

Abstract
Motivation: The analysis of gene coexpression is at the core of many types of genetic analysis. The coexpression between two genes can be calculated by using a traditional Pearson's correlation coefficient. However, unobserved confounding effects may cause inflation of the Pearson's correlation so that uncorrelated genes appear correlated. Many general methods have been suggested, which aim to remove the effects of confounding from gene expression data. However, the residual confounding which is not accounted for by these generic correction procedures has the potential to induce correlation between genes. Therefore, a method that specifically aims to calculate gene coexpression between gene expression arrays, while accounting for confounding effects, is desirable.
Results: In this article, we present a statistical model for calculating gene coexpression called mixed model coexpression (MMC), which models coexpression within a mixed model framework. Confounding effects are expected to be encoded in the matrix representing the correlation between arrays, the inter-sample correlation matrix. By conditioning on the information in the inter-sample correlation matrix, MMC is able to produce gene coexpressions that are not influenced by global confounding effects and thus significantly reduce the number of spurious coexpressions observed. We applied MMC to both human and yeast datasets and show it is better able to effectively prioritize strong coexpressions when compared to a traditional Pearson's correlation and a Pearson's correlation applied to data corrected with surrogate variable analysis (SVA).
Availability: The method is implemented in the R programming language and may be found at http://genetics.cs.ucla.edu/mmc.
Contact: nfurlott@cs.ucla.edu; eeskin@cs.ucla.edu
1 INTRODUCTION
The analysis of gene coexpression patterns has been of great interest in recent years due to the widespread availability of microarray datasets measuring thousands of genes. Gene coexpressions, evaluated by comparing the expression patterns of pairs of genes, have been utilized in order to identify loci responsible for regulating genes (Ghanzalpour et al., 2006; Lee et al., 2006), to evaluate the significance of known pathways (Subramanian et al., 2005) and to identify functionally related genes whose relationships have been conserved through evolution (Stuart et al., 2003). Unfortunately, gene expression data can be largely affected by technical bias such as a batch effects or plate effects (Johnson et al., 2007). Such non-biological effects have been shown to induce correlations between genes. For example, Balázsi et al. (2003) showed that spacial placement of microarray probes affected the correlation between gene expression patterns, causing genes to be more or less correlated depending on the proximity of their respective probes on the array. More generally, unobserved factors affecting gene expression have the potential to cause correlation between genes. When these factors are shared between gene expressions, they cause genes to have similar patterns of overall variation. Since these effects are not directly observed they are not incorporated into statistical models. The shared variation between genes is attributed to biological causes. This issue is referred to as expression heterogeneity and has been acknowledged as a general problem when analyzing expression datasets (Leek and Storey, 2008).
The detrimental effects of technical confounding on the results obtained from microarray analysis are well known. Qiu et al. (2005), while examining the effects of stochastic dependence between arrays on the correlation of test statistics used in determining differential expression, noted that the correlation structure of arrays induced through non-biological effects can lead to spurious correlation between genes. They note that microarray normalization procedures mitigate such phenomenon, but are unable to completely negate them. In fact, the presence of spurious correlations is a general problem that arises when analyzing many types of noisy high dimensional biological datasets, and has been examined in many different contexts (Clarke et al., 2008). In the context of gene coexpression, the cause of spurious correlations can be conceptualized by viewing a set of n microarrays measuring m genes as a m×n matrix. In this matrix, we expect that the microarrays represented by the columns are independent and that some of the rows, representing the genes will be correlated, indicating biological relationships. In the presence of technical confounding effects, such as batch effects, the columns will share characteristics that will cause the overall patterns of expression to be similar and thus the arrays will be statistically correlated. This increased correlation between columns induces correlation between rows, as it becomes more likely that two randomly selected rows will be correlated, given that the overall patterns of expression for each array are similar. In this way, the correlation between arrays, or inter-sample correlation, has the potential to induce correlations between genes.
