

Abstract
Complexes formed by DNA and polyethylenimine (PEI) are of great research interest because of their application in gene therapy. In this work, we carried out all-atom molecular dynamics simulations to study eight types of DNA/PEI complexes, each of which was formed by one DNA duplex d(CGCGAATTCGCG)2 and one PEI. We used eight different PEIs with four different degrees of branching and two protonation ratios of amine groups (23% and 46%) in the simulations to investigate how the branching degree and protonation state can affect the binding. We found that 46% protonated PEIs form more stable complexes with DNA, and the binding is achieved mainly through direct interaction between the protonated amine groups on PEI and the electronegative oxygens on the DNA backbone, with some degree of interaction with electronegative groove nitrogens/oxygens. For the 23% protonated PEIs, indirect interaction mediated by one or more water molecules plays an important role in binding. Compared with the protonation state, the degree of branching has a smaller effect on binding, which essentially diminishes at the protonation ratio of 46%. These simulations shed light on the detailed mechanism(s) of PEI binding to DNA, and may facilitate the design of PEI-based gene delivery carriers.
Introduction
Complexes formed by DNA and synthetic polymers are of great research interest because of their application in gene therapy, which involves delivering genetic materials into cells for therapeutic purposes (1,2). This approach offers tremendous hope for patients with cancer, hereditary disease, or viral infection, and has the potential to raise vaccination technology to a new level (1,2). Gene therapy uses carrier molecules such as viruses, synthetic polymers, and carbon nanotubes as vehicles to deliver nucleic acids into cells (2–4). Viruses are the most common and efficient delivery carriers. However, their toxicity and immunogenicity greatly limit their general use (5). Synthetic polymers are an alternative to viral carriers and have the advantages of less toxicity, low cost, ease of production, and versatility for different applications (1,2). Polyethylenimine (PEI) is one of the most effective synthetic polymers for delivering nucleic acids into cells through endocytosis (6,7). PEI can condense nucleic acids and form nanoparticles via electrostatic interactions between negatively charged nucleic acid phosphate groups and positively charged PEI amine groups. The nanoparticles thus formed can facilitate cellular uptake of the nucleic acids and protect the nucleic acids from degradation during the delivery path. However, the efficacy of PEI as a gene delivery vector has been found to depend on the structure and molecular mass of the PEI used (8,9). High molecular mass PEIs (e.g., 25 kDa) can yield a high transfection efficiency but also display high cytotoxicity, whereas low molecular mass (LMM) PEIs (e.g., 600–2000 Da) have low cytotoxicity but poor transfection efficiency. Cross-linked and grafted LMM PEIs, however, can overcome the high cytotoxicity of high molecular mass PEIs and low transfection efficiency of naked LMM PEIs (10). Although progress is being made in the development of better PEI-based gene delivery systems, a detailed understanding of the structure and properties of nucleic acids/PEI complexes is still lacking. It is critical to elucidate the interaction of DNA molecules with carriers at the atomistic level to understand the role of carrier molecules and design more effective DNA/polymer complexes.
To date, experimental studies have focused on studying transfection efficacy using PEIs of different sizes and with different chemical modifications (8,11,12). Utsuno and Uludağ (13) recently performed a thermodynamics analysis of PEI/DNA complexes in different solutions and at different pH values using isothermal titration calorimetry, and found that PEI at low pH had a greater tendency to form a complex with DNA. They also concluded that PEI has two modes of binding to DNA: one involving PEI binding to the DNA groove, and one involving external binding of PEI to the DNA phosphate backbone.
On the theoretical front, Ziebarth and Wang (14) performed all-atomic molecular-dynamics (MD) simulations of DNA/PEI complexes. They focused on the formed structures and PEI's ability to neutralize DNA, and made a comparison with the DNA complexed with the poly-L-lysine carrier. To the best of our knowledge, their study is the only one in which all-atom simulations have been performed for DNA/PEI complexes. The PEIs employed in Ziebarth and Wang's work were of the linear form; however, branched PEIs are also widely used as a gene delivery vector (9).
The protonability of PEI has been credited for its success as a gene delivery vector (6). Compared with other polymers, such as poly-L-lysine, PEI has a high buffer capacity over a broad range of pH values (6,15). It has been reported that PEI has a much higher protonation ratio of amine groups at low pH than at high pH (16). Experimentally, it has been also found that a low-pH environment can facilitate transfection (17), presumably affecting the protonation state of PEI. Hence, it is relevant to study the interaction between DNA and PEIs with different protonation ratios. In Ziebarth and Wang's work (14), two protonation ratios (100% and 50%) were investigated. As reported in most experimental works, the protonation ratio under physiological conditions ranged from 10% to 50% (16,18–21). In this work, we adopted two protonation ratios that are relevant to the gene delivery environment: 23% and 46%. Commercial PEIs have a large structural diversity in terms of branching. To elucidate whether PEIs with different architectures bind differently to DNA, we also studied the effects of PEI branching on the complex binding. In particular, we performed all-atom MD simulations with explicit water and counterions to study the structures formed by the DNA duplex d(CGCGAATTCGCG)2 and eight different PEIs. These PEIs have similar molecular masses of ∼570 Da, but they have four different degrees of branching and two protonation states of amine groups. We used LMM PEIs in the simulations not only because the size of the system that can be simulated in all-atom MD is rather limited, but also because LMM PEI-based gene delivery vectors are showing increasing promise for practical applications (10,22). Our results show how the degree of branching and protonation states affect DNA/PEI binding.
Methods
Initial structures
The DNA simulated in this study was a Drew-Dickerson dodecamer composed of 24 nucleotides, d(CGCGAATTCGCG)2, which carries a total charge of −22 (fully deprotonated) in physiological solution. The initial structure of this dodecamer was built to be a canonical B form using the AMBER NAB tool (23). Four structures of PEI with different degrees of branching were adopted in this work, as shown in Fig. 1. All four structures consisted of 13 amine groups and had a similar molecular mass (∼570 Da). To differentiate the four structures, we introduce the following terminology: purely linear (PL) PEI has 13 amine groups connected in a chain without any branching, representing a linear PEI structure. Semilinear (SL) PEI has three short chains, each containing one amine group, distributed nearly uniformly along the primary chain (we refer to the longest chain in the PEI structure as the primary chain), representing a nearly linear or lightly branched PEI structure. Moderately branched (MB) PEI has a short branch with one amine group and a longer branch with two amine groups on the primary chain, representing a moderately branched structure. Hyperbranched (HB) PEI has four branches, each of which contains one or two amine groups connected to the middle four nitrogens on the primary chain, representing a hyperbranched PEI structure.
