

Abstract
Summary: Transcription factor binding events are frequently associated with a pattern of nucleosome occupancy changes in which nucleosomes flanking the binding site increase in occupancy, while those in the vicinity of the binding site itself are displaced. Genome-wide information on enhancer proximal nucleosome occupancy can be readily acquired using ChIP-seq targeting enhancer-related histone modifications such as H3K4me2. Here, we present a software package, BINOCh that allows biologists to use such data to infer the identity of key transcription factors that regulate the response of a cell to a stimulus or determine a program of differentiation.
Availability: The BINOCh open source Python package is freely available at http://liulab.dfci.harvard.edu/BINOCh under the FreeBSD license.
Contact: cliff@jimmy.harvard.edu; xsliu@jimmy.harvard.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Although transcription factors are known to have a high affinity for specific DNA sequences, binding is often cell type and condition specific, therefore DNA sequence alone is a poor predictor of in vivo genome-wide binding locations (Carroll et al., 2006). When the relevant transcription factors are known and suitable antibodies are available, ChIP-chip and ChIP-seq technologies enable us to map their genome-wide binding locations. In many cases, however, the transcription factors governing a regulatory response are not known or antibodies suitable for ChIP-seq are not available. Without transcription factor-specific antibodies, it is nevertheless possible to infer transcription factor binding events based on two observations. First, transcription factor binding sites are often associated with certain types of post-translational histone modifications, in particular histone H3 lysine 4 mono- (H3K4me) and di-methylation (H3K4me2) (Heintzman et al., 2007). Second, transcription factor binding is frequently associated with a pattern of nucleosome occupancy changes in which nucleosomes flanking the binding site increase in occupancy, while those in the vicinity of the binding site itself are displaced (He et al., 2010). Transcription factor (TF) binding events can therefore be inferred by comparing nucleosome resolution H3K4me1/2 ChIP-seq data under treatment and control conditions. We present a software package, BINOCh, to allow biologists to carry out an analysis of nucleosome occupancy data to discover stimulus-induced transcription factor binding.
2 METHOD
2.1 Identification and scoring of candidate transcription factor binding regions
The first step in the analysis is to use histone-related ChIP-seq data to detect well-positioned nucleosomes, which we do with the nucleosome positioning from sequencing (NPS) package (Zhang et al., 2008) available at http://liulab.dfci.harvard.edu/NPS. The second step, shown in Figure 1A, is to find pairs of nucleosomes with center to center distances characteristic of nucleosomes flanking TF binding sites (between 250 and 450 bp) and to generate a table summarizing ChIP-seq tag counts in these regions. For every nucleosome pair, we count the number of sequence reads within 100 bp of the center of each positioned ‘flanking’ nucleosome under both treatment and control conditions, denoted nflank,t and nflank,c, respectively. We also count the number of sequence reads mapped to the ‘central’ region between two flanking regions, denoted ncentral,t and ncentral,c. To generate this table we provide an easy to use python script, tagtab. We measure nucleosome occupancy change with the nucleosome stabilization–destabilization (NSD) score,
where Nt and Nc are the total number of sequence reads in treatment and control. In the final step, using the python script binoch, we compute NSD scores and search through DNA motif libraries, TRANSFAC (Wingender et al., 2000), JASPAR (Sandelin et al., 2004) and UniPROBE (Berger and Bulyk, 2009), to detect motifs that are associated with the central regions of high NSD scoring regions.
Fig. 1.

(A) Pattern of nucleosome occupancy changes associated with transcription factor binding in the treatment condition. (B) Analysis of the tendency for transcription factor binding to be located toward the midpoint between paired nucleosomes. The midpoint defines the relative position 0, while the edges fix the relative position 1. Within each region the highest scoring DNA sequence element is associated with a relative position between 0 and 1. Sequence motifs detected in the flanking nucleosome positions are excluded from the calculation.
2.2 Motif position bias toward the midpoint of central regions
Our model for transcription factor binding is one in which transcription factor binding sites are located near the midpoint between paired nucleosomes. To assess a trend of DNA sequence motif occurrence toward this midpoint, we compute the mean relative location of motif hits. Statistical significance is derived using a null model where it is assumed that DNA motifs not associated with binding sites are uniformly distributed relative to the midpoint. We conduct this analysis on 600 bp DNA segments from the N highest NSD-scoring set of paired nucleosomes. Figure 1B shows how each 600 bp segment is derived from a 1 kb sequence centered at the midpoint between a nucleosome pair from which 200 bp centered on each of the pair of well-positioned nucleosomes is excluded. All DNA subsequences within these sequences are scored by a known motif position weight matrix using a low-order Markov model to account for the genomic background sequence composition. For each nucleosome pair i, we obtain two values: si, the maximum motif score for that pair and xi, a value between 0 and 1 representing the location of the motif position relative to the midpoint. Sorting paired regions by motif score from high to low, we compute a list of z scores, 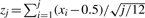 , that represent the positional bias of a motif toward the centers of these regions. We calculate the P-value for a motif from the minimum z-score, making an adjustment for N.
, that represent the positional bias of a motif toward the centers of these regions. We calculate the P-value for a motif from the minimum z-score, making an adjustment for N.
2.3 Motif enrichment in high-scoring NSD regions
The motif centrality statistic, while telling us that a motif is occurring in the DNA sequence between paired nucleosomes does not allow us to assess whether this motif is enriched in the high NSD-scoring regions. To make this assessment, BINOCh computes a P-value motif enrichment statistic where the frequency with which a motif is found within the set of high NSD-scoring regions is compared with its occurrence frequency within a control set of regions. We select the control regions as those having NSD scores closest to the median of the NSD-score distribution. The analysis is carried out on the 200 bp of DNA sequence centered on the midpoint between paired nucleosomes. Motifs that are significant by both enrichment and centrality measures are likely to be bona fide transcription factor motifs important to the system.
3 CASE STUDY
We applied this approach to nucleosome resolution H3K4me2 ChIP-seq data from the prostate cancer cell line LNCaP under the control condition and in response to stimulation by the androgen receptor (AR) agonist 5a-dihydrotestosterone (DHT) (He et al., 2010). The BINOCh position analysis of this data produces a ranked list of motifs with their associated P-values (Supplementary Table 1), showing the AR motif to be the most strongly associated with the midpoint of high NSD scoring nucleosome pairs; in this analysis TRANSFAC progesterone receptor motif which is the same as the AR motif obtains the most significant P-value (<1e−8). The FoxA1 motif is also biased toward the midpoint (P<1e−6). This observation is biologically meaningful as FoxA1 is thought to be a ‘pioneer factor’ facilitating the binding of AR. The BINOCh motif enrichment analysis (Supplementary Table 2) also finds a TRANSFAC AR motif to be the most significant (P<1e−48). See http://liulab.dfci.harvard.edu/BINOCh to download the data and further results of this analysis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jun Song, Tao Liu, Hyunjin Gene Shin and Bo Jiang for contributing elements of this software.
Funding: National Institutes of Health (R01 HG4069).
Conflict of Interest: none declared.
REFERENCES
- Berger M.F., et al. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J.S., et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- He H.H., et al. Nucleosome dynamics defines transcriptional enhancers. Nat. Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman N.D., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Sandelin A., et al. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E., et al. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., et al. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics. 2008;9:537. doi: 10.1186/1471-2164-9-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.