Many methods have been developed that aim to remove confounding effects from gene expression data. For example, in the case of known batch effects, a method such as ComBat (Johnson et al., 2007) may be employed. ComBat (Johnson et al., 2007) uses an empirical Bayes approach to estimate parameters associated with batch and produces corrected gene expression data. This corrected expression data can then be used in subsequent analysis. Unfortunately, technical confounding such as a batch effect may not be easily observable. In this case, a method that is able to identify possible confounding effects without prior information is of interest. For example, surrogate variable analysis (SVA) (Leek and Storey, 2007) is a method for correcting gene expression data in the absence of known confounding effects. In SVA, a set of surrogate variables are estimated and regressed out of the expression data. These surrogate variables represent the unknown confounding effects that cause expression heterogeneity. Expression heterogeneity is expected to be encoded by the inter-sample correlation matrix, which is the matrix representing the correlation between all pairs of arrays. Surrogate variables are estimated by iteratively weighting a subset of the principal components of this matrix. The SVA method is aimed at the general problem of correcting gene expression data and does not specifically target the problem of calculating pairwise gene correlations. Furthermore, SVA only utilizes the principal axes of the inter-sample correlation matrix in order to correct expression. We can reason that the full inter-sample correlation matrix contains more information than its principal components and therefore SVA is only utilizing a subset of the available correlation information in its correction procedure. When the patterns of confounding are complex, the estimated surrogate variables may not capture all of the structure encoded in the inter-sample correlation matrix and as a result the corrected expression data may contain residual correlation.
In this article, we propose a method for calculating pairwise gene correlations that utilizes the full inter-sample correlations matrix in order to correct for expression heterogeneity. Our proposed method, mixed-model coexpression (MMC), uses a linear mixed-model framework in order to adjust gene expression values and calculate pairwise gene correlations. Linear mixed-model frameworks have been successfully used in previous studies to remove confounding effects when performing eQTL analysis (Kang et al., 2008). The MMC procedure represents confounding as a random effect in a statistical model for coexpression. This approach allows us to more accurately calculate coexpression while removing the effects due to confounding. Unlike ComBat, our method does not require previous knowledge about the batch effects. MMC is also able to calculate coexpression without assuming and estimating some finite number of confounding effects, such as with SVA. These two properties give our method the advantage of being able to represent a wide range of unknown effects.
A caveat to our approach, as well as other approaches that utilize the inter-sample correlation matrix as a surrogate for confounding, is the potential to remove true biological signal, as expression heterogeneity may be caused by true biological effects. Consider one transcription factor whose activity marks the beginning of many possibly unrelated pathways. When this transcription factor exhibits high activity, the genes involved in the downstream pathways will appear to be highly differentially expressed. This high level of differential expression will cause the downstream genes to be statistically correlated. When this master regulator affects hundreds or even thousands of downstream genes, the global patterns of array variation become similar and thus arrays appear to be correlated. This correlation is represented in the inter-sample correlation matrix and is utilized in the correction procedure. Correlations between genes induced by such large-scale biological effects are not differentiable from correlations induced through large-scale technical confounding effects and therefore our method will ‘correct’ for both types of induced correlations. In this way, it is possible for our method or a method such as SVA that utilizes the inter-sample correlation matrix to remove a true biological signal. However, this caveat can also be a useful side effect, as the goal of many coexpression analyses is to find groups of genes that are tightly functionally related. Large-scale effects, whether true biological effects or technical confounding, may hinder the ability to find smaller gene modules. In this sense, our method can be seen as a complementary to current coexpression methods that identify large modules.
In order to evaluate MMC, we take advantage of the fact that microarrays contain many more probes than measured genes and that expression patterns for probes measuring the same gene should be among the most highly correlated within the set of all probes. We compare methods for computing coexpression by comparing their ability to highly rank these probe pairs in terms of correlation. Our results show that MMC is able to rank these pairs more highly when compared to SVA and a traditional Pearson correlation. We evaluate our method further by utilizing replicate gene expression datasets. We utilized two yeast gene expression datasets (Brem et al., 2002; Smith and Kruglyak, 2008) produced by the same lab, covering the same strains and same genes but produced 5 years apart using different microarray platforms. We applied our method to both datasets and show that it is able to produce coexpression results which are more concordant when compared to both traditional Pearson and SVA corrected coexpressions. Finally, we consider how coexpressions may be used in order to identify biologically meaningful groups of genes. Under the assumption that genes working together in the same complex or pathway should be highly coexpressed, we examined coexpression values for sets of genes belonging to known functional categories. Given a set of known gene functional modules, we evaluated the ability of MMC coexpressions to identify these modules as biologically significant. Compared to both the traditional Pearson correlation and with SVA corrected coexpressions, we show that MMC has higher power to detect biologically meaningful gene sets.
