

Abstract
Meiotic recombination predominantly occurs at discrete genomic loci called recombination hotspots, but the features defining these areas are still largely unknown (reviewed in1-5). To enable a comprehensive analysis of hotspot-associated DNA and chromatin characteristics we developed a direct molecular approach for mapping meiotic DNA double stranded breaks that initiate recombination. Here, we present the genome-wide distribution of recombination initiation sites in the mouse genome, constituting the first physical map of recombination hotspots in a multi-cellular organism. Hotspot centres are mapped with approximately 200-nucleotide precision that enables analysis of the fine structural details of the preferred recombination sites. We determine that hotspots share a centrally distributed consensus motif, possess a nucleotide skew that changes polarity at the centre of hotspots, and have an intrinsic preference to be occupied by a nucleosome. Furthermore, we find that the vast majority of recombination initiation sites in mouse males are associated with testis-specific trimethylation of lysine 4 on histone H3 that is distinct from histone H3 lysine 4 trimethylation marks associated with transcription. The recombination map presented here has been derived from a homogeneous mouse population with a defined genetic background and therefore, lends itself to extensive future experimental exploration. Importantly, the mapping technique developed here does not depend on availability of genetic markers and hence can be easily adapted for other species with complex genomes. Our findings uncover several fundamental features of mammalian recombination hotspots and underline the power of the new recombination map for future studies of genetic recombination, genome stability and evolution.
The vast majority of homologous recombination takes place in recombination hotspots – discrete regions of the genome with a recombination frequency significantly above the frequency in adjacent areas. Extensive studies of several individual hotspots in mammals have greatly advanced our understanding of hotspot biology (reviewed in1-6), but some critical features may be missed without examining the full ensemble of hotspots in a genome. Recently, remarkable progress has been made that culminated in the identification of more than 30,000 recombination hotspots in human7-11. Nevertheless, the relatively low resolution of the human map and the high variability of the recombination pattern between individuals still hinders fine structural analysis of hotspots. At the same time, it is clear that the primary DNA structure per se is a poor predictor of recombination activity and that hotspot position depends on additional factors, including epigenetic marks and most likely others1-4. Therefore, defining the complete set of variables that ultimately determine hotspot sites in mammals would be greatly facilitated if a hotspot map of a genetically homogeneous and malleable organism were available. We therefore embarked on generating a high-resolution physical map of recombination hotspots in the mouse using a direct molecular approach for identification of recombination initiation sites.
Meiotic recombination is initiated by the introduction of DNA double-stranded breaks (DSBs) by the SPO11 protein followed by resection of the ends to produce long single-stranded overhangs12. RAD51 and DMC1 proteins form nucleoprotein filaments at the ends of the breaks and carry out a search for a homologous chromosome that is used for repair12. We employed anti-DMC1 antibodies to localize recombination initiation sites in the male mouse genome by chromatin immunoprecipitation followed by high throughput sequencing (ChIP-Seq). To enrich for DSB-stage spermatocytes we utilised Hop2-/- mice that lack the cells of later spermatogenic stages due to meiotic arrest after DSB formation13 (Supplementary Text). While hotspots identified by our approach in wild type and in Hop2-/- mice correlate extensively (Supplementary Fig. 1), four times more hotspots can be identified using Hop2-/- mice due to a higher signal-to-noise ratio.
Consistent with colocalization of the RAD51 and DMC1 proteins to DSB sites12 we found that tag coverage in anti-RAD51 and anti-DMC1 ChIP-seq was highly correlated (Fig. 1). Furthermore, the mapping data were highly reproducible between biological replicates (R = 0.71–0.97) (Fig. 1c). We were able to identify 9,874 recombination hotspots (Supplementary Data Set 1 and Supplementary Figs. 2 and 3) (P value =10−4, FDR = 6.7%), although the number of hotspots could be higher when less strict parameters are used (Supplementary Fig. 4). Confirmation of several identified hotspots was carried out by two approaches including direct physical detection of DSBs as previously described14 (Supplementary Fig. 5). Furthermore, we found that the correlation of our DSB map with available genetic maps15,16 is almost as high as the correlation between the genetic maps themselves (Fig. 1d, Supplementary Fig. 6 and Supplementary Text). The correlation of these genetic maps with the DSB hotspot distribution from this study is an important validation of our hotspot mapping approach. Additional supporting evidence comes from our finding that the hottest cluster of DSB hotspots in the mouse genome is located in the pseudoautosomal region (PAR) – the only homologous region between the X and Y chromosomes (Fig. 1e and Supplementary Fig. 3). Despite the very short length of the PAR each spermatocyte undergoes an obligatory crossover (CO) in this area17. Estimates from our analysis show that this cluster is likely sufficient to ensure at least one DSB in the PAR of every spermatocyte (Supplementary Text).
