

Abstract
Clinically relevant studies of cell function in vitro require a physiologically-representative microenvironment possessing aspects such as a 3D extracellular matrix (ECM) and controlled biochemical and biophysical parameters. A polydimethylsiloxane (PDMS) microfluidic system with a 3D collagen gel has previously served for analysis of factors inducing different responses of cells in a 3D microenvironment under controlled biochemical and biophysical parameters. In the present study, applying the known commercially-viable manufacturing methods to a cyclic olefin copolymer (COC) material resulted in a microfluidic device with enhanced 3D gel capabilities, controlled surface properties, and improved potential to serve high-volume applications. Hot embossing and roller lamination molded and sealed the microfluidic device. A combination of oxygen plasma and thermal treatments enhanced the sealing, ensured proper placement of the 3D gel, and created controlled and stable surface properties within the device. Culture of cells in the new device indicated no adverse effects of the COC material or processing as compared to previous PDMS devices. The results demonstrate a methodology to transition microfludic devices for 3D cell culture from scientific research to high-volume applications with broad clinical impact.
Keywords: COC, microfluidics, 3D cell culture, hot embossing, thermal bonding, surface treatment
1 Introduction
The microenvironment surrounding a cell significantly influences cell function through both biochemical and biophysical parameters. Traditional platforms to study the biochemical and biophysical parameter influence on cell function often consist of culture wells, simple flow chambers, or stretchable substrates in which typically one, or a small number of factors, can be controlled and studied. As an alternative to typical methods, microfluidic systems display precise control over multiple factors and provide a wide range of capabilities including establishment and control of biochemical or thermal gradients, improved access for imaging, and control of communication among multiple cell types in a single in vitro device (Bowden, Weck et al. 2001; Whitesides, Ostuni et al. 2001; El-Ali, Sorger et al. 2006; Chung, Sudo et al. 2010). For example, recent work optimizes microfluidic systems to control (Vickerman, Blundo et al. 2008; Sudo, Chung et al. 2009) and investigate (Chung, Sudo et al. 2009; Chung, Sudo et al. 2009) angiogenesis arising from endothelial cells cultured within the device. The microfluidic platform, made of polydimethylsiloxane (PDMS), controls biochemical and biophysical factors in a 3D gel microenvironment, resulting in angiogenic sprouting and controlled cell migration within the 3D gel (Vickerman, Blundo et al. 2008; Chung, Sudo et al. 2009; Mack, Zhang et al. 2009; Sudo, Chung et al. 2009). Although the microfluidic platform provides advantages over traditional methods and demonstrates unique control over cell structure formation, opportunities exist to improve the platform further.
Material selection and fabrication methods largely dictate performance, applicability, and manufacturability of a microfluidic device. Despite the broad use of PDMS-based microfluidic systems, PDMS has limitations from both a materials and processing perspective. From a materials perspective, PDMS structures can absorb significant quantities of small molecules such as hormones (Regehr, Domenech et al. 2009), resulting in significant inaccuracy for any assay involving small molecules such as evaluation of a pharmaceutical compound. Since surface properties significantly alter protein adsorption, activity, and consequent function of cells bound to the proteins (Keselowsky, Collard et al. 2005), the inherently hydrophobic surface of PDMS may lead to an unknown and uncontrolled impact on cell function within the device. In these cases, PDMS could result in an altered concentration of a specific molecule which has a significant impact on the experimental result or an altered protein layer resulting in different cell signaling and differentiation. In addition, the low elastic modulus of PDMS may allow significant dimensional changes of the microfluidic structures due to the pressure used to induce flow within the system. From a processing perspective, PDMS fabrication methods limit mass production and automation. The soft lithography method of fabricating PDMS devices involves several sequential steps, including a time-dependent curing step, which limits the ability to reduce cycle time and restricts the processing to batch fabrication. Post-curing solvent extraction of uncured oligomers from PDMS requires additional cycle time and may result in leaching of solvents into the cell culture space. Alternative materials and manufacturing methods exist which do not possess the limitations of PDMS and therefore further improve microfluidic platforms as used for biological experimentation and cell culture.