Figure 1.
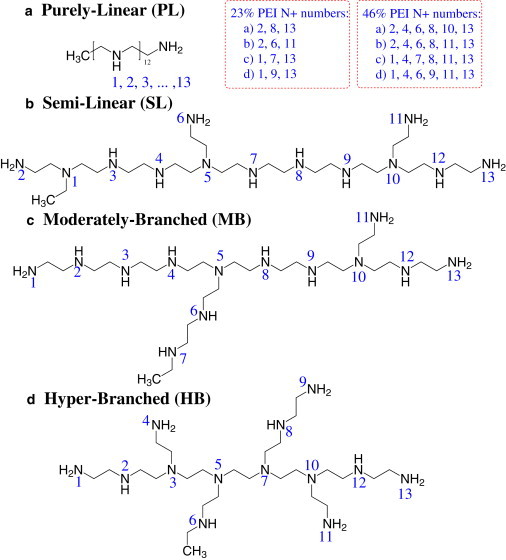
Molecular structures, nitrogen numbering (indexed by numbers near nitrogens), and protonation sites (specified in dashed squares) for four PEIs with similar molecular mass but different degree of branching. (a) Purely linear structure, (b) semilinear structure, (c) moderately branched structure, and (d) hyperbranched structure.
There is no conclusive value for the protonation ratio of PEI amine groups under physiological conditions. The protonation ratio reported in most experimental works ranged from 10% to 50% (16,18–21). Ziebarth and Wang (24) recently performed a Monte Carlo simulation of linear PEI, and reported that the protonation ratio of PEI amine groups was ∼55% under physiological conditions with a nearly alternating arrangement of protonated and unprotonated amines. In their thermodynamics study of 600 Da PEI binding to DNA, Utsuno and Uludağ (13) found that 47% of the PEI amine groups were protonated at pH 6 and 21% were protonated at pH 8. In the work presented here, we chose two protonation ratios close to these values, namely, one protonation state with three out of the total 13 amine groups protonated and one with six amine groups protonated. We assigned the protonation sites on the primary and secondary amines because they are more nucleophilic (e.g., with higher pKa) than the tertiary amines (16). In addition, we assigned the protonation sites as uniformly as possible and separated the neighboring protonation sites as far apart as possible to minimize thermodynamic interactions between the protonated amines. The uniform distribution of the protonation sites was previously confirmed theoretically (24). The PEI nitrogen index and protonation sites are illustrated in Fig. 1. Because the two protonation states correspond to ∼23% and ∼46% protonated amines, respectively, hereafter we will simply refer to them as 23% systems (or 23%) and 46% systems (or 46%).
We first conducted separate MD simulations for each individual PEI with explicit water and counterions, and then adopted the final configurations of these simulations as the initial configurations for PEIs in the complex formation. Details about the MD simulations are described further below.
Force field for PEI
The CHARMM 27 force field was used for all of the molecules in our simulations. However, the residues for PEI did not originally exist in the CHARMM force field. They were devised by adopting parameters from analogous residues available in the CHARMM force field following the CHARMM General Force Field methodology (25). We performed a comparison with Ziebarth and Wang's (14) study, in which the AMBER force field was used, by repeating a simulation with the same simulation procedure. Similar results were obtained, demonstrating the similarity of these two force fields for describing the DNA/PEI systems. We further validated the torsional parameters for PEIs by performing ab initio calculations and repeating two simulations using a different set of torsional parameters (26). Details about the development and validation of the force field for PEIs are given in the Supporting Material.
MD simulations
The MD simulations were performed using MD package NAMD (27) with the CHARMM 27 force field (28,29). The TIP3P water model (30), periodic boundary condition, and full electrostatics with the particle-mesh Ewald method (31) were used for all MD simulations. A cutoff of 12 Å was used for van der Waals interactions and electrostatics pairwise calculations. All bonds containing hydrogen atoms were constrained (SHAKE algorithm (32)) during each run, which allowed us to use a time step of 2 fs.
We minimized the configuration of each PEI residue in NAMD using the devised force field. We then manipulated and glued all of the residues for constructing each PEI using VMD (33), and minimized them using NAMD to generate a starting configuration for each PEI. This starting configuration was then solvated into a water box with a solvation shell of 16 Å thickness, and an adequate number of Cl− ions were added to the water box to neutralize the system. The system was minimized for 5000 steps to remove bad contact and then gradually heated from 0 K to 300 K in 20 ps. The heated system was equilibrated for 6 ns at 300 K and 1 bar. The final configuration of each PEI was used as the starting configuration for the corresponding PEI in the complex simulations.
To construct the initial system for each of the eight DNA/PEI complexes, we first separated the DNA and PEI by 30 Å and solvated them in a water box with a solvation shell of 18 Å thickness. We then added ions to the water box (three Cl− for 23% systems, six Cl− for 46% systems, and 22 Na+ for all systems) by randomly replacing the same number of water molecules. During the simulations, the systems that consisted of DNA, PEI, ions, and water were first minimized for 2000 steps with the solute atoms fixed, and then 2000 steps with the solute atoms harmonically restrained, followed by 1000 steps of unrestrained minimization. The systems were then heated to 300 K in 20 ps with 10 kcal/mol × Å2 harmonic restraint on the nonhydrogen solute atoms. The restraint was kept on for another 3 ns at 300 K and 1 bar to allow the ions to relax around the DNA and PEI. The restraint was then removed and an NPT ensemble simulation was performed for 60 ns for the 23% systems and 40 ns for the 46% systems. The simulation time for the 23% systems was longer than that for the 46% systems because we found it took more time for the complexes in the 23% systems to equilibrate and stabilize, as discussed in the next section. Trajectories were saved every 1000 steps. VMD (33) was used for visualization and trajectory analysis.
