.5]/[LPS] 50:1 weight % (10), shifting the therapeutic index to even higher values. Although these are preclinical data, obtained in galactosamine-sensitized mice, the application of this peptide to humans should render similar results. Humans can experience severe septic syndromes at LPS concentrations of <0.5 ng/ml in blood (40). The therapeutic dose of the peptide should be in the range of 100:1–1000:1 with respect to LPS, i.e., it should not exceed 1 μg/ml, in similarity to the situation in the animal model.
Acknowledgments
We thank Nina Hahlbrock and Christine Hamann for performing the infrared and FRET spectroscopic measurements, respectively.
This study was supported by the Ministerium für Bildung und Forschung (project No. 01GU0824). G.M.T. received grants from the Ministerio de Sanidad y Consumo (FIS-PI050768) and Proyectos de Investigación Universidad de Navarra (PIUNA-2008-11).
Supporting Material
References
- 1.Rietschel E.Th., Kirikae T., Di Padova Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 2.Rietschel E.T., Brade H., Schumann R.R. Bacterial endotoxin: chemical constitution, biological recognition, host response, and immunological detoxification. Curr. Top. Microbiol. Immunol. 1996;216:39–81. doi: 10.1007/978-3-642-80186-0_3. [DOI] [PubMed] [Google Scholar]
- 3.Springer J., Safley M., McGoey R.R. Histopathological findings in fatal novel H1N1: an autopsy case series from September-November 2009 in New Orleans, LA. J. LA. State Med. Soc. 2010;162:88–91. [PubMed] [Google Scholar]
- 4.Brandenburg K., Jürgens G., Koch M.H. Biophysical characterization of lipopolysaccharide and lipid A inactivation by lactoferrin. Biol. Chem. 2001;382:1215–1225. doi: 10.1515/BC.2001.152. [DOI] [PubMed] [Google Scholar]
- 5.Andrä J., Koch M.H.J., Brandenburg K. Biophysical characterization of endotoxin inactivation by NK-2, an antimicrobial peptide derived from mammalian NK-lysin. Antimicrob. Agents Chemother. 2004;48:1593–1599. doi: 10.1128/AAC.48.5.1593-1599.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrä J., Garidel P., Brandenburg K. Biophysical characterization of the interaction of Limulus polyphemus endotoxin neutralizing protein with lipopolysaccharide. Eur. J. Biochem. 2004;271:2037–2046. doi: 10.1111/j.1432-1033.2004.04134.x. [DOI] [PubMed] [Google Scholar]
- 7.Dankesreiter S., Hoess A., Miethke T. Synthetic endotoxin-binding peptides block endotoxin-triggered TNF-α production by macrophages in vitro and in vivo and prevent endotoxin-mediated toxic shock. J. Immunol. 2000;164:4804–4811. doi: 10.4049/jimmunol.164.9.4804. [DOI] [PubMed] [Google Scholar]
- 8.Garidel P., Brandenburg K. Current understanding of polymyxin B application in bacteraemia/sepsis therapy prevention: clinical, pharmaceutical, structural and mechanistic aspects. Anti-Infect. Agents Med. Chem. 2009;8:367–385. [Google Scholar]
- 9.Brandenburg K., Schromm A.B., Gutsmann T. Endotoxins: relationship between structure, function, and activity. Subcell. Biochem. 2010;53:53–67. doi: 10.1007/978-90-481-9078-2_3. [DOI] [PubMed] [Google Scholar]
- 10.Gutsmann T., Razquin-Olazarán I., Brandenburg K. New antiseptic peptides to protect against endotoxin-mediated shock. Antimicrob. Agents Chemother. 2010;54:3817–3824. doi: 10.1128/AAC.00534-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanos C., Lüderitz O., Westphal O. A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 1969;9:245–249. doi: 10.1111/j.1432-1033.1969.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 12.Hirschfeld M., Ma Y., Weis J.J. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J. Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 13.Garidel P., Rappolt M., Brandenburg K. Divalent cations affect chain mobility and aggregate structure of lipopolysaccharide from Salmonella minnesota reflected in a decrease of its biological activity. Biochim. Biophys. Acta. 2005;1715:122–131. doi: 10.1016/j.bbamem.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Schromm A.B., Brandenburg K., Seydel U. Lipopolysaccharide-binding protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS Lett. 1996;399:267–271. doi: 10.1016/s0014-5793(96)01338-5. [DOI] [PubMed] [Google Scholar]
- 15.Brandenburg K., Moriyon I., Seydel U. Biophysical investigations into the interaction of lipopolysaccharide with polymyxins. Thermochim. Acta. 2002;382:189–198. [Google Scholar]
- 16.Brandenburg K., Kusumoto S., Seydel U. Conformational studies of synthetic lipid A analogues and partial structures by infrared spectroscopy. Biochim. Biophys. Acta. 1997;1329:183–201. doi: 10.1016/s0005-2736(97)00109-0. [DOI] [PubMed] [Google Scholar]
- 17.Howe J., Andrä J., Brandenburg K. Thermodynamic analysis of the lipopolysaccharide-dependent resistance of gram-negative bacteria against polymyxin B. Biophys. J. 2007;92:2796–2805. doi: 10.1529/biophysj.106.095711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seydel U., Koch M.H.J., Brandenburg K. Structural polymorphisms of rough mutant lipopolysaccharides Rd to Ra from Salmonella minnesota. J. Struct. Biol. 1993;110:232–243. doi: 10.1006/jsbi.1993.1026. [DOI] [PubMed] [Google Scholar]