Figure 1.

DSB hotspots in the mouse genome. a. The ChIP-seq tag density profiles. DMC1 and RAD51: anti-DMC1 and anti-RAD51 ChIP. DMC1, Spo11-/-: anti-DMC1 ChIP from Spo11-/- mice that do not form DSBs. Control: IgG ChIP and input DNA pool. b. Close-up of a representative hotspot. c. Agreement between ChIP-Seq samples (correlations in 2 kb bins across genome). d. Correlation between the DSB hotspot map and the published genetic maps A15 and B16 for chromosome 1. The DSB map is generated from hotspot strengths (Supplementary Methods). All maps are generated in 5 Mb windows and normalized by area of the map. e. PAR contains a large cluster of overlapping hotspots. DMC1 ChIP-Seq tag coverage (smoothing window = 1 kb, step size = 100 bp).
The centres of the hotspots in our map are defined with an approximate precision of 200 nucleotides, at least an order of magnitude higher than that of other available recombination maps in multi-cellular organisms (Supplementary Methods). More than 250 of the identified hotspots are hotter than the previously known strong hotspot, H2Eα (Fig. 2a). Based on the estimated maximum recombination efficiency of the H2Eα hotspot of 2 cM18 the strength of the hottest hotspot in the mouse might be as high as 6 cM. Most of the hotspots lie 60-330 Kb apart with only a few recombination deserts of more than 3 Mb (Supplementary Fig. 7). The average strength of individual hotspots on different autosomes are similar, and on a chromosome scale, the slight variation in hotspot density between autosomes does not correlate with the density of genes, chromosomal GC content, abundance of DNA repeats or chromosome length (not shown).
Figure 2.

Characteristics of mouse DSB hotspots. a. The H2Eα hotspot and the PAR hotspot cluster are among the strongest in the mouse genome. The strength of the hottest individual hotspot identified in this study (Chromosome 15, ~91 Mb) is also indicated. b. Mouse DSB hotspots are significantly enriched in genes (one-sided binomial tests). Genic regions are defined from start to stop codons including introns. Error bars represent the 5th to 95th percentiles of the expected value distributions (n = 10,000 iterations). c. A purine-pyrimidine skew is apparent at DSB hotspots. Skew is calculated in 100 bp windows with a step size of 1 bp. d. Consensus hotspot motif is similar to the predicted binding site of PRDM9. e. Consensus motif is present in the centres of DSB hotspots. Distribution of hits to the consensus motif is shown in the 5 kb regions around hotspots (window: 200 bp, step: 1 bp). f. Both predicted (blue) and experimentally determined (red) nucleosome occupancy profiles peak at the centre of DSB hotspots. MNase-Seq coverage ratio is plotted as the whole fragment coverage ratio of micrococcal nuclease-digested chromatin to randomly fragmented chromatin in sliding 500 bp windows (step: 1 bp).
We found that mouse hotspots have a tendency to overlap genes (Fig. 2b), although only the hottest 40% of the hotspots contribute significantly to this correlation (Supplementary Fig. 8). We next examined association of DSB hotspots with different classes of DNA repeats and other genomic features. Significant correlations were found with GC content, SINEs, LINEs, LTRs, and other repeats, in agreement with those garnered from previous work1,2,4,5 (Supplementary Table 1 and Supplementary Text).