For commercial applications of microfluidics for cell culture, thermoplastics, such as polystyrene commonly used for cell culture, have several advantages. Thermoplastics have controllable surface properties to enable specific functions (Bhattacharyya and Klapperich 2006; Diaz-Quijada, Peytavi et al. 2007) and adapt well to simple, low-cost fabrication techniques (Martynova, Locascio et al. 1997; Narasimhan and Papautsky 2004). Among many thermoplastic materials, cyclic olefin copolymer (COC) has good optical properties, good chemical properties such as low water resistance (Tsao and DeVoe 2009), and bulk properties adjustable to ease fabrication (Leech 2009). Optical access to enable phase and fluorescent imaging is an important advantage of using microfluidic devices for cell culture and may necessitate good optical properties of the device material. Fortunately COC exhibits low autofluorescence (Hawkins and Yager 2003; Piruska, Nikcevic et al. 2005; Diaz-Quijada, Peytavi et al. 2007) an essential property particularly for fluorescent imaging. Additionally, COC has strong chemical resistance and low water absorption, (Diaz-Quijada, Peytavi et al. 2007) both factors critical for the success of devices often sterilized in chemical solvents and used in aqueous environments.
Fabrication of microfluidic devices for large sample sizes or commercial applications requires a molding process suitable to a low-cost, high-throughput format. Hot embossing provides a low-cost, high-throughput method to mold thermoplastics with control of feature dimensions in the nanoscale over a large area for thermoplastic cell culture materials (Charest, Eliason et al. 2005) or serial processing (Ahn and Guo 2009) formats. Hot embossing materials feature sizes of structures with significant influence over cells (Charest, Garcia et al. 2007). Several materials are available for hot embossing molds including etched silicon (Charest, Bryant et al. 2004) and electroformed nickel (Charest, Eliason et al. 2005), with epoxy masters widely used since they are durable and inexpensive to fabricate (Steigert, Haeberle et al. 2007; Desai, Freeman et al. 2009). With widespread use in plastics manufacturing and proven control of micro- and nanoscale features, hot embossing can ease the transition of microfluidic cell culture devices from low-volume research applications to clinically relevant sample sizes and the commercial market.
Better material selection and more commercially-relevant manufacturing methods can improve upon the already established use of in vitro microfluidic systems for analysis of biomechanical and biochemical factors on cell function. In this paper, we describe a method to fabricate microfluidic platforms for cell culture using thermoplastic COC as the device material, hot embossing to mold the microfluidic structures, and a roller-lamination process to seal the devices. In addition, we use an energetic oxygen plasma to treat the COC surfaces to control surface hydrophobicity and improve the roller-lamination bonding process. Finally, we demonstrate functionality of the device by injecting and properly locating a 3D gel into the device and culturing cells within the device. The technology described here provides control of device parameters to better regulate in vitro conditions leading to elucidating mechanisms of cell interactions with biophysical and biochemical parameters. In addition, manufacturing the microfluidic system through a low-cost, high-throughput method extends the reach of the technology beyond the research setting to commercially-relevant applications such as pharmaceutical screening for safety, efficacy, and optimal patient-specific drug interactions in pre-clinical or clinical settings.
2 Materials and Methods
2.1 Material Selection
Cyclic olefin copolymer served as the base material of the hot embossed microfluidic devices for its low water and chemical absorptivity and wide spectrum of optical transmission. Manufacturers offer several types of COC with different glass transition temperatures, allowing optimal COC material selection depending on device requirements and processing constraints. The COC material used in this study was 2.0 mm thick Zeonor 1060R (Tg 100°C) from Zeon Chemicals (Louisville, KY, USA) as the plate for embossing, and 100 μm thick Topas 8007 (Tg 77°C) from Topas (Tokyo, Japan) as the laminated thin film layer.
2.2 Microfluidic Device Molding
Hot embossing from an epoxy master, schematically illustrated in Fig. 1, was used to form the microfluidic channels and strucutres. A durable master, which can withstand high temperature and pressure, was created from epoxy to hot emboss the COC material. Fabrication of the epoxy master required a series of steps. A first mold was produced consisted of a 4-inch silicon wafer with a SU8 photoresist (MicroChem, MA, USA) layer, patterned using standard photolithography techniques as described in detail previously (Borenstein, Terai et al. 2002). Briefly, SU8 was applied to clean, pre-baked silicon wafers, spin-coated at 2000 rpm for 30 seconds twice, exposed to UV light using a mask aligner (Karl Suss MA-6; Suss America, Waterbury, VT), developed for 12 minutes in developer (Shipley AZ400K) and baked at 150°C for 15 minutes, resulting in a110 μm ± 10 μm height SU8 pattern. The patterned SU8 photoresist served as a mold to create the second mold, a negative replica cast mold made of polydimethyl siloxane (PDMS, Sylgard 184, Dow Chemical, MI, USA). The PDMS base elastomer and curing agent were mixed in a 10:1 ratio by mass, poured on the patterned SU8 wafer, placed under house vacuum for 30 minutes to degas, and cured in an oven at 80°C for more than 2 hours. The durable epoxy master mold was created by mixing Conapoxy (FR-1080, Cytec Industries Inc., Olean, NY, USA) in a 3:2 volume ratio of resin and curing agent, pouring it into the PDMS mold, and curing it at 120°C for 6 hours.
