

Abstract

A promising method for the high-throughput synthesis of linear C-hydroxyalkylamido peptidomimetics and β-turn cyclic peptidomimetics via “volatilizable” aminoalkyl functionalized silica gels is presented. Boc amino acids and carboxylic acids were coupled on functionalized aminoalkyl silica gels using a standard DIC/HOBt coupling protocol. After peptide synthesis, the resin bound peptide was cleaved using a two-step process to obtain the linear C-hydroxyalkylamido peptidomimetics. β-Turn cyclic peptidomimetics were generated by intramolecular SNAr cyclization in an aqueous solution. Both the linear and the cyclic peptidomimetics were obtained with good to excellent yields and purities through a “one-pot” reaction.
Protein–protein interactions are known to play a critical role in many biological processes such as immune responses, protein enzyme inhibitors, signal transduction, and apoptosis. Protein–protein interactions are becoming important targets in medicinal chemistry.1 The distinct secondary structural elements such as helices, sheets, and turns contribute to protein–protein complex interfaces. Among these secondary structural motifs, β-turns have received the most attention for mediating a myriad of protein–protein interactions.2 There is an increasing interest in designing and synthesizing peptidomimetics based on the β-turn structure.3 Recently, macrocyclic peptidomimetics of structures A were reported to mimic the turn regions of proteins.4 Intramolecular SNAr reaction is often used for the construction of these macrocycles via formation of an endo alkyl-aryl ether bond. Solid- and solution-phase synthetic methods have been investigated to synthesize similar structures. By using the solid-phase synthesis approaches, an amino acid with protected side-chain hydroxyl (X=O) or amino (X=N) group was first anchored on resin. The side-chain amino or hydroxyl group was used as the nucleophile to conduct the on resin SNAr cyclization.5 The solution-phase strategies involved intramolecular SNAr cyclization usually promoted by TBAF or CsF in an anhydrous organic solvent.6 However, a variable amount of intermolecular SNAr byproduct was reported by using these catalysts. To develop a new methodology to these promising structures, herein we report an efficient “one-pot” synthetic method, which incorporate the solid-phase synthesis of linear peptides on “volatilizable” supports7 and cyclization in aqueous solution.

Aminopropyl and aminobutyl functionalized silica gels were synthesized and utilized as the “volatilizable” supports.8 Parallel solid-phase synthesis was carried out using the “teabag” approach.9 Boc-amino acids were coupled on the aminoalkyl silica gel 1 by standard Boc-peptide synthesis chemistry to yield the resin-bound peptides 2, as shown in Scheme 1. Resin-bound peptides 2 were N-acylated with carboxyl acids to yield resin-bound N-acylated peptides 3. The resin bound N-acylated peptides 3 were treated with aqueous HF or anhydrous HF (the side-chain protecting groups of amino acids were removed by use of anhydrous HF) to decompose the silica gel. Following the removal of the solvent by lyophilization, H2O2 and DMF were added to further oxidatively cleave the Si–C bond10 producing the C-hydroxyalkylamido peptidomimetics 4 with high yields and purities (Table 1).
Scheme 1.
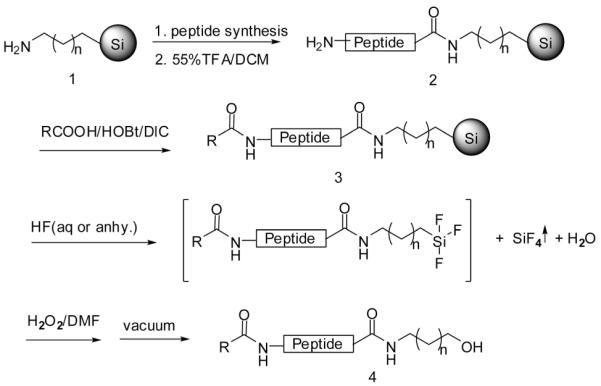
Synthesis of C-Hydroxyalkylamido and N-Acyl Peptidomimetics on the “Volatilizable” Aminoalkyl Silica Gel
TABLE 1.