In search for additional features that might define hotspot locations we analysed the nucleotide composition of the hotspot regions, as characteristic nucleotide skews have been found at some functional genomic elements including replication origins and transcription start sites19. Examination of either single strand of the double stranded DNA in a 5′- to - 3′ direction reveals that the sequence 5′ to the hotspot centres is enriched for purines, but the polarity of the bias changes in the middle of hotspots such that the sequence 3′ to the hotspot centre is more pyrimidine-rich (Fig. 2c and Supplementary Fig. 9). Since replication- and transcription-related skews have been attributed to mutational biases acting asymmetrically on complementary DNA strands, it is conceivable that the skew detected at hotspots is the result of mutational asymmetry as well (Supplementary Text and Supplementary Fig. 9c). It is also possible that the skew represents some unknown functional feature of the genome that favours DSB formation. Importantly, we were able to detect the same signature of nucleotide usage in human hotspots (Supplementary Fig. 9d), indicating that the purine/pyrimidine skew is an intrinsic property of recombination hotspots in mammals. Interestingly, we also noticed a slight increase in the overall GC content in the middle of hotspots (not shown), which might indicate the presence of a gene conversion bias20.
The PRDM9 protein is a meiosis-specific methyltransferase responsible for trimethylation of lysine 4 on histone H3 (H3K4me3)21. In addition to its well-known role in transcription22 this histone modification is associated with increased recombination activity in yeast and in mice23,24. The PRDM9 protein is highly polymorphic in its multi-Zn2+ finger DNA binding domain, and recent studies have implicated PRDM9 in determining the positions of recombination hotspots through the different binding specificities of its alleles25-27. Approximately 40% of human recombination hotspots possess a consensus motif28 that matches the predicted binding site of human PRDM9. We were able to identify a consensus motif specific for mouse hotspots (Fig. 2d and 2e). Sequences with better alignment scores to the motif consensus are more strongly over-represented in the hotspot regions with the best hits showing almost 180-fold enrichment (Supplementary Fig. 10). Overall, hotspots containing consensus sequences are stronger than those without, and the quality of the motif alignment within hotspots is positively correlated with hotspot strength (Supplementary Fig. 11). Importantly, the motif shows a strong match to the predicted binding site of the Prdm9 allele present in our mouse strain (Fig. 2d and Supplementary Fig. 12) and it is present at the centre of at least 73% of hotspots. This indicates that PRDM9 is a determinant for many more hotspots than previously thought and can explain the stronger than expected correlation between hotspot activity and Prdm9 allelic variation found in recent association27 and sperm typing29 studies.
Since posttranslational histone modification has been implicated in the regulation or maintenance of recombinational activity we asked whether DNA in the hotspot regions is assembled into nucleosomes. Strikingly, we found that both predicted30 and actual nucleosomal occupancy are co-centred with recombination hotspots (Fig. 2f), reflecting a previously unknown, intrinsic property of hotspot DNA to assemble a nucleosome. We next examined the distribution of the H3K4me3 marks in germ (testis) and somatic (liver) tissues. We found that 94% of hotspots overlap H3K4me3, with the majority of hotspots overlapping testis specific H3K4me3 marks (87%), and practically none overlapping the marks specific to the liver (Fig. 3a, Supplementary Fig. 13). While the association of H3K4me3 with DSB hotspots is not surprising in light of recent work23-27, the extent of the overlap revealed here is such that H3K4me3 can be considered the first global feature of DSB sites in multi-cellular organisms. Importantly, unlike in S. cerevisiae23 where gene promoters and hotspots seem to share the same H3K4me3 mark, H3K4me3 marks at mouse hotspots are hotspot-specific (Fig. 3b). Even when a hotspot is located very close to a transcription start site, the corresponding H3K4me3 marks are clearly spatially distinct (Fig. 3c), indicating that different mechanisms are involved in the histone modifications at these sites. (Fig. 3c, Supplementary Fig. 13, Supplementary Text).
Figure 3.