2 METHODS
We first highlight the relationship between a traditional Pearson's correlation coefficient and a basic linear model. We demonstrate the mathematical connection between the Pearson correlation and hypothesis testing under a linear model, and use this intuition when developing the MMC.
2.1 Pearson correlation as a linear model
The coexpression between two genes is often estimated by using the traditional Pearson correlation coefficient. The Pearson correlation gives an absolute value ranging from 0 to 1. If the absolute value of the correlation is close to 1, then we say that the pair of genes is significantly coexpressed. The threshold for significance is usually domain dependent and set on a case by case basis. The Pearson correlation can be calculated for any two genes, y1 and y2, by using Equation (1).
| (1) |
In this case, y1 and y2 are both gene expression vectors of size n and the Pearson correlation is the ratio of the their sample covariance to the product of their sample standard deviations. Here 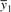 and
and 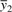 are the sample means for each gene. The Pearson correlation can be represented concisely using matrix notation.
are the sample means for each gene. The Pearson correlation can be represented concisely using matrix notation.
| (2) |
We use 1n to represent a n×1 vector of 1s.
When the elements of y1 and y2 are sampled IID from a bi-variate normal distribution and are truly uncorrelated, the following relation holds (Weatherburn, 1949).
| (3) |
where t has a Student t-distribution with n−2 degrees of freedom. In order to test the hypothesis that rP=0, we test the equivalent hypothesis that t=0, while evaluating t using the observed rP.
To understand how this relationship arises, let us consider the general purpose of the correlation coefficient. The correlation coefficient gives a measure of how linearly related one variable is to another. Another way to evaluate the linear dependence between two variables is by adopting a linear regression framework. Within this framework, the linear dependence between two variables is tested by first defining a linear model, in which one variable is used as a predictor of the other variable, which is called the response. We evaluate the magnitude of the linear dependence, by testing the hypothesis that the predictor variable has no effect on the response variable. With this in mind we define the two following linear models, in which we assume that each gene is a function of its mean, some random error and the observed expression value of another gene. We use ŷi to represent the observed gene expression vector for yi.
| (4) |
| (5) |
In order to evaluate the significance of the effect that gene 1 has on gene 2, we test the hypothesis that β2=0 in the model in Equation (5). Under the null hypothesis that β2=0, we have that the computed t-statistic follows a central Student t-distribution with n−2 degrees of freedom (McCulloch and Searle, 2001). The computed t-statistic is a function of both the estimate 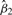 and the sample variance for y1. Through a series of algebraic manipulations we can show that the computed t-statistic has the relationship observed in Equation (3) with the Pearson's correlation (Rao, 1973). We briefly summarize this relationship here as follows.
and the sample variance for y1. Through a series of algebraic manipulations we can show that the computed t-statistic has the relationship observed in Equation (3) with the Pearson's correlation (Rao, 1973). We briefly summarize this relationship here as follows.
| (6) |
| (7) |
t1 and t2 correspond to the t-statistics computed for the estimates of β1 and β2, respectively. Equation (7) shows that there is a direct relationship between the Pearson correlation and a linear model of the type in Equations (4) and (5). Under the null hypothesis, we assume that both t1 and t2 asymptotically follow the t-distribution. Implicit in this assumption is the assumption that the variance of the residuals e1 and e2 is of the form σe2I. More specifically, we assume that both y1 and y2 are normally distributed with means μ1 and μ2, respectively, and variances σ12I and σ22I, respectively. When these assumptions do not hold, such as when the residuals are not independent, we might experience overdispersion of the test statistics (McCulloch and Searle, 2001). In other words, the variance of the test statistics will be greater than expected and thus our assumed null distribution will be incorrect. This phenomenon has been observed in many cases, for example, when the effects of population structure are not accounted for when computing association statistics (Devlin et al., 2001). Since overdispersion leads to inflation of test statistics and the Pearson correlation coefficient is directly proportional to the t-statistics for the models in Equations (4) and (5), this implies that overdispersion may lead to inflation of the Pearson correlation.