Results
In this section, we present our simulation results regarding the flexibility of PEI, the formation of complexes from eight different PEI molecules, and how the PEIs bind to the DNA at atomic level. How the molecular structure of PEI and its protonation ratio affect its binding with DNA is discussed.
PEI flexibility
Fig. 2 shows the radius of gyration, Rg, of the eight PEIs in the single PEI simulations over the 6 ns simulation time. It can be seen that HB is the most compact of the four structures and has the smallest Rg, which remains almost constant during the entire simulation. Furthermore, the degree of ionization does not affect the Rg of the HB PEI. This is because the atoms in the highly branched structure are distributed closer to its center of mass (COM). SL and MB PEIs have similar Rg values, which fluctuate more than that of HB PEI, demonstrating that the SL and MB PEIs are more flexible than the HB PEI. Of the four PEIs, PL shows the most fluctuation in Rg. This is as expected because its linear chain configuration makes it the most flexible structure. Intuitively, one would expect the 46% PEIs in general to have a larger Rg than the 23% PEIs because the former have a higher charge density and presumably possess a more extended structure. Although this is true for the SL and MB structures, our results show that HB PEI has similar Rg values at 23% and 46% protonation ratios. This can be attributed to the dendritic structure of the HB PEI, which results in a mechanically stiff molecule. Even though the electrostatic repulsion at 46% is larger, it is not sufficient to cause a clear increase in Rg. In addition, the 23% PL appears to have a larger Rg than the 46% PL. This may be caused by configuration sampling, as the flexible PL PEI can adopt many equilibrium configurations that may not be sufficiently sampled during the 6 ns MD run.
Figure 2.
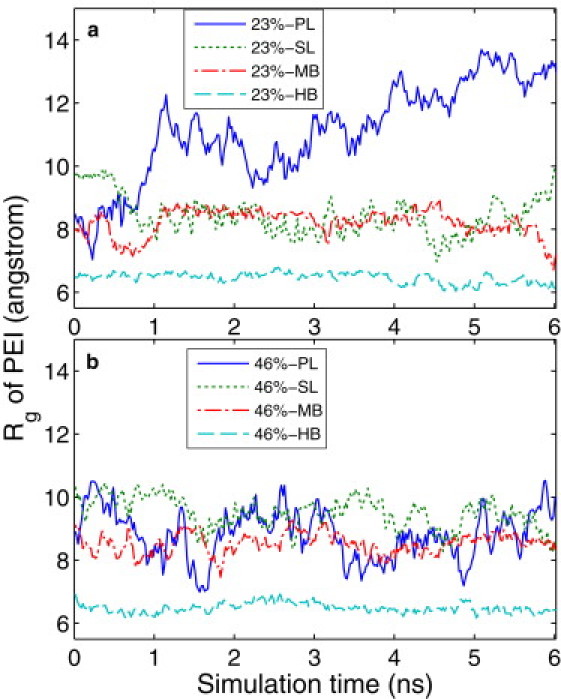
Rg of PEIs in the single-PEI simulations: (a) 23% PEIs and (b) 46% PEIs.
The PEI structure after 6 ns of equilibration was used to form a complex with DNA. Because of the fluctuation shown in Fig. 2, the initial PEI configuration for complexation would be different if it were taken at a different time during this equilibration period. However, we do not think the initial configurations of PEIs in the complex simulations would affect the general results reported here. In fact, we performed simulations for the 23%-PL system with different initial PEI structures, and obtained similar binding results. Furthermore, we repeated the simulation for the 50%-PEI system described by Ziebarth and Wang (14) and obtained similar results regarding the binding structure, ion distribution, and radial distribution function of PEI nitrogens around DNA (see Supporting Material). The initial PEI structures in these two works were very unlikely to be the same, because the simulations were run separately and with different force fields.
Complex formation
Fig. 3 shows the configurations of the eight complexes at the last stage of the simulations. The PL, SL, and MB PEIs mainly interact with one strand of DNA, and a significant part the PEI aligns with the DNA backbone. The HB PEIs tend to stay in the DNA major groove and interact with both strands of the DNA. In all of the simulations, the DNA preserved its B form with distinguishable minor and major grooves. The Watson-Crick DNA basepairs at the middle of the DNAs remained intact; however, in five of the eight cases (46% SL, 23% MB, 46% MB, 23% HB, and 46% HB), one terminal basepair at one end or two terminal basepairs at two ends of the DNA are broken, as can be seen in Fig. 3, d–h. The broken bases can in turn attach to the PEIs, as shown in Fig. 3, d and g. However, this does not have a significant effect on the overall binding pattern, as will be discussed further below.
Figure 3.
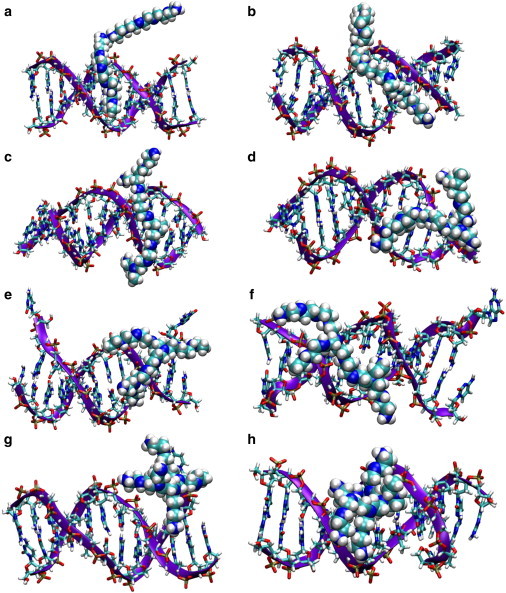
Snapshots for each complex at the last stage of the simulations: (a) 23% PL, (b) 46% PL, (c) 23% SL, (d) 46% SL, (e) 23% MB, (f) 46% MB, (g) 23% HB, and (h) 46% HB.