Fig. 1.

Hot embossing generated the devices. Patterned SU8 photoresist on a silicon wafer (a) served as a mold to create a negative replica in PDMS (b) to permit pouring and curing of epoxy (c) to create the durable epoxy master mold (d). The master mold formed the microfluidic features in the COC plate under load and elevated temperature through hot embossing (e). The resulting embossed COC plate was trimmed (f), drilled for fluid connections (g), then sealed with a thin COC laminate layer to complete the array of finished devices (h)
The epoxy master formed the microfluidic features in the COC plate through hot embossing using a custom built press, which controlled temperature via a thermocouple and heater control system and embossing pressure by applying compressed air and vacuum. The COC plate was placed on the epoxy master, loaded into the press, and embossed at 100 kPa and 120°C for one hour. The resulting embossed plates were cooled to 60°C under 100 kPa pressure, then unloaded from the press and separated from the epoxy master mold. The embossed plates were then trimmed and drilled to create holes to fluidically access the channels. After cleansing, the COC embossed plates received an oxygen plasma treatment using a Technics plasma etcher (Technics Inc., Dublin, CA, USA) for 30 seconds at 100 W intensity and 13 Pa pressure. To seal the devices, a roller-lamination process was used to bond a thin COC film to the embossed microfluidic plate. Preheating the embossed plate with the thin film COC on top covering the microfluidic channels occurred on hot plate for 20 minutes at 77°C. After preheating, the embossed plate and film were run between two rollers heated to 120°C for lamination by thermal fusion bonding. After assembly of the device was complete, the devices were sterilized using ethylene oxide (ETO) for 24 hours. To facilitate adhesion of the collagen gel to the COC as well as cell attachment within the device, the inner surfaces of the device were soaked in 1 mg/ml poly-d-lysine (PDL, Sigma-Aldrich, St. Louis, MO, USA) coating solution for a minimum of 3 hours as described previously (Chung, Sudo et al. 2009).
For cell viability tests, PDMS devices used as controls were fabricated as described in detail previously (Chung, Sudo et al. 2009). Briefly, a PDMS mixture was cured in a mold consisting of patterned SU8 on a silicon wafer, removed from the mold, then perforated with a biopsy punch to create ports for fluidic access and autoclaved for 20 minutes on the wet cycle followed by 20/10 minutes on the dry cycle. The PDMS devices and glass cover slips were oxygen plasma treated, irreversibly bonded together to seal the fluidic channels, and later coated with PDL solution.
2.3 Metrology
Scanning electron microscopy (SEM)
The microchannel features of the SU8 on silicon molds, PDMS molds, epoxy master molds, and embossed COC devices were observed using SEM. All samples were cleaned using compressed nitrogen gas to remove dust particles, mounted onto SEM stubs using carbon tape, then sputter coated with 50 angstroms of gold in argon plasma under vacuum. The images were captured with 5kV acceleration using a Hitachi S-3500N (Tokyo, Japan).
Profilometry
The profile of the epoxy master molds and embossed COC device were traced using a contact profilometer (P-16 KLA-Tencor, Milpitas, CA, USA) with a triangular tip probe. The samples were cleaned with compressed nitrogen gas prior to the scan. The scan speed was 50 μm/sec with a 50 Hz sampling rate. The linear scan was performed over a length of 0.5 mm per scan for each sample.
Three-point bending tests using Dynamic Mechanical Analyzer (DMA)
Bond strength between the embossed plates and the laminated thin film created during the thermal fusion process was evaluated by a three-point bending test using a dynamic mechanical analyzer (Q800 DMA, TA Instruments, New Castle, DE, USA). A 55 mm (L) × 12 mm (W) × 2 mm (thick) COC plate test sample base plate was created to fit the dimensions of the three-point bending test and bonded to a 50 mm (L) × 4 mm (W) × 100 μm (thick) COC film using the roller-laminator procedure described above. For each test group, different plasma treatment conditions were applied to the materials before bonding. All plasma indicates plasma treatment to both COC plate and film, half plasma indicates plasma treatment only to the COC plate, and no plasma indicates no plasma treatment prior to bonding. After thermal bonding, another base plate was bonded to the previously thermally bonded film-plate sample using an adhesive carefully applied to the film using a sharp utensil to spread the adhesive uniformly as a thin layer and restrict adhesive to the film area. The samples were dried for at least 24 hours before testing to ensure that the adhesive was completely dried. The final configuration of the test sample consisted of two 2 mm thick COC plates sandwiching the thin film, with one side having a thermally bonded plate-film bond and the other side having an adhesively bonded plate-film bond. In the three-point bending test, the displacement of the sample was recorded as load increased at rate of 2.0 N/min up to a maximum load of 18N. During load ramping, a discontinuity in displacement versus load output was observed as the bond failed. A student t-test was used for statistical comparisons with p-values less than 0.03 considered significant.