2.2 Coexpression as a linear mixed model
In the previous section, we illustrated the relationship between a traditional Pearson's correlation and a linear model. We concluded from this relationship that when the variance of the residuals is misspecified, we have the potential to observe overdispersion, which leads to inflation of the test statistics and subsequently the Pearson's correlation. In the presence of expression heterogeneity, we expect that shared confounding between arrays will make them correlated. When this is the case, we no longer expect that the residuals for the models in Equations (4) and (5) will be independent. Therefore, the assumption of independent residuals is incorrect and this misspecification might lead to overdispersion and subsequently to inflation of the Pearson's correlation.
One way to deal with overdispersion is to account for the source of overdispersion with a random variable (McCulloch and Searle, 2001). Therefore, we propose the following two linear models that have an additional random variable, which accounts for confounding effects.
| (8) |
| (9) |
In these models, we assume that var(e1)=var(e2)=σe2I, var(u1)=var(u2)=σu2K and that cov(ei,uj)=0 ∀ i,j, where K represents the inter-sample correlation matrix. Given a set of n arrays each measuring m genes, we define the inter-sample correlation matrix as the n×n sample covariance matrix for the m×n matrix of the complete array data. In other words, the matrix K is a matrix containing all pairwise covariances for all pairs of arrays. The key assumption here is that the additional variance due to systematic confounding effects is proportional to the correlation between arrays.
When gene 1 and gene 2 are truly uncorrelated (β1=β2=0), the Pearson's correlation should be zero. However, when the models in Equations (8) and (9) hold, the observed Pearson's correlation will be inflated due to correlation between the elements of u1 and u2. Subtracting the true values of u1 and u2 from y1 and y2, will produce corrected vectors for which the observed Pearson's correlation will not be inflated. However, the true values of these variables are unknown and in order to obtain estimates of them, we would have to make further assumptions and restrictions on the model. Instead, we only make an assumption about the distributions of both u1 and u2. With knowledge of these distributions, we estimate the total variance of y1 and y2.
Under the null hypothesis, β1=β2=0, we have the following.
| (10) |
| (11) |
where
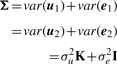 |
When the gene expression vectors follow the distributions in Equations (10) and (11), the traditional Pearson's correlation will be inflated due to overdispersion. That is, when computing the Pearson's correlation, we assume that Σ=σc2I, for some σc2. In order to remove the effects of overdispersion in each gene expression vector, we need to transform the gene expression vectors so that they have the same variance–covariance structure assumed when computing the Pearson's correlation. Then using these transformed vectors we apply the definition for a traditional correlation coefficient. This is accomplished by utilizing the following rule, which is applicable to random variables with a multivariate normal distribution with mean μ and positive semi-definite variance–covariance matrix Σ (Kariya and Kurata, 2004).
| (12) |
| (13) |
Using this rule we obtain the distribution for y1* and y2* defined as follows.
| (14) |
| (15) |
When the Pearson's correlation is calculated in this transformed space (i.e. using the transformed vectors), we expect that the assumptions of independent residuals will hold and thus the correlation will not be subject to inflation. Given the true Σ and the observed gene expression vectors y1 and y2, we transform the observed vectors and calculate a corrected Pearson's correlation.
| (16) |
We expect that this adjusted correlation coefficient will have a mean of zero when gene 1 and gene 2 are uncorrelated. Simplifying we obtain the following.
| (17) |
We are not given the true values of the means μ1 and μ2 or the true value of Σ. In order to calculate rMMC between two given gene expression vectors, we must estimate these parameters from the data. Substituting the estimates for μ1, μ2 and Σ, we arrive at the final formula.
| (18) |
The t-statistics corresponding to the βs from the models in Equations (8) and (9) maintain the relationship illustrated in Equation (6), while rMMC has been substituted for rP.