Fig. 4 shows the COM distances between the DNA and the PEIs during the complexation process, with time zeroed at the moment the restraints were removed from the solutes. The COM distances all start from 30 Å, as the COMs of the PEIs were separated by 30 Å from the DNA COM at the beginning of the simulations. For the 23% systems, the COM distances decrease to a series of plateaus after 20 ns, indicating the formation of DNA/PEI complexes. We further define the bound state as a state in which a significant part of the PEI is in close contact with the DNA, i.e., there is at most a monolayer of water molecules between the PEI and the DNA. By visually checking the complex structures, we ascertained that all 23% PEIs bound to the DNA within 20 ns. Compared with the 23% protonated PEIs, the 46% protonated PEIs (except for the 46%-SL PEI) were faster in moving toward the DNA. By visually checking the complex structures, we found that all of the 46% PEIs bound to the DNA within 7 ns. This is as expected because the electrostatic force, the main driving force for binding, is larger in the 46% systems than in the 23% systems. In addition, the curves for the 23% systems fluctuate more than their 46% counterparts, indicating that the complexes that form in the 23% systems are less stable. Note that a shorter COM distance in these plots does not necessarily indicate tighter binding. This is because the PEIs in our simulations are short molecules compared with the DNA, and their locations along the DNA axis can greatly affect the COM distances. This is clear from Fig. 3, b and d, where the horizontal location of the 46% PL is much closer to the DNA COM compared with the 46% SL. This explains the much larger DNA-PEI COM distance for the 46% SL shown in Fig. 4 b.
Figure 4.
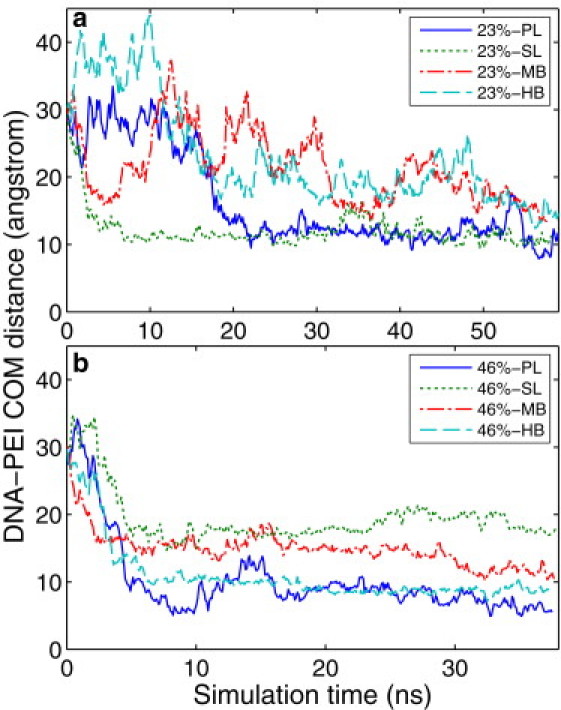
COM distance between the DNA and each PEI as a function of simulation time. Time is zeroed at the moment when the restraints were removed from the solutes. Shown are (a) 23% systems and (b) 46% systems.
Binding pattern
We next examined how PEIs bind to DNA at the atomic level. As previously shown in MD studies by Korolev et al. (34,35) on polyamines, including spermine, spermidine, putrescine, and diaminopropane, the amine groups interact mainly with DNA phosphate groups but can also interact with other electronegative atoms in the DNA grooves. In this work, we sought to determine where and how the PEIs bind to the electronegative atoms of DNA (oxygens and nitrogens), and the stability of such binding.
Table 1 summarizes the average number of PEI nitrogens in close contact (within 4 Å) with DNA electronegative nitrogen/oxygen (N/O) atoms, averaged over the last 20 ns of the simulations. It can be seen that PEI nitrogens in the 46% systems are more likely to be in close contact with the DNA. Except for the 46%-MB PEI, the average number of PEI nitrogens within 4 Å of DNA N/O in the 46% systems is more than twice that of their counterparts in the 23% systems. Moreover, the PEI predominantly interacts with the DNA backbone oxygens, although it also interacts with the DNA base N/O. Note that the summation of numbers in the Backbone O and Base N/O columns is usually higher than the number in the All N/O column. This is because some PEI nitrogens can be simultaneously in close contact with the DNA backbone and the base N/O, whereas we only counted such nitrogens when calculating the number of PEI nitrogens in close contact with all DNA N/O. In Table 1, we further distinguish the PEI nitrogens that interact with O1P and O2P, O3′ and O5′, and O4′ in the DNA backbone oxygens. It can be seen that for the 46% systems, the PEI nitrogens are much more likely to interact with O1P and O2P atoms than with O3′ and O5′. The PEI nitrogens in the 23% systems tend to be almost equally likely to interact with O1P and O2P atoms and with O3′ and O5′ atoms. Except for the 23%-PL PEI, the PEI nitrogens are very unlikely to be in close contact with O4′ atoms on the DNA sugar rings, and some nitrogens can interact with multiple backbone oxygens simultaneously.
Table 1.