C-Hydroxyalkylamido and N-Acyl Peptidomimetics
| entry | product | purityc (%) |
yieldd (%) |
|---|---|---|---|
| 4a a | C6H5(CH2)3CO-Leu-NH(CH2)3OH | 90 | 92 |
| 4b a | BnCO-Phe-Gly-Ala-NH(CH2)3OHe | 85 | 90 |
| 4c a | BnCO-Asn-Leu-Phe-Ala-NH(CH2)3OH | 81 | 77 |
| 4d b | BnCO-Phe-Asp-Ala-NH(CH2)4OH | 74 | 78 |
| 4e b | CH3CO-Phe-Phe-Ile-Arg-NH(CH2)4OH | 84 | 70 |
Cleave with 35% aqueous HF at RT for 0.5 h.
Cleave with anhydrous HF at 0 °C for 2 h.
HPLC purity (in %) was determined by the peak area at 214 nm.
Yields (in %) are based on the weight of crude material and are relative to the substitution of the resin (see Supporting Information).
Bn = benzyl.
β-Turn cyclic peptidomimetics were designed and synthesized based on the intramolecular SNAr macrocyclization depicted in Scheme 2. 2-Fluoro-5-nitrobenzoic acid was used as the N-acylating reagent. After oxidative cleavage, the cyclic precursor 5 was obtained. Following removal of DMF and H2O2 in vacuum, the precursors 5 were dissolved in 10% aqueous acetonitrile at a concentration of 0.5 mg/mL. Mixed bed ion-exchange resins were then added to promote the SNAr macrocyclization. To our knowledge this is the first time reporting the utilizing of mix bed ion-exchange resins to catalyze SNAr cyclization in aqueous solution. The macrocyclization proceeded to completion within 12 h at room temperature to yield 14- or 15-membered β-turn cyclic peptidomimetics depending on the aminoalkyl functionalized silica gel used. Mixed bed ion-exchange resins consist of equivalent amounts of strong anion (OH−) and strong cation (H+) exchange resin. The resins serve both as a support-bound base to promote the SNAr macrocyclization and as a scavenging/desalting agent. No intermolecular SNAr byproducts were found in solution during the cyclization. After filtration of the ion-exchange resins and removal of the solvent, the desired cyclic products were left in the vessel in good-to-excellent purities and yields as shown in Table 2. The byproducts from the synthesis were mainly generated from incomplete coupling. The incomplete coupling is most likely due narrow pores caused by the rigid and nonswelling nature of silica.
Scheme 2.
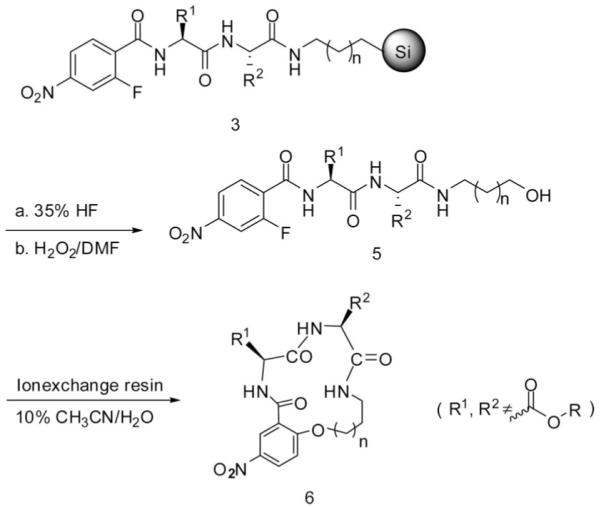
Synthesis of Cyclic Peptidomimetics
TABLE 2.
Cyclic Peptidomimetics
| entry | R1 | R2 | n | puritya (%) |
yieldb (%) |
|---|---|---|---|---|---|
| 6a | CH3 | CH2Ph | 1 | 90 | 92 |
| 6b | CH3 | H | 1 | 81 | 77 |
| 6c | H | CH2CH(CH3)2 | 1 | 88 | 89 |
| 6d | Cyo(CHCH2CH2CH2N) | CH3 | 1 | 85 | 88 |
| 6e | CH2CH2CONH2 | H | 1 | 76 | 80 |
| 6f | CH(CH3)OBzl | CH3 | 1 | 82 | 85 |
| 6g | CH2Ph | CH3 | 2 | 80 | 78 |
| 6h | CH(CH3)2 | CH3 | 2 | 88 | 70 |
HPLC purity (in %) was determined by the peak area at 214 nm.
Yields (in %) are based on the weight of crude material and are relative to the weight and purity of linear precursors.