Contact angle measurement
Contact angles of distilled water on the COC plate were measured before and after surface treatment using an optical goniometer (Model 190, Rame-hart instrument co., Netcong, NJ, USA) by sessile drop method. Water droplets of 10 μl in volume were released from a syringe above the sample surface, and the images of droplet formation captured by high-resolution camera were analyzed using image analysis software (DROPImage, Rame-hart instrument co., Netcong, NJ USA) to calculate the contact angle. For each time point, a minimum of 3 locations on each sample were tested and contact angles were averaged.
In order to evaluate the change of hydrophilicity as a function of plasma treatment duration, the contact angles measurements were recorded 10 minutes after each treatment. In order to quantify the recovery of hydrophilicity over time, contact angles were measured at different time points starting before plasma treatment (pre-plasma), after plasma treatment (0.0 hours), and over time for a period up of to 168 hours (remaining time points). The samples were placed in a petri dish and under vacuum during the storage phase of the experiment. The effect of thermal treatment of COC on hydrophilicity recovery over time was also studied by repeating the above process to a COC sample that was heat-treated. COC heated samples were first heated at 77°C for 30 minutes and later at 120°C for 2 minutes after plasma treatment, and the contact angle measurement (0.0 hours) began after completing the heat treatment.
2.4 Cell culture
The protocol for human microvascular endothelial cells (hMVECs) culture was identical to that described previously (Vickerman, Blundo et al. 2008). In brief, hMVECs (Lonza, NJ, USA) were cultured in endothelial growth medium (EGM-2MV, Lonza, NJ, USA), and maintained at 37°C and 5% CO2. The medium was changed every two days until an 80% confluent monolayer was formed before passaging or seeding. Collagen type I (BD Biosciences, San Jose, CA, USA) gel solution with 2.0 mg/ml density was inserted in the gel region of the device using a 10 μl pipette and was incubated in a humidity box to polymerize the gel. The EGM-2MV was inserted into the rest of the channels. To seed samples, hMVECs were detached with trypsin (Invitrogen, Carlsbad, CA, USA) and resuspended in the EGM-2MV at a density of 2 million cells/ml. A 40 μl volume of cell suspension was introduced to PDL-coated PDMS and COC devices and the hMVECs were cultured for the prescribed amount of time. All experiments were conducted using cells of passage 8 or lower.
A live/dead assay kit (Invitrogen, Carlsbad, CA, USA) was used to assess cell viability. In live cells, the intracellular esterase activity transformed calcein AM to fluorescent calcein, resulting in green fluorescence. In dead cells, the entry of ethidium homodimer-1 (Ethd-1) into damaged membranes of dead cells and subsequent binding to nucleic acid resulted in red fluorescence. The samples were washed with Hank’s balanced saline solution (HBSS), and incubated for 15 minutes with 500 μl HBSS solution containing 1 μl calcein AM solution and 1 μl EthD-1 solution. Fluorescent microscope images were acquired with a microscope and a Nikon TEH100 camera with OPENLAB 4.0.4 software, and processed using ImageJ software (National Institute of Health, http://rsb.info.nih.gov/ij/) to false-color the gray scale images. The total cell population was quantified by counting the total number of green and red cells, and the percentage of live cell population was calculated. A student t-test was used for statistical comparisons with p-values greater than 0.1 considered not significant.
3 Results and discussion
3.1 Replication
The durable epoxy master mold used in the hot embossing process enabled molding of numerous microfluidic devices with good feature replication. A single epoxy master generated over 20 samples without degradation. Release of the device plate from the master required no special techniques or solvents. The process steps conveniently produced high quality replicas of the original microfluidic design pattern. Fig. 2a shows the replicated pattern configured in 4-device arrays at each step in the mold and device fabrication process. The images on the left side show 100 mm diameter molds which were either a positive or negative relief replica of the original microfluidic pattern in a different material. Cautious handling of PDMS mold was necessary during the replication step because the PDMS mold required a flat support to supply sufficient rigidity during the epoxy casting step to ensure planarity of the resulting epoxy mold.