Average number of PEI nitrogens within 4 Å of the DNA electronegative atoms (oxygens and nitrogens) in the last 20 ns of simulations
| Systems | All |
Backbone |
Backbone O |
Base |
||
|---|---|---|---|---|---|---|
| N/O | O | O1P and O2P | O3′ and O5′ | O4′ | N/O | |
| 23% PL | 2.25 | 2.08 | 1.00 | 0.83 | 0.79 | 0.71 |
| 46% PL | 6.19 | 6.10 | 6.09 | 0.72 | 0 | 0.09 |
| 23% SL | 2.26 | 2.10 | 1.71 | 1.20 | 0.25 | 0.29 |
| 46% SL | 5.07 | 4.72 | 4.39 | 0.99 | 0.28 | 0.47 |
| 23% MB | 2.34 | 1.96 | 1.46 | 1.00 | 0.23 | 0.46 |
| 46% MB | 3.14 | 2.97 | 2.94 | 0.31 | 0.02 | 0.22 |
| 23% HB | 2.68 | 1.82 | 1.56 | 0.76 | 0.02 | 0.98 |
| 46% HB | 5.51 | 5.12 | 5.11 | 0.64 | 0 | 0.40 |
Fig. 5 shows the percentage of time in which the individual PEI nitrogens are in close contact (within 4 Å) with the DNA electronegative atoms in the last 20 ns of the simulations. A value of 100% means that a nitrogen is within 4 Å of at least one DNA electronegative atom at all times during the last 20 ns of the simulations, and 0% means that a nitrogen is not within 4 Å of any DNA electronegative atoms at all during the last 20 ns of the simulations. Nitrogens in protonated amine groups are marked with a plus symbol (+) for the 23% systems and a star symbol (∗) for the 46% systems in Fig. 5. Several observations can be made from the figure: First, nitrogens in protonated amine groups are generally more likely to be in close contact with the DNA. In fact, of the 23 nitrogens that are in close contact with the DNA for >50% of the time, only six are not protonated. Second, nitrogens in the 46% systems are generally more likely to be in close contact with the DNA than their counterparts in the 23% systems. For 23% PL, SL, MB, and HB PEIs, the average percentages of time the PEI nitrogens were in close contact with DNA are 17%, 17%, 18%, and 21%, respectively. In contrast, the corresponding percentages for the 46% systems are 48%, 39%, 24%, and 42%, respectively. Third, for 46% PL and 46% SL, unprotonated nitrogens sandwiched by two protonation sites have a higher probability to be in close contact with DNA. Specifically, all three of the unprotonated nitrogens that are within 4 Å of the DNA for >50% of the time are located between two protonated nitrogens. Such an observation is not so clear for 46% MB and 46% HB, since because of their branched structure, nitrogens with neighboring indices may not be located next to each other. Nor is this seen in the 23% systems, because the few protonated nitrogens are located too far apart to strongly affect the unprotonated nitrogens in between.
Figure 5.
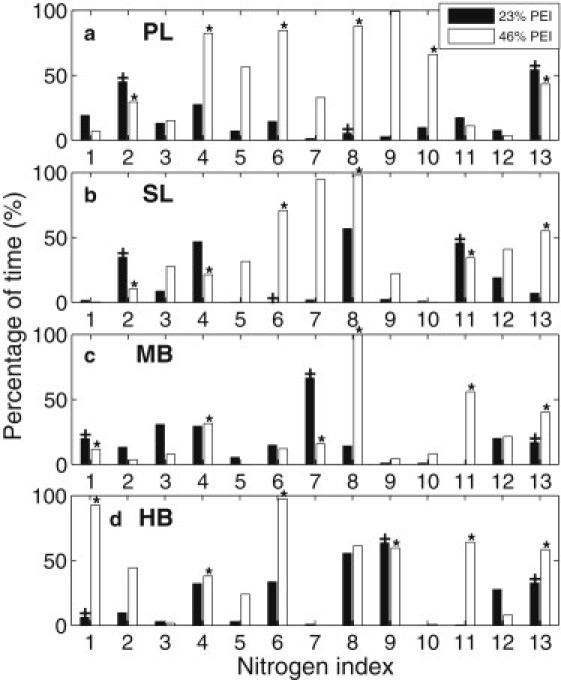
Percentage of time in which each individual PEI nitrogen was in close contact (within 4 Å) with any DNA electronegative atoms during the last 20 ns of the simulations. Nitrogen numbering is the same as that in Fig. 1. Nitrogens in protonated amine groups are marked with + for 23% systems and ∗ for 46% systems. (a) PL, (b) SL, (c) MB, (d) HB.
Let us examine Fig. 5 together with Fig. 1 to further explore how the location of the nitrogens might affect their contact with the DNA. For 46%-PL PEI, the PEI nitrogens in the middle of the polymer chain are more likely to be in close contact with the DNA than are those at the two ends. For 23%-PL PEI, the PEI nitrogens at the two ends are more likely to be in close contact with the DNA than are those in the middle. The same phenomenon was also observed for SL PEI. A possible explanation for such behavior is that for the 23% PEIs, the electrostatic interaction is not strong enough to cause a large part of the PL or SL chain to be in close contact with the DNA. Having the two ends in close contact with the DNA allows the majority of the charges (two out of three) to bind, while also giving some flexibility to the middle part of the PEI molecule. In the 46% systems, however, the electrostatic interaction is sufficiently large to cause the majority of the nitrogens in the 46% PEIs, which are located in the middle, to be in close contact with the DNA, leaving the end nitrogens with more fluctuation. On the basis of this observation, we can make the following conjecture: If an LMM PL or SL PEI forms a complex with a DNA at a high protonation ratio (e.g., 46%), the complex might be more stable for longer PEIs because of its low percentage of end nitrogens. At a low protonation ratio (e.g., 23%), shorter PEIs might form a tighter complex with DNA because a higher percentage of end nitrogens are available. This phenomenon becomes less pronounced as the degree of branching is increased to MB, and disappears for HB, because all of the protonation sites are located at the branch ends.
Because the PEI nitrogens interact mainly with the DNA backbone oxygens, we plot the radial distribution function (RDF) of the PEI nitrogens around the DNA backbone oxygens in Fig. 6. Fig. 6, a and b, are respectively the RDF plots for all PEI nitrogens and for protonated PEI nitrogens around the DNA backbone oxygens in the 23% systems. Fig. 6, c and d, are the same RDF plots for the 46% systems. These RDF plots were generated from the trajectories of the last 20 ns of the simulations. In all cases, a step distance of 0.2 Å was used and the curves were normalized by the total number of PEI nitrogens (n = 13). For almost all of the RDF curves, there are two predominant peaks: one at ∼3 Å and one at ∼5 Å from the oxygens. The first peak corresponds to the expected distance for direct contact between the PEI amine groups and the DNA oxygens through hydrogen bonding. The second peak corresponds to the distance for an indirect interaction, such as hydrogen bonding mediated by one water molecule. For some RDF curves, there is a less distinct third peak at ∼7 Å. This third peak corresponds to weak indirect interactions, such as hydrogen bonding mediated by two or more water molecules. For the 23% systems, it can be seen clearly from Fig. 6 a that the second peaks are predominant over the first peaks, whereas in Fig. 6 b the first peaks are more pronounced than the second peak. This indicates that the protonated PEI nitrogens are more likely to be in direct contact with the DNA, whereas the majority of nitrogens are in indirect contact with the DNA. From the RDF plots of the 46% systems shown in Fig. 6, c and d, it can be seen the profiles of the first peaks from the two figures are almost identical. This indicates that the majority of the PEI nitrogens in direct contact with the DNA are from the protonated amine groups.