The 20 natural L-amino acids were examined in the synthesis of the above linear and cyclic peptidomimetics. Protecting groups such as OBzl, Tos, 4-MeOBzl, and 2-Cl-Z are stable to oxidative cleavage when aqueous HF is used in the first cleavage step. Cysteine and methionine were found to be oxidized during the oxidative cleavage. Byproducts were also generated when histidine was used in the sequence. Other than noted above, amino acids were unchanged in the linear peptidomimetics synthesis.
Unfortunately, by using this synthetic scheme, no cyclic petidomimetics containing a carboxyl side chain could be generated. For example, while peptidomimetics made with glutamic acid and aspartic acid were stable in oxidative cleavage, their ester-protected side chains were catalytically hydrolyzed by the ion-exchange resin. These carboxyl groups then attached to the ion-exchange resin, resulting in the complete loss of any cyclic peptidomimetics containing these carboxyl groups.
Larger rings incorporating 3- and 4-amino acid residues (cyclic peptidomimetics 7 and 8) were also synthesized by this method. The cyclization took a longer time to complete. The 17/18- and 20/21-membered cyclic peptidomimetics were obtained with satisfactory purity and yield (data shown in Supporting Information).
Circular dichroism spectra (CD) of compounds 6a were also recorded in 20% aqueous methanol. Compounds 6a gave an ellipticity minimum at approximately 203 nm, characteristic of a type-I β-turn.
In summary, we present here a novel method to produce β-turn cyclic peptidomimetics and C-hydroxyalkyamido linear peptide analogues on “volatilizable” aminoalkyl functionalized silica gels. The desired C-hydroxyalkylamido-terminated linear peptide analogues were obtained by tandem cleavage with aqueous HF or anhydrous HF and hydrogen peroxide in DMF. With the use of 2-fluoro-5-nitrobenzoic acid as the N-capping reagent, β-turn cyclic peptidomimetics were obtained from the linear precursor in 10% aqueous acetonitrile through SNAr cyclization. Mixed bed ion-exchange resins served both as a base and as a scavenger in the cyclization. 14- and 15-Membered cyclic peptidomimetics were obtained with high purity through the “one-pot” reaction. The methodology described is promising for high-throughput synthesis of C-hydroxyalkylamido linear peptides and β-turn cyclic peptidometics.
Experimental Section
General Procedure for Synthesis of Linear C-Hydroxyalkylamido Peptide (4)
A polypropylene mesh packet was sealed with 100 mg of functionalized silica gel resin 1 (loading 0.2 mmol/g). After washing with DCM, 5% DIEA in DCM and DCM, the resin was coupled with N-Boc-protected amino acids using DIC and HOBt (6 equiv each, 0.1 M) in DMF for 2 h to generate the desired resin-bound peptides. Boc deprotection was performed using 55% TFA in DCM for 30 min, followed by washing with DCM (3 times). Following neutralization with 5% DIEA in DCM, the resin was sequentially coupled with Boc-protected amino acids using the DIC/ HOBt approach for 2 h to yield the resin-bound peptides. After the terminal Boc group was removed using 55% TFA in DCM for 30 min, the resin-bound peptide 2 was acylated with a carboxyl acid using DIC and HOBt (10 equiv each, 0.1 M) in DMF overnight to yield resin-bound 3. The cleavage of compound 4 was performed by first adding either 1 mL of 35% aqueous HF at room temperature for 30 min or with anhydrous HF at 0 °C for 2 h to decompose the silica, forming tetrafluorosilane and water. Note that when using anhydrous HF the side chain protecting groups are removed. After removing the solvent and tetrafluorosilane by lyophilization, DMF (1 mL) and 30% hydrogen peroxide (1 mL) were added to oxidatively cleave the Si–C bond at room temperature for 40 h providing the C-hydroxyalkylamido peptide 4.
C6H5(CH2)3CO-Leu-NH(CH2)3OH (4a)
1H NMR (500 MHz, DMSO-d6) δ 0.85 (dd, 6H, J = 19.8, 6.6 Hz), 1.31–1.45 (m, 2H), 1.50–1.58 (m, 3H), 1.74–1.80 (m, 2H), 2.11–2.16 (m, 2H), 2.53 (t, 2H, J = 7.5 Hz), 3.04–3.13 (m, 2H), 3.38 (t, 2H, J = 6.3 Hz), 4.23–4.27 (m, 1H), 7.16–7.18 (m, 3H), 7.26–7.29 (m, 2H), 7.85 (t, 1H, J = 5.6 Hz), 7.89 (d, 1H, J = 8.3 Hz); 13C NMR (125 MHz, DMSO-d6) δ 21.6, 23.0, 24.3, 27.2, 32.3, 34.6, 34.7, 35.7, 41.0, 50.9, 58.4, 125.7, 128.2, 128.3, 141.8, 171.7, 172.1. MS calcd for C19H30N2O3, 334.2; (ESI) m/z found, 335.1 (M + H)+.
