

Abstract
The primary mechanism of copper transport to the brain is unknown, although this process is drastically impaired in Menkes disease, an X-linked neurodevelopmental disorder caused by mutations in an evolutionarily conserved copper transporter, ATP7A. Potential central nervous system entry routes for copper include brain capillary endothelial cells that originate from mesodermal angioblasts and form the blood-brain barrier, and the choroid plexuses, which derive from embryonic ectoderm, and form the blood-cerebrospinal fluid barrier. We exploited a rare (and first reported) example of somatic mosaicism for an ATP7A mutation to shed light on questions about copper transport into the developing brain. In a 20-month-old Menkes disease patient evaluated before copper treatment, blood copper and catecholamine concentrations were normal, whereas levels in cerebrospinal fluid were abnormal and consistent with his neurologically severe phenotype. We documented disparate levels of mosaicism for an ATP7A missense mutation, P1001L, in tissues derived from different embryonic origins; allele quantitation showed P1001L in approximately 27% and 88% of DNA samples from blood cells (mesoderm-derived) and cultured fibroblasts (ectoderm-derived), respectively. These findings imply that the P1001L mutation in the patient preceded formation of the three primary embryonic lineages at gastrulation, with the ectoderm layer ultimately harboring a higher percentage of mutation-bearing cells than mesoderm or endoderm. Since choroid plexus epithelia are derived from neuroectoderm, and brain capillary endothelial cells from mesodermal angioblasts, the clinical and biochemical findings in this infant support a critical role for the blood-CSF barrier (choroid plexus epithelia) in copper entry to the developing brain.
Keywords: Somatic mosaicism, Menkes disease, ATP7A, copper metabolism, choroid plexus
INTRODUCTION
Menkes disease is an X-linked recessive neurodegenerative disease associated with abnormal copper metabolism [Menkes et al., 1962]. This disorder is caused by loss-of- function mutations in ATP7A, a P-type ATPase with a critical role in copper transport from the gastrointestinal lumen to the bloodstream and subsequently into the brain [Vulpe et al., 1993; Mercer et al., 1993; Chelley et al., 1993; Liu et al., 2005], as well as in the intracellular delivery of copper to the trans-Golgi compartment, for loading onto secreted copper enzymes such as tyrosinase, dopamine-β-hydroxylase, peptidylglycine alpha-amidating monooxygenase, and lysyl oxidase [Kaler, 1998; Lutsenko and Petris, 2003]. Numerous clinical manifestations of Menkes disease reflect these copper transport defects and include failure to thrive, temperature instability, seizures, hypotonia, global developmental delay, light pigmentation, coarse hair, lax skin, osteopenia, vascular tortuousity, arterial and venous aneurysms, and bladder diverticula [Kaler, 1994; Kanumakala et al., 2002; Godwin et al., 2006; Price et al., 2007]. Death by three years of age is typical although very early diagnosis and treatment with copper injections improves survival and neurological outcomes [Kaler et al., 2008].
The precise mechanisms that facilitate delivery of copper and other metals into the brain are not completely understood [Choi and Zheng, 2009; Nischwitz et al., 2008; Rouault et al., 2009], although the blood-brain [Risau and Wolburg, 1990] and blood-cerebrospinal fluid [Segal, 2000] barriers are assumed to control these processes. Brain capillary endothelial cells comprise the blood-brain barrier throughout the central nervous system and derive from angioblasts of mesodermal lineage [Noden, 1989; Drake, 2000]. In contrast, the epithelia of choroid plexuses, which abut the two lateral, the third, and the fourth ventricles of the brain, represent the blood-cerebrospinal fluid barrier, and clearly derive from neuroectoderm [Catala, 1998; Awatramani, 2003; Wolburg and Paulus 2010]. Choroid plexus epithelial cells are polarized, with their basolateral surfaces facing a dense core of capillaries and loose connective tissue stroma, and their apical surfaces projecting into the cerebrospinal fluid-filled ventricles. These specialized cells regulate the chemical composition of the cerebrospinal fluid [Johanson, 1988; Brill et al., 2008].
Several known copper transport genes (ATP7A, ATP7B, CTR1) are expressed in mammalian cells that comprise the blood-brain and blood-cerebrospinal fluid barriers [Choi and Zheng, 2009; Kaler and Schwartz, 1998; Kuo et al., 1997; Kuo et al., 2001; Scheiber IF et al., 2010]. Animal models support the importance of ATP7A and CTR1 for copper delivery to brain; mice with defects in these genes show markedly reduced brain copper content [Phillips et al., 1986; Kuo et al., 2001]. Despite this knowledge, many questions remain about copper intake, export, and utilization within both the central and peripheral nervous systems.
Somatic mosaicism, the occurrence of two or more genetically distinct cell lines in an organism, has been noted in many single gene disorders [Hall, 1988]. This phenomenon may occur via chromosomal abnormalities, epigenetic alterations of DNA, post-zygotic DNA mutations, and spontaneous reversion of inherited mutations [Youssoufian and Pyeritz, 2002; Hirschhorn, 2003]. Selection in favor or against cells harboring the mutant allele may impact clinical and biochemical phenotypes [Kesari et al., 2009; Kvittingen, 1994; Leuer, 2001]. Somatic mosaicism has not been reported in Menkes disease; here we describe and characterize the first known example which, in correlation with the biochemical and clinical phenotype, sheds light on the normal process of copper entry to the developing brain.