Figure 6.
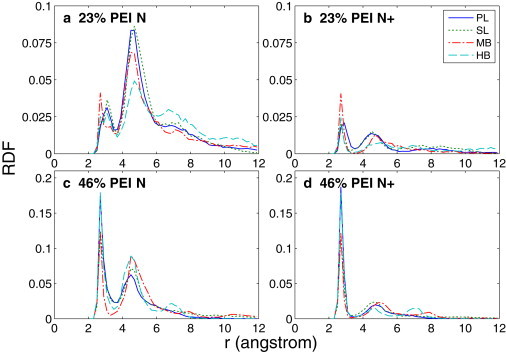
RDF of the PEI nitrogens around the DNA backbone oxygens based on the last 20 ns trajectory of the simulations: (a) 23% all PEI nitrogens, (b) 23% protonated PEI nitrogens, (c) 46% all PEI nitrogens, and (d) 46% protonated PEI nitrogens.
To quantify the number of PEI nitrogens involved in each peak of the RDF, we plotted the cumulative number of PEI nitrogens around the DNA backbone oxygens in Fig. 7. From Fig. 7 a (for all PEI nitrogens in the 23% systems), it can be seen that approximately two PEI nitrogens are within 4 Å of the DNA backbone oxygens for all four PEI structures, which corresponds to the first peak in Fig. 6 a. There are approximately six PEI nitrogens at 4–6 Å from the DNA backbone oxygens. These PEI nitrogens account for approximately half of the total number of PEI nitrogens and make the second peaks in Fig. 6 a dominant. Therefore, for the 23% systems, the majority of the PEI nitrogens are in indirect interaction with the DNA at 4–6 Å. The cumulative number in Fig. 7 b shows that for the 23% systems, there is approximately one protonated PEI nitrogen within 4 Å of DNA backbone oxygens and approximately one protonated PEI nitrogen at 4–6 Å from the DNA backbone oxygens. Note that in most of the cases shown in Fig. 7 b, the cumulative numbers sharply increase from zero to a plateau at 2.3 Å. This indicates that the direct contact between protonated amine groups and the DNA is strong hydrogen bonding. For the 46% systems, the fact that the majority of the PEI nitrogens in direct contact with the DNA are from the protonated amine groups is further confirmed by the cumulative number curves in Fig. 7, c and d. Specifically, the cumulative number of all PEI nitrogens within 3 Å of the DNA backbone oxygens is very close to that for the protonated PEI nitrogens. About three out of six protonated PEI nitrogens are in direct contact with DNA, whereas most unprotonated PEI nitrogens are in indirect interaction with DNA.
Figure 7.
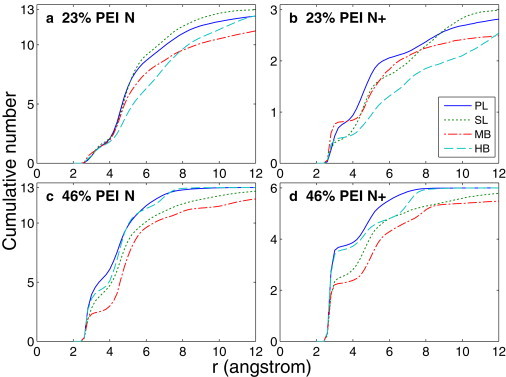
Cumulative number of the PEI nitrogens around the DNA backbone oxygens based on the last 20 ns trajectory of the simulations: (a) 23% all PEI nitrogens, (b) 23% protonated PEI nitrogens, (c) 46% all PEI nitrogens, and (d) 46% protonated PEI nitrogens.
To further demonstrate the stability of the formed complexes, we plotted the RDF and cumulative number curves based on trajectories within different time windows in the simulations (see Supporting Material). For the 23% systems, the figures show that even after 49 ns of simulation, the curves are still evolving with time, and the order of the curves corresponding to different PEI structures does not remain the same at all times. This indicates that the complexes formed in the 23% systems are not stable, which is consistent with the fact that the majority of the nitrogens bind to DNA through indirect interactions. Compared with the 23% systems, the RDF and cumulative number curves for the 46% systems demonstrate more stability (i.e., less variation among different simulation windows). Moreover, the curves corresponding to different PEI structures are closer to each other compared with the 23% systems. In fact, after 40 ns of simulations, these curves essentially overlap one another. This indicates that the degree of branching has a vanishingly small effect on binding at a protonation ratio of 46%.
Discussion
Implications
To our knowledge, this is the first MD study to investigate the effects of the degree of branching and protonation state on PEI binding of DNA. The results shed light on the detailed mechanism(s) of PEI binding to DNA, and will help investigators better understand and design PEI-based gene carriers. A clear outcome of this study is the beneficial effect of a higher PEI protonation state on DNA binding, resulting from the shorter complex formation time, and more-intimate contact of PEI nitrogens with DNA at the higher protonation state. Changing the pH of a PEI solution is a practical way to enhance the protonation state of PEI, and we previously observed that pH changes from 6.0 to 9.0 changed the percentage of protonated amines from 47% to 13% (13). Consistent with the MD results presented here, a better DNA binding was observed when PEI interaction to DNA was investigated at low pH (13,17), and the PEI molecule became highly protonated without significantly affecting the charge of the DNA. These experimental studies were conducted with ∼600 Da branched PEI (13) or ∼25,000 Da linear PEI (17), but the role of protonation on DNA binding should be independent of the size and the architecture of the PEI molecule employed. The initial binding constant K1 (estimated after fitting the titration heat with a single set of identified sites model) was found to be enhanced at lower-medium pH for such an interaction (13). A stronger binding is likely to result in a better ability of PEI to deliver extracellular DNA molecules into cells, resulting in better gene expression (17). Tailoring for a stronger binding is also beneficial when one considers the use of such complexes in vivo, where highly bound complexes have been shown to be more resilient against degradation (36). Although the predominant PEI-DNA interaction is expected to be between the electronegative oxygen atoms on the DNA backbone and protonated PEI nitrogens, our simulations also predict interactions with the DNA base oxygens and nitrogens, implying DNA groove binding of the PEI. This was experimentally shown to be the case in our hands (13) as well as in independent studies (37).