Fig. 2.

Products of each replication step of the process demonstrated good replication of features. (a) Photographs of 4-device arrays (left) and SEMs of the post and channel structure of a device (right) illustrate results of each replication step. (b) A single device schematic with the gel area expanded shows the SEM image area outlined.
Inspection of the microfluidic device features by SEM confirmed the quality of replication showing faithful reproduction of the post and channel structures. Fig. 2b shows a single device schematic with expanded gel region and indicates the SEM-imaged area with a dashed line. Since the area contained both a protruding post and recessed channel, it served as an excellent region to gage replication quality for these two feature types within the device. Surfaces of master molds and embossed devices possessed roughness less than 5 μm and did not exhibit signs of voids or other molding defects.
Profilometry quantified feature depth and surface finish for the epoxy master and the final embossed COC plate, indicating good replication accuracy during the hot embossing step. Table 1 shows profilometry data for height and roughness of features for scans taken across the gel-region channel. Feature heights were 110.16 ± 9.56 μm for the epoxy master and 109.30 ± 10.66 μm for the COC plate. The roughness of mold and plate as measured by profilometry were 1.01±0.11 μm and 1.17±0.19 μm respectively, which agrees with observations from SEM images and indicates no irregularity, coarseness, voids or other artifacts of incomplete replication during the hot embossing step. The differences in height and roughness of the channel between epoxy master and COC plate were within the standard error. Feature height measurements taken at various locations on both epoxy mold and embossed device were uniform across the device area.
Table 1.
Profilometer results for the epoxy master and COC plate confirmed accurate hot embossed replication of the microfluidic structure in COC devices. Ra is the roughness of the surface.
| epoxy master | Height (μm) | 110.16 ± 9.56 |
| Ra (μm) | 1.01±0.11 | |
| COC plate | Height (μm) | 109.30±10.66 |
| Ra (μm) | 1.17±0.19 |
These results indicate pattern replication by the hot embossing process produces accurately formed devices with minimal surface defects. Using the fabrication process described above, we can produce a durable epoxy master for hot embossing. The fabrication process of an epoxy master is not limited to the particular method mentioned here, and a hot embossing master could also be made of a different material such as micro-milled brass or electroformed nickel (Charest, Eliason et al. 2006; Hupert, Guy et al. 2007). The fabrication of microfluidic devices using hot embossing is advantageous from its higher rate of production than soft lithography method for PDMS devices, and is a mass-manufacturable yet flexible process (Becker and Locascio 2002).
3.2 Bond strength
Laminating a thin COC film to the embossed microfluidic structures resulted in a sealed device with controlled laminated layer bond strength. Fig. 3a illustrates the double roller laminator thermally bonding the thin COC film to the device resulting in a system with sealed fluidic channels. Properly bonded areas were easily distinguished from non-bonded areas by visual inspection. In addition, green dye introduced into the channels was contained within the sealed structures, further confirming proper sealing of the fluidic channels by the laminated layer.
Fig. 3.

Laminating a thin COC film resulted in a sealed device with controlled laminate bond strength. The lamination process used roller-applied pressure and thermal bonding to seal the complete device, which showed no leakage of green dye introduced in the channels (a). Three-point bending tests using a DMA determined laminated bond strength of samples of thin COC film bonded to COC plates, and the representative DMA output showed a displacement discontinuity where the thin COC film laminate bond failed (b insert). The bond strength depended on the plasma treatment applied to the COC components before lamination (b). Plasma treatment was used on both the COC plate and film (“all plasma”), only the COC plate (“half plasma”), or neither the COC plate nor the film (“no plasma”) before lamination. Shear stress at the bond calculated from the load at failure during the 3-point bending test quantified the bond strength for each sample. Shear stresses were average values (n = 3) with standard error of mean. * indicates statistical significance when half plasma and all plasma were compared to no plasma (p < 0.03)
Three-point bending tests using a DMA determined the strength of the thermal fusion bond between the thin COC film and COC plates created by the lamination process (Fig. 3b insert). The displacement versus force plot for each sample possessed a discontinuity at the point when the thin COC film laminate bond fails (see e.g., Fig. 3b insert). Bond strength, determined by the shear stress calculated from the load occurring at laminate bond failure, depended on the plasma treatment applied to the COC components before lamination. The load at the discontinuity of the displacement versus force plot was used to determine the critical load at delamination and used to calculate the shear stress at failure, assuming the thermal bond would fail before the adhesive bond. This assumption was proved correct from observation after the test when the bonding by adhesive was still intact and the thermal fusion bond was no longer intact. The stress levels were 252.3 ± 18.6 kPa for no plasma, 414.0 ± 74.5 kPa for half plasma, and 685.6 ± 47.2 kPa for all plasma (Fig. 3b). Half plasma and all plasma data were compared to no plasma data, and both pairs were found to be statistically significant with p < 0.03. Other tests confirmed that a bond strength of 290 kPa was sufficient to withstand the pressures encountered curing normal filling and operation of the device (Eddings, Johnson et al. 2008).