In order to determine the value of rMMC, we must first determine the value of 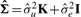 . This means that we need to estimate the two variance components,
. This means that we need to estimate the two variance components, 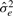 and
and 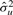 . When estimating only one variance component, the estimates are obtained analytically through a maximum likelihood (ML) or restricted maximum likelihood (REML) approach. However, there does not exist a general analytical method for estimated more than one variance component. Therefore, we must incorporate a numerical search strategy in order to obtain optimal estimates. Such solutions are computationally intensive. In order to estimate these variance components, we employ a method described by (Kang et al., 2008). This method reduces the computational complexity at each search step from O(n3), using the basic Newton–Raphson algorithm, to O(n) by re-formulating the problem so that the singular value decomposition of K can be reused. The method combines grid search with the Newton–Raphson algorithm and can be applied, in order to find the optimal variance components.
. When estimating only one variance component, the estimates are obtained analytically through a maximum likelihood (ML) or restricted maximum likelihood (REML) approach. However, there does not exist a general analytical method for estimated more than one variance component. Therefore, we must incorporate a numerical search strategy in order to obtain optimal estimates. Such solutions are computationally intensive. In order to estimate these variance components, we employ a method described by (Kang et al., 2008). This method reduces the computational complexity at each search step from O(n3), using the basic Newton–Raphson algorithm, to O(n) by re-formulating the problem so that the singular value decomposition of K can be reused. The method combines grid search with the Newton–Raphson algorithm and can be applied, in order to find the optimal variance components.
For each pair of genes, i and j, we use the numerical search method to find the optimal estimates for the variance components iσe2, iσu2, jσe2 and jσu2. We use the left subscript to identify the gene for which the component belongs to. For example, iσe2 and iσu2 are estimated using the model for gene i [refer to Equations (8) and (9)]. Using the estimated variance components, we obtain 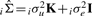 and
and 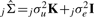 , the variance–covariance matrices for the models corresponding to yi and yj. These variance–covariance matrices are used to obtain the observed MMC coexpression values corresponding to gene i and gene j, irMMC and jrMMC. Ideally, these MMC coexpressions are equal. However, in practice this is not the case, so we average them to define the final corrected correlation rMMC, in order to ensure the symmetry of coexpression. It is also possible to calculate rMMC by using the corrected vectors
, the variance–covariance matrices for the models corresponding to yi and yj. These variance–covariance matrices are used to obtain the observed MMC coexpression values corresponding to gene i and gene j, irMMC and jrMMC. Ideally, these MMC coexpressions are equal. However, in practice this is not the case, so we average them to define the final corrected correlation rMMC, in order to ensure the symmetry of coexpression. It is also possible to calculate rMMC by using the corrected vectors 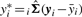 and
and 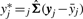 and then applying the definition of the Pearson's correlation from Equation (16). When
and then applying the definition of the Pearson's correlation from Equation (16). When 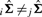 , we found the solution to be very concordant with that obtained by averaging irMMC and jrMMC.
, we found the solution to be very concordant with that obtained by averaging irMMC and jrMMC.
3 RESULTS
3.1 Prioritizing probe pairs targeting the same gene
In order to evaluate the ability of MMC to prioritize true coexpressions, we leverage the fact that microarrays typically contain many more probes than there are genes to measure, meaning that most genes are targeted by more than one probe. We assume that the expression levels of any two probes targeting the same gene should be highly correlated, and thus when ranked against all other pairwise coexpressions, these probe pairs should be among the most highly ranked. We compare the relative ranking of coexpressions for probes targeting the same gene between different methods, in order to determine which method is better able to prioritize strong coexpressions. It may be noted that when certain forms of alternative splicing occur or when some genes are simply not expressed, the results of this evaluation strategy may fail to differentiate the methods for calculating coexpression.
We utilized a set of 732 probe pairs obtained from the Human HapMap gene expression arrays (International HapMap Consortium, 2003). The gene expression data represents 60 unrelated individuals of European descent (http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GSE6536). The probe set corresponds to those probes that target known RefSeq genes and that could be coupled with at least one other probe targeting the same gene. For each probe pair, we calculate the MMC coexpression, the traditional Pearson correlation and an SVA corrected Pearson correlation. Each expression value is ranked with respect to all pairwise coexpressions for each method. Smaller ranks indicate higher coexpression. We expect that when examining the coexpression ranks for the set of 732 probes, the method that performs best should have an abundance of low ranks.