It is known that both linear and branched forms of PEI can complex with DNA and form particles suitable for cell uptake and gene expression. Independent laboratories that compared DNA binding with linear versus branched PEI experimentally observed stronger DNA binding by the branched PEI (38–41). The functional consequence of the stronger binding could be better gene expression due to increased cellular uptake and/or better protection against degradation; however, less stable complexes (i.e., complexes formed with linear PEI) may result in better gene expression under some conditions because less stable complexes are also more prone to free the DNA inside the cells and make it available for transcription (42). In our simulations, we did not observe a clear trend for how the binding of the protonated amines with the DNA backbone oxygens was affected by the degree of branching. It remains to be investigated whether this is also the case for PEIs with higher molecular mass. The PEI molecules chosen for this study had similar molecular sizes, and it is well known that DNA binding is significantly influenced by the size of the PEI as well as its architecture (42). We will address this issue in future studies to better understand the role of architecture in combination with the molecular size.
Limitations
The PEI molecules simulated in this work are small, LMM molecules. Experiments have shown that PEIs with higher molecular mass (e.g., ∼25 kDa) are the most effective for gene delivery. However, it is not practical to simulate such large molecules by MD, even with state-of-the-art computation capacities. The results we obtained with LMM PEIs are still expected to shed light on binding in the DNA/PEI complexes, because we believe that the binding mechanism at the atomistic level is the same for PEIs of any molecular mass. In addition, the high toxicity of larger PEI molecules limits their use in practical situations, whereas recent successful attempts to deliver nucleic acids with modified LMM PEIs (10,22) encouraged us to study DNA interactions with LMM PEIs.
In this work we focused on single PEI binding with a single DNA molecule. When more than one PEI and DNA molecules are present, multiple PEIs can bind to a single DNA segment, and a single PEI can bridge multiple DNA molecules. Interactions between multiple formed complexes can also occur. These are interesting and practically related problems to be investigated in future work.
Finally, it is known that counterion release during complex formation can play an important role in binding. Counterion release is clearly observed in our simulations on longer PEIs (see Supporting Material). However, for smaller PEIs with fewer charges, we found no distinct correlation between binding and counterion release. Whether increasing the salt concentration, i.e., by adding more ions in the simulation, would change the scenario remains to be determined.
Conclusions
In this work, we performed all-atom MD simulations of a DNA duplex (d(CGCGAATTCGCG)2) with PEIs of four different architectural structures and two protonation ratios. Our results provide insight into how the degree of branching and the protonation state of the PEI affect binding to DNA. We report the following findings: 1), The PEIs primarily bind to the DNA backbone through the formation of hydrogen bonding with the backbone oxygens. 2), The 46% protonated PEIs bind to the DNA mainly through direct hydrogen bonding, whereas for the 23% protonated PEIs, indirect interaction mediated by water molecules plays an important role in binding. This results in less stable complex formation for the 23% protonated PEIs. These findings are also consistent with experimental results indicating that more stable binding is found at low pH (13), because a higher protonation ratio is expected at lower pH values. 3), At the 23% protonation ratio, the RDF and cumulative number of PEI nitrogens around DNA backbone oxygens show some difference between the different PEI structures, but we did not observe a systematic trend for such a difference, and the less stable complexation also leads to fluctuations in the behavior of these curves. At the 46% protonation ratio, the effect of PEI structure essentially diminishes. In general, our results show that for the LMM PEI structures investigated here, the degree of branching has a smaller influence on the DNA binding than does the protonation state of the polymers.
Acknowledgments
We acknowledge the computing resources and technical support from WestGrid and the high performance computing facility at the National Institute for Nanotechnology. This work was supported by the National Science and Engineering Research Council of Canada, Alberta Innovates—Technology Futures and Canada Foundation for Innovation.
Supporting Material
References
- 1.Ledley F.D. Nonviral gene therapy: the promise of genes as pharmaceutical products. Hum. Gene Ther. 1995;6:1129–1144. doi: 10.1089/hum.1995.6.9-1129. [DOI] [PubMed] [Google Scholar]
- 2.Luo D., Saltzman W.M. Synthetic DNA delivery systems. Nat. Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- 3.Mulligan R.C. The basic science of gene therapy. Science. 1993;260:926–932. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 4.Pantarotto D., Singh R., Bianco A. Functionalized carbon nanotubes for plasmid DNA gene delivery. Angew. Chem. Int. Ed. Engl. 2004;43:5242–5246. doi: 10.1002/anie.200460437. [DOI] [PubMed] [Google Scholar]
- 5.Lehrman S. Virus treatment questioned after gene therapy death. Nature. 1999;401:517–518. doi: 10.1038/43977. [DOI] [PubMed] [Google Scholar]
- 6.Boussif O., Lezoualc'h F., Behr J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godbey W.T., Wu K.K., Mikos A.G. Poly(ethylenimine) and its role in gene delivery. J. Control. Release. 1999;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 8.Godbey W.T., Wu K.K., Mikos A.G. Size matters: molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999;45:268–275. doi: 10.1002/(sici)1097-4636(19990605)45:3<268::aid-jbm15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.Wightman L., Kircheis R., Wagner E. Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. J. Gene Med. 2001;3:362–372. doi: 10.1002/jgm.187. [DOI] [PubMed] [Google Scholar]
- 10.Jere D., Jiang H.L., Cho C.S. Degradable polyethylenimines as DNA and small interfering RNA carriers. Expert Opin. Drug Deliv. 2009;6:827–834. doi: 10.1517/17425240903029183. [DOI] [PubMed] [Google Scholar]