3.3 Collagen gel retention and placement
Oxygen plasma treatment helped to control hydrophilicity on COC devices which enabled confinement of collagen gel to specific regions in the devices. Hydrophilicity as measured by water contact angle, depended on plasma treatment duration at a fixed plasma intensity of 100W. Without plasma treatment, the COC material was nearly hydrophobic with a contact angle 84.5 ± 3.6°. In contrast, after a plasma treatment for as short as 10 seconds, the COC material became hydrophilic with a contact angle of 29.7 ± 1.2°. For longer durations of plasma treatment, the resulting hydrophilicity increased as measured by decreased contact angle measurements. The hydrophilicity change due to plasma treatment was more stable over time for COC devices versus PDMS devices (Fig. 4a). Contact angles prior to plasma treatment (pre-plasma), immediately after treatment for 30 seconds at 100W intensity plasma treatment (0.0 hours), and over time following treatment (remaining time points) indicated the long-term stability of COC surface energy compared to PDMS. PDMS rapidly returned to an almost hydrophobic state after plasma treatment. Within the first hour after plasma treatment the contact angle on PDMS recovered from 11.2 ± 2.9° to 50.8 ± 4.8°, and by 3 hours the contact angle was 80.0 ± 3.1° and leveled off to 83.6 ± 4.2° by 168 hours. In contrast, the COC material maintained a hydrophilic state for several days after plasma treatment with the contact angle remaining below 40° for 24 hours and only reaching 50.6 ± 0.5° after 168 hours. Thermal treatment of the COC reduced its hydrophilicity after plasma treatment, although the hydrophilicity remained more stable than that of PDMS. The contact angle for heated COC was 53.5 ± 1.7° at 24 hours after plasma treatment, and remained below 65° for 168 hours. Both time and thermal treatment of COC altered the hydrophilicity after plasma treatment, although the changes occurred over a long timescale with the material remaining hydrophilic for the duration of data collection. This indicates that a lower contact angle can be preserved longer on COC than PDMS.
Fig. 4.

Oxygen plasma treatment of COC created controlled, stable hydrophilicity compared to PDMS to enable collagen gel control in the devices. (a) Contact angles prior to 30 seconds at 100W intensity plasma treatment (pre-plasma), after the plasma treatment (0.0 hours), and over time following the plasma treatment (remaining time points) indicated the long-term stability of COC surface energy compared to PDMS. Thermal treatment of COC (heated) altered hydrophilicity as compared to COC not thermally treated (no heat). Thermal treatment consisted of 30 minutes at 77°C and 2 minutes at 120°C after the plasma treatment. (b) Controlling hydrophilicity of the COC device enabled gel containment to the appropriate areas within the square posts. The triangle markers in the channels point to the gel/channel interface. The collagen gel mimics extracellular environment in this COC microfluidic device
Reducing the hydrophilicity of the COC device relative to the non-heated sample enabled gel containment as shown in Fig. 4b. The triangular markers are located in the channels and point to the gel/channel interface. The three channels were separated by collagen gels inserted in two gel regions up to the square posts, with the gel contained by surface tension. Collagen gel was inserted using a pipette so as not to overfill the gel region. For COC devices not thermally treated, gel inserted into the device spread beyond the square posts and into the channels, indicating a lack of control of gel confinement. However, for COC devices with more hydrophobic surfaces, either due to shorter plasma treatment or longer post-plasma treatment thermal treatment, gel could be predictably contained within the outer surface of the square posts indicating good control of gel placement.
Collagen gel mimics an in vivo extracellular environment, providing a key feature of the device: to culture and observe cells in a 3D environment. To our knowledge, this is the first demonstration of a COC device with a collagen gel inserted as a scaffold for observation of cells in a 3D environment. From our experience, retaining the gel in position requires two main elements: mechanical features to hold the formed gel in place in the event of small fluctuation in pressure difference across it, such as retaining posts molded into the device, and controlled device material surface properties so that the gel solution can be contained within specified boundaries. The COC device with plasma treatment provides both elements, in a user-defined, predictable, and relatively stable format. In addition, although not demonstrated here, the stability of COC surface treatments allows for patterning of different levels of surface hydrophobicity. This may be done by masking off selected area prior to plasma treatment, and will result in further improved gel placement and retention. It is also important to note that altered surface parameters with different surface energy can drive cells to different levels of function (Keselowsky, Collard et al. 2004; Keselowsky, Collard et al. 2005). Therefore, control of microfluidic device material surface parameters enhances control of cell experiments and could provide an additional, controlled experimental variable. The controlled hydrophobicity in the COC device presents potential improvements for both 3D and 2D cell culture.