- 19.Roessle M., Klaering R., Svergun D. Upgrade of the small-angle X-ray scattering beamline at the European Molecular Biology Laboratory, Hamburg. J. Appl. Cryst. 2007;40:190–194. [Google Scholar]
- 20.Andrä J., Howe J., Brandenburg K. Mechanism of interaction of optimized Limulus-derived cyclic peptides with endotoxins: thermodynamic, biophysical and microbiological analysis. Biochem. J. 2007;406:297–307. doi: 10.1042/BJ20070279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrä J., Lamata M., Brandenburg K. Cyclic antimicrobial peptides based on Limulus anti-lipopolysaccharide factor for neutralization of lipopolysaccharide. Biochem. Pharmacol. 2004;68:1297–1307. doi: 10.1016/j.bcp.2004.05.054. [DOI] [PubMed] [Google Scholar]
- 22.Galanos C., Freudenberg M.A., Reutter W. Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc. Natl. Acad. Sci. USA. 1979;76:5939–5943. doi: 10.1073/pnas.76.11.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandenburg K., Wiese A. Endotoxins: relationships between structure, function, and activity. Curr. Top. Med. Chem. 2004;4:1127–1146. doi: 10.2174/1568026043388213. [DOI] [PubMed] [Google Scholar]
- 24.Gutsmann T., Howe J., Brandenburg K. Structural prerequisites for endotoxic activity in the Limulus test as compared to cytokine production in mononuclear cells. Innate Immun. 2010;16:39–47. doi: 10.1177/1753425909106447. [DOI] [PubMed] [Google Scholar]
- 25.Vaara M., Vaara T. Polycations sensitize enteric bacteria to antibiotics. Antimicrob. Agents Chemother. 1983;24:107–113. doi: 10.1128/aac.24.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrä J., Gutsmann T., Brandenburg K. Mechanisms of endotoxin neutralization by synthetic cationic compounds. J. Endotoxin Res. 2006;12:261–277. doi: 10.1179/096805106X118852. [DOI] [PubMed] [Google Scholar]
- 27.Rosenfeld Y., Sahl H.G., Shai Y. Parameters involved in antimicrobial and endotoxin detoxification activities of antimicrobial peptides. Biochemistry. 2008;47:6468–6478. doi: 10.1021/bi800450f. [DOI] [PubMed] [Google Scholar]
- 28.Rosenfeld Y., Papo N., Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 2006;281:1636–1643. doi: 10.1074/jbc.M504327200. [DOI] [PubMed] [Google Scholar]
- 29.Tobias P.S., Soldau K., Weiss J. Lipopolysaccharide (LPS)-binding proteins BPI and LBP form different types of complexes with LPS. J. Biol. Chem. 1997;272:18682–18685. doi: 10.1074/jbc.272.30.18682. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburg K., Koch M.H.J., Seydel U. Phase diagram of deep rough mutant lipopolysaccharide from Salmonella minnesota R595. J. Struct. Biol. 1992;108:93–106. doi: 10.1016/1047-8477(92)90010-8. [DOI] [PubMed] [Google Scholar]
- 31.Brandenburg K., Koch M.H.J., Seydel U. Phase diagram of lipid A from Salmonella minnesota and Escherichia coli rough mutant lipopolysaccharide. J. Struct. Biol. 1990;105:11–21. doi: 10.1016/1047-8477(90)90093-r. [DOI] [PubMed] [Google Scholar]
- 32.Richter W., Vogel V., Brandenburg K. Morphology, size distribution, and aggregate structure of lipopolysaccharide and lipid A dispersions from enterobacterial origin. Innate. Immun. 2010 doi: 10.1177/1753425910372434. Aug 3 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Tobias P.S., Gegner J., Ulevitch R.J. LPS binding protein and CD14 in the LPS dependent activation of cells. Prog. Clin. Biol. Res. 1994;388:31–39. [PubMed] [Google Scholar]
- 34.Tobias P.S., Soldau K., Ulevitch R.J. Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J. Biol. Chem. 1995;270:10482–10488. doi: 10.1074/jbc.270.18.10482. [DOI] [PubMed] [Google Scholar]
- 35.Kirkland T.N., Finley F., Tobias P.S. Analysis of lipopolysaccharide binding by CD14. J. Biol. Chem. 1993;268:24818–24823. [PubMed] [Google Scholar]
- 36.Seydel U., Oikawa M., Brandenburg K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 2000;267:3032–3039. doi: 10.1046/j.1432-1033.2000.01326.x. [DOI] [PubMed] [Google Scholar]
- 37.Calhoun W.I., Shipley G.G. Sphingomyelin—lecithin bilayers and their interaction with cholesterol. Biochemistry. 1979;18:1717–1722. doi: 10.1021/bi00576a013. [DOI] [PubMed] [Google Scholar]
- 38.Untrach S.H., Shipley G.G. Molecular interactions between lecithin and sphingomyelin. Temperature- and composition-dependent phase separation. J. Biol. Chem. 1977;252:4449–4457. [PubMed] [Google Scholar]
- 39.Gandhavadi M., Allende D., McIntosh T.J. Structure, composition, and peptide binding properties of detergent soluble bilayers and detergent resistant rafts. Biophys. J. 2002;82:1469–1482. doi: 10.1016/S0006-3495(02)75501-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Opal S.M., Scannon P.J., Lemke J.H. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J. Infect. Dis. 1999;180:1584–1589. doi: 10.1086/315093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.