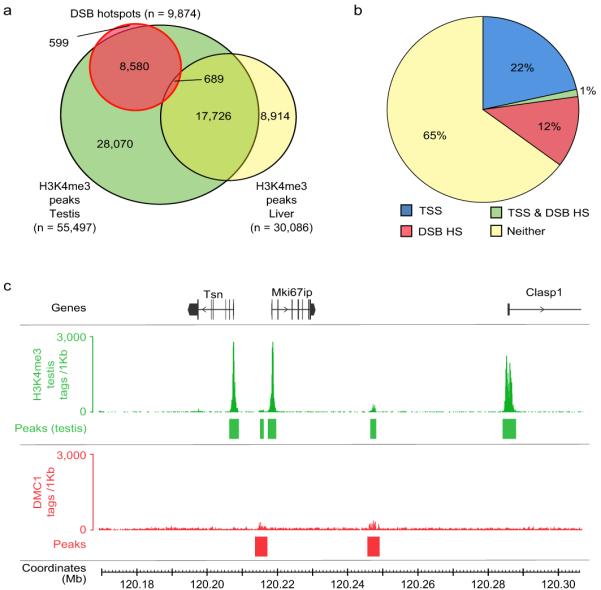
Specific H3K4me3 marks are associated with DSB hotspots. a. The vast majority (93.9%) of DSB hotspots overlap H3K4me3 marks, most of which (86.9%) are testis-specific. The six DSB hotspots that overlap liver-specific H3K4me3 marks are not shown. Peak calling for each dataset was performed using an equal number of tags. b. DSB hotspots are associated with a set of H3K4me3 marks that are distinct from those at transcription start sites (TSSs). c. H3K4me3 marks at DSB hotspots are generally weaker than TSS-associated marks, and are also spatially distinct despite being sometimes in very close proximity. Tag coverage is displayed in 100 bp steps.
Importantly, H3K4me3 per se is not a sufficient mark for DSB formation. Why meiotic DSBs are correlated with only a small fraction (16.7%) of the ~55,000 H3K4me3 marks in testis remains a mystery. An attractive possibility is that some component of the DSB machinery directly associates with PRDM9 or PRDM9-containing complex and is therefore delivered to the potential DSB sites. Subsequent trimethylation of the H3K4 might be required to set the stage for recombination initiation and progression. Other possibilities may also be considered: i) in addition to H3K4me3 other epigenetic marks may be present that require H3K4me3 to allow DSB formation; ii) H3K4me3 introduced by PRDM9 may be different from other H3K4me3 marks (e.g. PRDM9, unlike other methyltransferases, might modify one rather than both H3 histones in the same nucleosome, or the other way around); iii) specific histone variants could be present in the PRDM9-modified nucleosomes or be substrates of PRDM9; iv) transcription factors or other proteins bound to “non-PRDM9” trimethylation marks may interfere with the DSB machinery.
Methods Summary
Hop2-/- mice13 on a [C57Bl/10.S × C57Bl/10.F] F1 genetic background, and wild-type mice of the same background were used for making a map of DSB hotspots. ChIP and high-throughput sequencing were performed according to manufacturer-provided protocols (Upstate and Illumina, respectively) with minor modifications. For each sample, 36 bp end sequences were aligned to the mm9 reference genome using the Illumina GAII analysis pipeline. Only quality filtered reads that mapped uniquely to the genome were retained for downstream analyses. DSB hotspots and H3K4me3 peaks were identified by comparing the sequence tag coverage for each ChIP sample with that of tag-count matched control sample using MACS. The hotspot consensus motif was identified from 9-mers enriched near hotspot centres using a bespoke analysis pipeline. Additional details are available in Supplementary Information.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Michael Lichten (NCI, NIH) and Peggy Hsieh (NIDDK, NIH) for helpful comments and discussion. We are thankful to Shaila Sharmeen for her help with high-throughput sequencing. This work was supported in part by Basil O’Connor Starter Scholar Research Award Grant No. 5-FY07-667 from the March of Dimes Foundation (GP), NIH grant 1R01GM084104-01A1 from NIGMS (GP), New Investigator Start-up Grants FS71HU, R071HU, FS71HU from USUHS (GP), and the NIDDK (NIH) Intramural Research Program (RDC-O).