3.4 Cell viability
Cell viability after 72 hours of culture in COC devices was similar to that in PDMS devices and standard polystyrene (PS) well plates. Quantified live/dead assay data for hMVECs showed near 80 percent cell survival rates for cells cultured in COC devices, PDMS devices, and standard PS well plates (Fig. 5a). Green fluorescent signal indicated active (live) cells that are undergoing intracellular esterase activity and red indicated permeable (dead) cells (Fig. 5b). Cell culture conditions included no perfusion of media other than a daily change of media, standard incubator conditions as mentioned previously, and no pre-treatment of the media. Percent cell population data plotted was the average value of n = 3 replicates for each condition with the standard error of mean shown. A paired Student t-test showed no statistical difference for viability rates in PDMS and COC. Under fluorescence microscopy, the optical clarity of the COC device equaled that of the PDMS device.
Fig. 5.
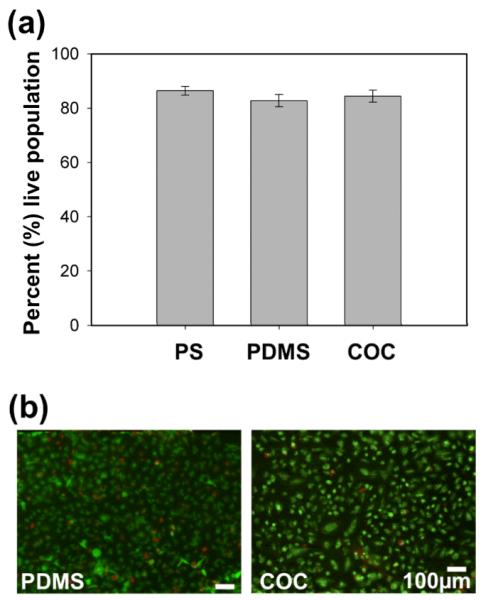
Cell viability after 72 hours of culture was similar in sealed COC devices as compared to PDMS devices and standard PS cell culture plates. (a) Quantified live /dead assay data verified similar cell survival rates in COC, PDMS, and PS culture conditions. The percent of cell populations were average values (n = 3) with standard error of mean. There was no statistical difference for PDMS and COC data using a paired student t-test (p > 0.05) (b) Green fluorescent signal indicated active (live) cells and red indicates permeable (dead) cells in PDMS and COC microfluidic device
One major concern regarding transition from PDMS to a COC device for cell culture may be the inherent lower oxygen permeability of hard plastics as compared to PDMS. To resolve this issue, a device employing both PDMS and hard plastic could be used, (Mehta, Lee et al. 2009) however some of the inherent limitations of PDMS will still be observed. Here, viability of the hMVECs indicates culture of cells is not limited by the COC properties, with nothing more than a regular change of media required over a 72 hour period. Thermoplastics, specifically COC, present an alternative to PDMS for microfluidic devices intended for cell culture.
4 Conclusion
We have presented a low-cost and high-throughput fabrication method for a microfluidic device with an enclosed 3D gel scaffold as a cell culture platform. Hot embossing with epoxy masters accurately replicated features in the COC material, and thermal bonding laminated a thin COC film to the embossed COC to seal the device. Plasma and thermal treatments enhanced bonding of the thin film and device layers. The combination of plasma and thermal treatments also controlled hydrophobicity levels of the COC surface, ensuring proper placement of the 3D collagen gel and providing known conditions for adherent cell growth. Finally, evaluation of cell viability in the completed COC device indicated no adverse effects as compared to previous PDMS devices. With improved properties and a more commercially-viable manufacturing approach, the microfluidic device offers further opportunities to study cell function in a 3D environment with the added potential to serve high-throughput/high-volume applications ranging from pharmaceutical screening to personalized medicine.
Acknowledgement
The research is supported by Award Number R21CA140096 from the National Cancer Institute and Draper Laboratories Inc. (IR&D Grant). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institue of Health.