General Procedure for Cyclic Peptidomimetics (6, 7, and 8)
The method for preparation of the linear peptide 5 was the same as described in the above text. After removal of the solvent by lyophilization, the product was dissolved in 10% acetonitrile in water at a concentration of 0.5 mg/mL. Mixed bed ion-exchange resins were then added to promote the cyclization. The mixture was shaken overnight to yield the cyclic peptidomimetics 6, 7, and 8.
Cyclic Peptidomimetics (6a)
1H NMR (500 MHz, DMSO-d6) δ 0.99 (d, 3H, J = 7.5 Hz), 1.91–1.93 (m, 2H), 2.71 (dd, 1H, J = 13.8, 11.5 Hz), 3.21–3.25 (m, 1H), 3.30 (dd, 1H, J = 13.9, 3.9 Hz), 3.48–3.51 (m, 1H), 3.87–3.90 (m, 1H), 4.19–4.21 (m, 1H), 4.39–4.41 (m, 1H), 4.48–4.52 (m, 1H), 7.13–7.18 (m, 3H), 7.20–7.23 (m, 2H), 7.30 (dt, 2H, J = 9.5, 5.5 Hz), 8.05 (d, 1H, J = 9.5 Hz), 8.27 (d, 1H, J = 2.8 Hz), 8.36 (dd, 1H, J = 9, 3 Hz), 8.89 (d, 1H, J = 4.5 Hz); 13C NMR (125 MHz, DMSO-d6) δ 16.0, 28.2, 36.7, 38.5, 52.1, 52.8, 70.4, 112.8, 123.7, 126.0, 126.1, 127.2, 127.8, 129.2, 138.4, 139.7, 161.4, 165.6, 170.2, 172.2. MS calcd for C15H18N4O6, 440.2; (ESI) m/z found, 441.1 (M + H)+.
Cyclic Peptidomimetics (7)
1H NMR (500 MHz, DMSO-d6) δ 0.67–0.69 (m, 1H), 0.97–1.03 (m, 1H), 1.32 (d, 3H, J = 7 Hz), 1.32–1.35 (m, 1H), 2.07–2.11 (m, 1H), 2.71–2.73 (m, 1H), 2.89–2.94 (m, 1H), 3.13 (dd, 1H, J = 14, 3.5 Hz), 3.27 (dd, 2H, J = 14, 5.5 Hz), 4.00–4.08 (m, 3H), 4.24–4.26 (m, 1H), 4.91 (m, 1H), 6.99–7.00 (m, 1H), 7.03–7.07 (m, 4H), 7.10–7.13 (m, 1H), 7.34 (d, 1H, J = 10 Hz), 8.37 (dd, 1H, J = 9, 3 Hz), 8.71 (d, 1H, J = 7 Hz), 8.75 (dd, 1H, J = 8, 4.5 Hz), 8.87 (d, 1H, J = 3 Hz), 9.10 (d, 1H, J = 2.6 Hz); 13C NMR (125 MHz, DMSO-d6) 16.0, 23.7, 26.5, 37.7, 40.6, 42.4, 50.7, 52.8, 71.1, 114.0, 120.6, 126.3, 126.8, 127.7, 128.5, 130.1, 135.4, 140.8, 160.6, 161.8, 169.3, 171.2, 172.5. MS calcd for C25H29N5O7, 511.2; (ESI) m/z found, 512.3 (M + H)+.
Supplementary Material
Acknowledgment
This work was supported by the National Science Foundation (RAH CHE 0455072), 1P41GM079590, 1P41GM081261, the State of Florida, Executive Office of the Governor’s Office of Tourism, Trade, and Economic Development, and U54HG03916-MLSCN. Y.Y. would like to thank the National Science Foundation of China (NSFC20572098) and Zhejiang Science and Technology Programme (2005c3401).