REFERENCES
- 1.Arnheim N, Calabrese P, Tiemann-Boege I. Mammalian meiotic recombination hot spots. Annu Rev Genet. 2007;41:369–399. doi: 10.1146/annurev.genet.41.110306.130301. doi:10.1146/annurev.genet.41.110306.130301. [DOI] [PubMed] [Google Scholar]
- 2.Buard J, de Massy B. Playing hide and seek with mammalian meiotic crossover hotspots. Trends Genet. 2007;23:301–309. doi: 10.1016/j.tig.2007.03.014. doi:S0168-9525(07)00117-5 [pii] 10.1016/j.tig.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 3.Lichten M. Meiotic Chromatin: The Substrate for Recombination Initiation. Genome Dyn Stab. 2008;3:165–193. [Google Scholar]
- 4.Paigen K, Petkov P. Mammalian recombination hot spots: properties, control and evolution. Nat Rev Genet. 2010;11:221–233. doi: 10.1038/nrg2712. doi:nrg2712 [pii] 10.1038/nrg2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark AG, Wang X, Matise T. Contrasting methods of quantifying fine structure of human recombination. Annu Rev Genomics Hum Genet. 2010;11:45–64. doi: 10.1146/annurev-genom-082908-150031. doi:10.1146/annurev-genom-082908-150031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kauppi L, May CA, Jeffreys AJ. Analysis of meiotic recombination products from human sperm. Methods Mol Biol. 2009;557:323–355. doi: 10.1007/978-1-59745-527-5_20. [DOI] [PubMed] [Google Scholar]
- 7.A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. doi:10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi:nature09534 [pii] 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frazer KA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. doi:nature06258 [pii] 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong A, T. G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, Gudjonsson SA, Frigge ML, Helgason A, Thorsteinsdottir U, Stefansson K. ine-scale recombination rate differences between sexes, populations and individuals. Nature. 2010;467:1099–1103. doi: 10.1038/nature09525. [DOI] [PubMed] [Google Scholar]
- 11.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. doi:310/5746/321 [pii] 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 12.Neale MJ, Keeney S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature. 2006;442:153–158. doi: 10.1038/nature04885. doi:nature04885 [pii] 10.1038/nature04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petukhova GV, Romanienko PJ, Camerini-Otero RD. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell. 2003;5:927–936. doi: 10.1016/s1534-5807(03)00369-1. doi:S1534580703003691 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Qin J, Richardson LL, Jasin M, Handel MA, Arnheim N. Mouse strains with an active H2-Ea meiotic recombination hot spot exhibit increased levels of H2-Ea-specific DNA breaks in testicular germ cells. Mol Cell Biol. 2004;24:1655–1666. doi: 10.1128/MCB.24.4.1655-1666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paigen K, et al. The recombinational anatomy of a mouse chromosome. PLoS Genet. 2008;4:e1000119. doi: 10.1371/journal.pgen.1000119. doi:10.1371/journal.pgen.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox A, et al. A new standard genetic map for the laboratory mouse. Genetics. 2009;182:1335–1344. doi: 10.1534/genetics.109.105486. doi:genetics.109.105486 [pii] 10.1534/genetics.109.105486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burgoyne PS. Genetic homology and crossing over in the X and Y chromosomes of Mammals. Hum Genet. 1982;61:85–90. doi: 10.1007/BF00274192. [DOI] [PubMed] [Google Scholar]
- 18.Khambata S, Mody J, Modzelewski A, Heine D, Passmore HC. Ea recombinational hot spot in the mouse major histocompatibility complex maps to the fourth intron of the Ea gene. Genome Res. 1996;6:195–201. doi: 10.1101/gr.6.3.195. [DOI] [PubMed] [Google Scholar]
- 19.Francino MP, Ochman H. Strand asymmetries in DNA evolution. Trends Genet. 1997;13:240–245. doi: 10.1016/S0168-9525(97)01118-9. doi:S0168-9525(97)01118-9 [pii] 10.1016/S0168-9525(97)01118-9. [DOI] [PubMed] [Google Scholar]
- 20.Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. doi:10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- 21.Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science. 2009;323:373–375. doi: 10.1126/science.1163601. doi:1163601 [pii] 10.1126/science.1163601. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127–134. doi: 10.1016/j.gde.2009.02.001. doi:S0959-437X(09)00034-3 [pii] 10.1016/j.gde.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borde V, et al. Histone H3 lysine 4 trimethylation marks meiotic recombination initiation sites. EMBO J. 2009;28:99–111. doi: 10.1038/emboj.2008.257. doi:emboj2008257 [pii] 10.1038/emboj.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buard J, Barthes P, Grey C, de Massy B. Distinct histone modifications define initiation and repair of meiotic recombination in the mouse. EMBO J. 2009;28:2616–2624. doi: 10.1038/emboj.2009.207. doi:emboj2009207 [pii] 10.1038/emboj.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. doi:science.1181495 [pii] 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers S, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. doi:science.1182363 [pii] 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baudat F, et al. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. doi:science.1183439 [pii] 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. doi:10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 29.Berg IL, et al. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet. 2010;42:859–863. doi: 10.1038/ng.658. doi:ng.658 [pii] 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaplan N, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. doi:nature07667 [pii] 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.