- 11.Kircheis R., Wightman L., Wagner E. Design and gene delivery activity of modified polyethylenimines. Adv. Drug Deliv. Rev. 2001;53:341–358. doi: 10.1016/s0169-409x(01)00202-2. [DOI] [PubMed] [Google Scholar]
- 12.Schaffert D., Wagner E. Gene therapy progress and prospects: synthetic polymer-based systems. Gene Ther. 2008;15:1131–1138. doi: 10.1038/gt.2008.105. [DOI] [PubMed] [Google Scholar]
- 13.Utsuno K., Uludağ H. Thermodynamics of polyethylenimine-DNA binding and DNA condensation. Biophys. J. 2010;99:201–207. doi: 10.1016/j.bpj.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebarth J., Wang Y. Molecular dynamics simulations of DNA-polycation complex formation. Biophys. J. 2009;97:1971–1983. doi: 10.1016/j.bpj.2009.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang M.X., Szoka F.C. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 1997;4:823–832. doi: 10.1038/sj.gt.3300454. [DOI] [PubMed] [Google Scholar]
- 16.Suh J., Paik H., Hwang B. Ionization of poly(ethylenimine) and poly(allylamine) at various pHs. Bioorg. Chem. 1994;22:318–327. [Google Scholar]
- 17.Fukumoto Y., Obata Y., Yamaguchi N. Cost-effective gene transfection by DNA compaction at pH 4.0 using acidified, long shelf-life polyethylenimine. Cytotechnology. 2010;62:73–82. doi: 10.1007/s10616-010-9259-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagaya J., Homma M., Minakata A. Relationship between protonation and ion condensation for branched poly(ethylenimine) Biophys. Chem. 1996;60:45–51. [Google Scholar]
- 19.von Harpe A., Petersen H., Kissel T. Characterization of commercially available and synthesized polyethylenimines for gene delivery. J. Control. Release. 2000;69:309–322. doi: 10.1016/s0168-3659(00)00317-5. [DOI] [PubMed] [Google Scholar]
- 20.Koper G., van Duijvenbode R., Borkovec A. Synthesis and protonation behavior of comblike poly(ethyleneimine) Macromolecules. 2003;36:2500–2507. [Google Scholar]
- 21.Choosakoonkriang S., Lobo B.A., Middaugh C.R. Biophysical characterization of PEI/DNA complexes. J. Pharm. Sci. 2003;92:1710–1722. doi: 10.1002/jps.10437. [DOI] [PubMed] [Google Scholar]
- 22.Alshamsan A., Haddadi A., Uludağ H. Formulation and delivery of siRNA by oleic acid and stearic acid modified polyethylenimine. Mol. Pharm. 2009;6:121–133. doi: 10.1021/mp8000815. [DOI] [PubMed] [Google Scholar]
- 23.Case D., Darden T., Kollman P. University of California; San Francisco: 2008. AMBER 10. [Google Scholar]
- 24.Ziebarth J.D., Wang Y. Understanding the protonation behavior of linear polyethylenimine in solutions through Monte Carlo simulations. Biomacromolecules. 2010;11:29–38. doi: 10.1021/bm900842d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanommeslaeghe K., Hatcher E., MacKerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong H., Hyun J., Wheeler R. Molecular dynamics simulations and structural comparisons of amorphous poly(ethylene oxide) and poly(ethylenimine) models. Polymer (Guildf.) 2001;42:7809–7817. [Google Scholar]
- 27.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks B., Bruccoleri R., Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 29.MacKerell A.D., Jr., Brooks B., Karplus M. CHARMM: the energy function and its parameterization with an overview of the program. In: Schleyer P.v.R., editor. The Encyclopedia of Computational Chemistry. Vol. 1. John Wiley & Sons; Chichester: 1998. pp. 271–277. [Google Scholar]
- 30.Jorgensen W. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers—application to liquid water. J. Am. Chem. Soc. 1981;103:335–340. [Google Scholar]
- 31.Darden T., York D., Pedersen L. Particle mesh Ewald—an n.log(n) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 32.Ryckaert J., Ciccotti G., Berendsen H. Numerical-integration of Cartesian equations of motion of a system with constraints—molecular-dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 33.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 34.Korolev N., Lyubartsev A.P., Nordenskiöld L. On the competition between water, sodium ions, and spermine in binding to DNA: a molecular dynamics computer simulation study. Biophys. J. 2002;82:2860–2875. doi: 10.1016/S0006-3495(02)75628-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korolev N., Lyubartsev A.P., Nordenskiöld L. A molecular dynamics simulation study of polyamine- and sodium-DNA. Interplay between polyamine binding and DNA structure. Eur. Biophys. J. 2004;33:671–682. doi: 10.1007/s00249-004-0410-7. [DOI] [PubMed] [Google Scholar]
- 36.Mullen P.M., Lollo C.P., Carlo D.J. Strength of conjugate binding to plasmid DNA affects degradation rate and expression level in vivo. Biochim. Biophys. Acta. 2000;1523:103–110. doi: 10.1016/s0304-4165(00)00104-5. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Y.L., Li Y.Z. The interaction of poly(ethylenimine) with nucleic acids and its use in determination of nucleic acids based on light scattering. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004;60:377–384. doi: 10.1016/s1386-1425(03)00243-9. [DOI] [PubMed] [Google Scholar]
- 38.Seib F.P., Jones A.T., Duncan R. Comparison of the endocytic properties of linear and branched PEIs, and cationic PAMAM dendrimers in B16f10 melanoma cells. J. Control. Release. 2007;117:291–300. doi: 10.1016/j.jconrel.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 39.Intra J., Salem A.K. Characterization of the transgene expression generated by branched and linear polyethylenimine-plasmid DNA nanoparticles in vitro and after intraperitoneal injection in vivo. J. Control. Release. 2008;130:129–138. doi: 10.1016/j.jconrel.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahn L.D., Kong H., Mooney D.J. Polycation structure mediates expression of lyophilized polycation/pDNA complexes. Macromol. Biosci. 2010;10:1210–1215. doi: 10.1002/mabi.201000067. [DOI] [PubMed] [Google Scholar]
- 41.Kwok A., Hart S.L. Comparative structural and functional studies of nanoparticle formulations for DNA and siRNA delivery. Nanomedicine. 2011;7:210–219. doi: 10.1016/j.nano.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Bertschinger M., Backliwal G., Wurm F.M. Disassembly of polyethylenimine-DNA particles in vitro: implications for polyethylenimine-mediated DNA delivery. J. Control. Release. 2006;116:96–104. doi: 10.1016/j.jconrel.2006.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.