Figure 1 shows the distribution of the coexpression ranks obtained with each method. The total number of genes considered was over 26 000, meaning that there were over 300 million pairwise coexpressions (26 000 choose 2) to consider. Subsequently, there are over 300 million possible rankings for each coexpression. Each method places ~96% of the 732 probe pairs within the top 1 million ranks. The figure shows that the MMC method consistently ranks these probe pairs higher than either of the other methods. For example, MMC places 79 of the 732 probe pairs within the top 100 ranks, while SVA and Pearson place only 63 and 76, respectively. In the top 10 000 ranks, MMC places 216 probe pairs, while Pearson and SVA place only 177 and 191. If we assume that each of the 732 probe pairs should have a correlation of 1, then their ranks should be in the top 732 choose 2. MMC places 415 of the 732 probe pairs within this range, while Pearson and SVA place only 370 and 366, respectively. These results suggest that MMC is more accurately calculating the coexpressions of these probe pairs, which we assumed to be truly coexpressed.
Fig. 1.

The distributions of coexpression ranks for a set of 732 probe pairs, for which both probes in a pair target the same gene. The coexpression values for each probe pair are ranked with respect to all other pairwise coexpression values. Smaller ranks indicate higher coexpression. We expect that probes targeting the same gene should be highly coexpressed and therefore should have very low rank. The MMC method consistently ranks these coexpressions lower when compared to the other two methods.
3.2 Concordance between replicated data sets
Replicate datasets are great resources to use in order to validate experimental findings. When considering coexpression, we expect that genes found to be highly coexpressed in replicate dataset 1 would also be highly coexpressed in replicate dataset 2. However, due to confounding effects, we may observe a high level of discordance between coexpressions found using two separate replicate datasets. Methods that remove confounding may alleviate this problem and cause coexpressions to be more concordant between replicate datasets. In order to evaluate the performance of MMC in this respect, we obtain two yeast gene expression datasets produced by the same lab and measuring the same genes over the same strains of yeast but conducted 5 years apart (Brem et al., 2002; Smith and Kruglyak, 2008). For both datasets, we calculate coexpressions using MMC, traditional Pearson and SVA corrected Pearson. We then compare the concordance of coexpression values between the two datasets.
In order to compare coexpression values between two replicate datasets, we compare their relative rankings and compute the proportion that are common. We are considering a total of 6143 genes, so there are over 18 million gene pairs and thus over 18 million coexpressions. We expect that the most highly coexpressed genes will be the same within both datasets. Given this, we define a measure of concordance between two datasets in which we calculate the proportion of genes that are common within the top n most highly ranked coexpressions. For example, consider the top 100 coexpressions from dataset one, we might see that of these coexpressions only half appear in the top 100 when considering dataset two. In this case, we determine that the proportion in common is 50% for n=100. By calculating the proportion in common for every n, we obtain a concordance at the top (CAT) plot, as shown in Figure 2.
Fig. 2.

Comparison of the concordance between two yeast datasets for both methods Concordance between two sets of coexpressions is compared by looking at the proportion of coexpressions in common for the top ranking coexpressions. The x-axis represents the number of top ranked coexpressions considered, while the y-axis represents the proportion of those coexpressions that are common between the new and old dataset.
The CAT plot in Figure 2, illustrates the differences between concordance for each of the methods considered. Ideally, at each point on the x-axis the y-value would be 1, meaning that 100% of the coexpressions would be in common. Although, this is not the case, we do see that both MMC and SVA are concordant ~30–40% of the time when considering the top 200 ranks. However, when considering the ranks ranging from 300 to 50 000, our method out performs both methods by estimating coexpressions which are concordant 20–40% of the time. This result strongly suggests that MMC is more effective in removing confounding effects which may cause coexpressions to be discordant across datasets.