References
- Ahn SH, Guo LJ. ACS Nano. 2009;3(8):2304–2310. doi: 10.1021/nn9003633. [DOI] [PubMed] [Google Scholar]
- Becker H, Locascio LE. Talanta. 2002;56(2):267–287. doi: 10.1016/s0039-9140(01)00594-x. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, Klapperich CM. Anal Chem. 2006;78(3):788–792. doi: 10.1021/ac051449j. [DOI] [PubMed] [Google Scholar]
- Borenstein JT, Terai H, et al. Biomedical Microdevices. 2002;4(3):167–175. [Google Scholar]
- Bowden NB, Weck M, et al. Accounts Chem Res. 2001;34(3):231–238. doi: 10.1021/ar0000760. [DOI] [PubMed] [Google Scholar]
- Charest JL, Bryant LE, et al. Biomaterials. 2004;25(19):4767–4775. doi: 10.1016/j.biomaterials.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Charest JL, Eliason MT, et al. Biomaterials. 2006;27(11):2487–2494. doi: 10.1016/j.biomaterials.2005.11.022. [DOI] [PubMed] [Google Scholar]
- Charest JL, Eliason MT, et al. J Vac Sci Tech B. 2005;23(6):3011–3014. [Google Scholar]
- Charest JL, Garcia AJ, et al. Biomaterials. 2007;28(13):2202–2210. doi: 10.1016/j.biomaterials.2007.01.020. [DOI] [PubMed] [Google Scholar]
- Chung S, Sudo R, et al. Lab. Chip. 2009;9(2):269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- Chung S, Sudo R, et al. Ann. Biomed. Eng. 2010;38(3):1164–1177. doi: 10.1007/s10439-010-9899-3. [DOI] [PubMed] [Google Scholar]
- Chung S, Sudo R, et al. Adv. Mater. 2009;21(47):4863. doi: 10.1002/adma.200901727. + [DOI] [PubMed] [Google Scholar]
- Desai SP, Freeman DM, et al. Lab. Chip. 2009;9(11):1631–1637. doi: 10.1039/b822081f. [DOI] [PubMed] [Google Scholar]
- Diaz-Quijada GA, Peytavi R, et al. Lab. Chip. 2007;7(7):856–862. doi: 10.1039/b700322f. [DOI] [PubMed] [Google Scholar]
- Eddings MA, Johnson MA, et al. J Micromech. Microeng. 2008;18(6) [Google Scholar]
- El-Ali J, Sorger PK, et al. Nature. 2006;442(7101):403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- Hawkins KR, Yager P. Lab. Chip. 2003;3(4):248–252. doi: 10.1039/b307772c. [DOI] [PubMed] [Google Scholar]
- Hupert ML, Guy WJ, et al. Microfluid. Nanofluid. 2007;3(1):1–11. [Google Scholar]
- Keselowsky BG, Collard DM, et al. Biomaterials. 2004;25(28):5947–5954. doi: 10.1016/j.biomaterials.2004.01.062. [DOI] [PubMed] [Google Scholar]
- Keselowsky BG, Collard DM, et al. Proc. Natl. Acad. Sci. U. S. A. 2005;102(17):5953–5957. doi: 10.1073/pnas.0407356102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech PW, Micromech J. Microeng. 2009;19(5) [Google Scholar]
- Mack PJ, Zhang Y, et al. J. Biol. Chem. 2009;284(13):8412–8420. doi: 10.1074/jbc.M804524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynova L, Locascio LE, et al. Anal. Chem. 1997;69(23):4783–4789. doi: 10.1021/ac970558y. [DOI] [PubMed] [Google Scholar]
- Mehta G, Lee J, et al. Anal. Chem. 2009;81(10):3714–3722. doi: 10.1021/ac802178u. [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Papautsky I. J. Micromech. Microeng. 2004;14(1):96–103. [Google Scholar]
- Piruska A, Nikcevic I, et al. Lab. Chip. 2005;5(12):1348–1354. doi: 10.1039/b508288a. [DOI] [PubMed] [Google Scholar]
- Regehr KJ, Domenech M, et al. Lab. Chip. 2009;9(15):2132–2139. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steigert J, Haeberle S, et al. J. Micromech. Microeng. 2007;17(2):333–341. [Google Scholar]
- Sudo R, Chung S, et al. FASEB J. 2009;23(7):2155–2164. doi: 10.1096/fj.08-122820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CW, DeVoe DL. Microfluid. and Nanofluid. 2009;6(1):1–16. [Google Scholar]
- Vickerman V, Blundo J, et al. Lab. Chip. 2008;8(9):1468–1477. doi: 10.1039/b802395f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesides GM, Ostuni E, et al. Annu. Rev. Biomed. Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]