Footnotes
Supporting Information Available: Experimental detail, compound characterization, and copies of 1H NMR, 13C NMR spectra, and LC-MS for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Betzi S, Restouin A, Opi S, Arold ST, Parrot I, Guerlesquin F, Morelli X, Collette Y. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19256–19261. doi: 10.1073/pnas.0707130104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]; (c) Rush TS, Grant JA, Mosyak L, Nicholls A. J. Med. Chem. 2005;48:1489–1495. doi: 10.1021/jm040163o. [DOI] [PubMed] [Google Scholar]
- (2).Peczuh MW, Hamilton AD. Chem. Rev. 2000;100:2479–2494. doi: 10.1021/cr9900026. [DOI] [PubMed] [Google Scholar]
- (3) (a).Maliartchouk S, Feng Y, Ivanisevic L, Debeir T, Cuello AC, Burgess K, Saragovi HU. Mol. Pharmacol. 2000;57:385–391. [PubMed] [Google Scholar]; (b) Bondebjerg J, Xiang Z, Bauzo RM, Haskell-Luevano C, Meldal M. J. Am. Chem. Soc. 2002;124:11046–11055. doi: 10.1021/ja0123913. [DOI] [PubMed] [Google Scholar]; (c) Fink BE, Kym PR, Katzzenellenbogen JA. J. Am. Chem. Soc. 1998;120:4334–4344. [Google Scholar]; (d) Kitagawa O, Vander Velde D, Dutta D, Morton M, Takusagawa F, Aube J. J. Am. Chem. Soc. 1995;117:5169–5178. [Google Scholar]; (e) Eguchi M, Lee MS, Nakanishi H, Stasiak M, Lovell S, Kahn M. J. Am. Chem. Soc. 1999;121:12204–12205. [Google Scholar]; (f) Boger DL, Zhou J, Borzilleri RM, Nukui S, Castle SL. J. Org. Chem. 1997;62:2054–2069. doi: 10.1021/jo961346o. [DOI] [PubMed] [Google Scholar]; (g) Beugelmans R, Singh PG, Bois-Choussy M, Chastanet J, Zhu J. J. Org. Chem. 1994;59:5535–5542. [Google Scholar]; (h) Virgilio AA, Schurer SC, Ellman JA. Tetrahedron Lett. 1996;37:6961–6964. [Google Scholar]; (i) Zaccaro MC, Lee HB, Pattarawarapan M, Xia Z, Caron A, L’Heureux P-J, Bengio Y, Burgess K, Saragovi HU. Chem. Biol. 2005;120:1015–1028. doi: 10.1016/j.chembiol.2005.06.015. [DOI] [PubMed] [Google Scholar]; (j) Souers AJ, Virgilio AA, Rosenquist S, Fenuik W, Ellman JA. J. Am. Chem. Soc. 1999;121:1817–1825. [Google Scholar]
- (4).Feng Y, Wang Z, Jin S, Burgess K. J. Am. Chem. Soc. 1998;120:10768–10769. [Google Scholar]
- (5) (a).Feng Y, Burgess K. Chem.–Eur. J. 1999;5:3261–3272. [Google Scholar]; (b) Kohn WD, Zhang L, Weigel JA. Org. Lett. 2001;3:971–974. [PubMed] [Google Scholar]
- (6) (a).Boger DL, Borzilleri RM, Nukui S, Beresis RT. J. Org. Chem. 1997;62:4721–4736. doi: 10.1021/jo961346o. [DOI] [PubMed] [Google Scholar]; (b) Beugelmans R, Bois-Choussy MB, Vergne C, Bouillon JP, Zhu J. Chem. Commun. 1996:1029–1030. [Google Scholar]; (c) Laib T, Zhu J. Tetrahedron Lett. 1998;29:283–286. [Google Scholar]
- (7).Houghten RA, Yu Y. J. Am. Chem. Soc. 2005;127:8582–8583. doi: 10.1021/ja0402396. [DOI] [PubMed] [Google Scholar]
- (8).Li Y, Yu Y, Giulianotti M, Houghten RA. Tetrahedron Lett. 2008;49:3632–3633. [Google Scholar]
- (9).Houghten RA. Proc. Natl. Acad. Sci. U.S.A. 1985;82:5131–5135. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).Yang A, Li T. Anal. Chem. 1998;70:2827–2830. [Google Scholar]; (b) Stork G, Chan TY, Breault GA. J. Am. Chem. Soc. 1992;14:7578–7579. [Google Scholar]; (c) Tamao K, Alita H, Iwahara T, Kanatanl R, Yoshida J, Kumada M. Tetrahedron. 1983;39:983–990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.