3.3 Gene module significance
One intention behind the calculation of coexpression is to quantify the strength of the biological relatedness between genes. For example, if two genes code for signaling proteins that act together in one particular pathway, we expect that these genes will be expressed together and that their coexpression value will be quite high. If we assume that the coexpression between two genes reflects the strength of their biological relationship, it is possible to utilize coexpressions in order to predict how biologically relevant a group of genes may be. Consider a group of genes that all code for proteins that work together in a complex. It is reasonable to assume that each pair of these genes will be coexpressed. In this case, by comparing each of the pairwise coexpressions for the genes within this group we should see an abundance of significant coexpressions. In general, we can assume that a group of genes that are functionally related should all be significantly coexpressed. We can then use this assumption to test the significance of a group of genes in order to determine if it is biologically relevant. In practice, by using such an approach we will likely find many groups of genes which will appear to be biologically relevant, but in fact their high level of inter-coexpression is due to confounding.
We tested our ability to detect biologically significant groups of genes using MMC coexpressions. We define the statistic found in Equation (19), which is simply the sum of the logged coexpression ranks. rankij represents the relative ranking of the coexpression between gene i and gene j, with respect to all other pairwise coexpressions. When genes within a group are highly coexpressed, the value of this statistic will be high and when they are not it will be very low. To obtain a set of gene groups which are known to be functionally related, we chose to use yeast, as it has some of the most well-characterized genes. The MIPS comprehensive yeast genome database contains detailed functional data for all yeast genes (Mewes et al., 1999). We used this resource in order to construct 233 gene modules ranging in size from 2 to 20. Modules were chosen such that the number of modules was maximized while the modules in each size category did not overlap. We chose sizes of 2–20, assuming that smaller modules would represent more closely functionally related gene sets and thus the overall coexpressions within modules would be higher. For each of the 233 modules, we calculated the statistic T using coexpressions estimated with MMC, traditional Pearson and SVA corrected Pearson. We estimate the null distribution for T under each method and each module size n, by repeatedly selecting n random coexpressions and calculating the statistic T. Each null distribution was approximated with 1 million values. Using this null approximation we calculated P-values for each known module.
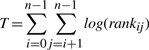 |
(19) |
Figure 3 shows the distribution of the P-values for all modules. Module P-values obtained when using our method tend to be smaller than the Pearson and SVA module P-values. For example, ~40% of the tested gene modules were significant at a level of 0.05 when using MMC, while ~25% and 30% were significant when using Pearson and SVA, respectively. This result suggests that MMC is able to produce coexpression values which were better able to predict real biological relationships.
Fig. 3.

Distribution of gene-module P-values for Pearson, SVA and MMC. We used a set of 233 known functional modules consisting of sets of genes of size 2 to 20. For each of these modules, a P-value representing the biological significance is calculated. This figure plots the distributions of these P-values. Since the P-values were calculated for gene sets known to be functionally related, we expect that there should be an inflation of significant P-values. It can be seen that the MMC method produces a larger number of significant P-values when compared to both the traditional Pearson and SVA-corrected coexpressions.
4 DISCUSSION
In this article, we present a statistical model for the calculation of gene coexpression called MMC. Our method calculates gene coexpressions that are robust to confounding effects. We calculate the coexpression between two genes by utilizing a mixed-model framework. Unknown confounding effects are represented as a random variable in a mixed-model formulation of coexpression. We use the inter-sample correlation to estimate the variance of the random variable representing unknown confounding and incorporate this variance into the model of coexpression.
We compare the coexpressions obtained with our method with those obtained using the traditional Pearson correlation and those obtained using SVA corrected expression data. Although, rank based correlation methods, such as the Spearman correlation, have been used to reduce the prevalence of spurious correlations due to deviations from assumptions of normality in expression data, we have observed in practice that the Spearman correlation coefficient performs similarly to the Pearson when comparing with MMC (data not shown). When probe pairs target the same gene, we expect that their coexpressions will be highly ranked when compared with all other pairwise coexpressions. For probe pairs of this type, MMC is shown to produce coexpressions that are more highly ranked when compared with the other two methods. We also show that MMC produces coexpressions that are more concordant across replicate datasets generated by the same lab using the same strains but generated at different times. Operating under the assumption that biologically and functionally meaningful groups of genes will be highly coexpressed, we create a simple statistic which is used to assess the functional significance of groups of genes. Our method shows increased power to discover sets of genes which are known to be biologically significant.
Although our method is able to calculate coexpression while removing the effects of confounding, it might also remove effects which are biologically meaningful. Technical confounding effects, such as a batch effect, typically have a global effect on the data. That is, these effects will increase the expression variation in a large number of genes. This shared variation within genes causes them to appear to be significantly coexpressed. Our method estimates global patterns of shared variation through the inter-sample correlation and effectively removes the effects causing the variation from the calculation of coexpression. A problem arises when we consider the case in which one gene has a large biological effect on hundreds of other genes. The effect that master regulators have on expression data as a whole is indistinguishable from the unwanted global confounding effects. That is, the variation in gene expression caused by master regulators quite closely resembles patterns of variation caused by confounding effects and will therefore be removed by our method. In this case, MMC may over-correct true biological signal and cause true coexpressions to be lost.
The drawback to our method may also be seen as a beneficial side effect. When master regulators target many genes, traditional coexpression analyses employing clustering will yield many large sized gene modules. By removing the effects of master regulators, MMC essentially enables coexpression clustering analysis to produce smaller gene modules conditional on the large modules. Large gene modules discovered through the use of standard coexpression analysis may be seen as representative of large-scale cellular functionality. Small modules discovered through clustering with MMC will be subsets of these large modules. By intersecting results, it may be possible to more fully understand the detailed circuitry of the cell.
Funding: National Institute of Health training grant T32-HG002536 (to N.F.). National Science Foundation (No. 0513612, No. 0731455 and No. 0729049) (to N.F., H.M.K., C.Y., E.E.); and National Institutes of Health (1K25HL080079 and U01-DA024417); Samsung Scholarship, the National Human Genome Research Institute (Grants No. HG00521401 to H.M.K.); National Institute for Mental Health NIMH No. NH084698, and GlaxoSmithKline (in part). UCLA subcontract of contract N01-ES-45530 from the National Toxicology Program/National Institute of Environmental Health Sciences to Perlegen Sciences (in part).
Conflict of Interest: none declared.
REFERENCES
- Balázsi G., et al. Spurious spatial periodicity of co-expression in microarray data due to printing design. Nucleic Acids Res. 2003;31:4425–4433. doi: 10.1093/nar/gkg485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem R.B., et al. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Clarke R., et al. The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat. Rev. Cancer. 2008;8:37–49. doi: 10.1038/nrc2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B., et al. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 2001;60:155–166. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- Ghanzalpour A., et al. Integrating genetic and network analysis to characterize gene related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Johnson W.E., et al. Adjusting batch effects in microarray expression using empirical bayes methods. Biostatistics. 2007;8:118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kang H.M., et al. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics. 2008;180:1909. doi: 10.1534/genetics.108.094201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya T., Kurata H. Generalized least squares. Chichester, England: John Wiley & Sons Inc.; 2004. The Atrium. [Google Scholar]
- Leek J.T., Storey J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J.T., Storey J.D. A general framework for multiple testing dependence. Proc. Natl Acad. Sci. USA. 2008;105:18718–18723. doi: 10.1073/pnas.0808709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.I., et al. Identifying regulatory mechanisms using individual variation reveals key role for chromatin modification. Proc. Natl Acad. Sci. USA. 2006;103:14062–14067. doi: 10.1073/pnas.0601852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch C., Searle S. Generalized, Linear, and Mixed Models. New York: Wiley-Interscience; 2001. [Google Scholar]
- Mewes H., et al. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 1999;27:44. doi: 10.1093/nar/27.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X., et al. The effects of normalization on the correlation structure of microarray data. BMC Bioinformatics. 2005;6:120. doi: 10.1186/1471-2105-6-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C. Linear Statistical Inference and Applications. NY: Wiley; 1973. [Google Scholar]
- Smith E.N., Kruglyak L. Gene-environment interaction in yeast gene expression. PLoS Biol. 2008;6:e83. doi: 10.1371/journal.pbio.0060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart J.M., et al. Gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Subramanian A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherburn C. A First Course Mathematical Statistics. Cambridge, England: Cambridge University Press; 1949. [Google Scholar]