PATIENT AND METHODS
All patients and patient specimens were studied under protocols approved by an Institutional Review Board (IRB) following informed parental consent.
Patient
The proband was a 20-month-old male infant referred to the Eunice Kennedy Shriver National Institute of Child Health after being diagnosed with Menkes disease. He was evaluated under NIH protocol #09-CH-0059 approved by the NICHD Institutional Review Board. He was born to a 30-year-old gravida 2 para 2 mother (first pregnancy was twin) after an uneventful pregnancy. Normal spontaneous vaginal delivery occurred at 35 weeks gestation with birth weight 2.35 kg. By 7 months of age, clinical seizure activity was suspected and global developmental delays were documented. Comparative genomic hybridization, standard karyotyping, fragile X testing, and DNA methylation analysis for Prader-Willi syndrome [Butler et al., 1982] were normal. A brain MRI at 8 months of age showed evidence of delayed myelination. Physical stigmata of Menkes disease [Kaler 1994], osteopenia, and bladder diverticula later suggested Menkes disease. The diagnosis was delayed because the typical biochemical markers of the disease including low copper and abnormal catecholamine levels in blood [White et al., 1992; Kaler et al., 1993, Kaler et al., 2008] were not evident. At 18 months of age, mutation screening of the ATP7A locus in genomic DNA from whole blood cells revealed a sequence alteration in the heterozygous state at base 3002 in exon 15. Both cytosine (normal) and thymidine bases were detected, indicating somatic mosaicism for a c.3002C>T mutation which predicts a proline to leucine amino acid substitution (p.P1001L). The patient was referred to the National Institute of Child Health and Human Development (NICHD).
Cell Culture
A primary fibroblast culture was established from a skin biopsy obtained from the patient. Cells were grown in Dulbecco’s modified Eagles medium containing 10% fetal bovine serum, 2mM glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin.
Other Patients
Unused cerebrospinal fluid (CSF) from 11 normal control patients undergoing lumbar puncture for medical reasons was obtained under a protocol approved by the Children’s Research Institute IRB, Children’s National Medical Center, Washington, DC. Baseline cerebrospinal fluid (n=10) and urine (n=9) specimens from Menkes disease patients studied under protocol 90-CH-0149 (ClinicalTrials.gov Identifier: NCT00001262), approved by the National Institute of Neurological Disorders and Stroke IRB, were obtained prior to initiating copper treatment.
Cerebrospinal Fluid Copper Determinations
Copper levels in cerebrospinal fluid were determined via atomic absorption spectrometry in a graphite furnace (Model AAnalyst 800, Perkin-Elmer Corporation, New Jersey). Prior to analysis, cerebrospinal fluid samples were thawed from storage at −70°C to room temperature. Samples were diluted two-fold with 0.1% HNO3 (JT Baker). All chemicals were of analytical-reagent grade or higher purity. Deionized water from a Milli-Q system (Millipore) was used for the analyses. The method for determining the calibration curve (linear, through zero) used certified standard reference materials at 2ppb, 5ppb, and 10ppb from High Purity Standards (Charleston, SC). Calibration curves had a correlation coefficient of at least 0.9989. Two dilutions were made for each specimen, and each dilution was measured in triplicate. Measurements were used only if the standard deviation of the three measurements was less than 12%. The resulting data represent an average of two triplicate measurements.
Catecholamine Levels
Catecholamine levels in plasma and cerebrospinal fluid from the proband were measured by high performance liquid chromatography, as previously described [Eisenhofer et al., 1986] and the ratios of dihydroxyphenylalanine: dihydroxyphenylglycol (DOPA:DHPG) and dihydroxyphenylacetic acid: dihydroxyphenylglycol (DOPAC:DHPG) were compared to results in normal pediatric and adult controls [Kaler et al., 1993].
Molecular Analyses of Somatic Mosaicism
The c.3002C>T mutation creates a novel BsmAI site in exon 15, which allowed for quantification of normal and mutant cells in tissue specimens from the patient, using two approaches.
Semi-quantitative assessment
The polymerase chain reaction (PCR) was used to amplify genomic DNA isolated from whole blood and fibroblasts using primers (forward: 5′-AATACGATTTGCTTTCCAAGCCTC-3′; reverse: 5′-AGCACCTACTCCTGTACCC-3′) specific for ATP7A exon 15. The 294 bp PCR products generated were purified using a DNA isolation kit (QIAGEN, Valencia, CA, USA), digested with BsmAI (NEB, Beverly, MA) and electrophoresed through 2% agarose. In the presence of P1001L, BsmAI digestion produces two fragments of sizes 158 bp and 136 bp. To estimate the relative quantities of normal and mutant alleles, the 294 bp and 158 bp fragments were digitized and quantitated by densitometry using ImageJ (http://rsb.info.nih.gov/ij) for each sample. The digestion and quantitation was performed in triplicate for both tissues.
Quantitative real-time PCR (Q-RT-PCR)
Q-RT-PCR was carried out in triplicate using SYBR® Green JumpStart™ Taq ReadyMix™ (Sigma, Saint Louis, MO, USA) on a DNA Engine Opticon® (Model CFD-3200). Genomic DNA (350 ng) from the patient and a normal male control was digested with 5 units of BsmAI or mock-digested (no enzyme added) for 2 hours at 55°C. Following digestion, the genomic DNA was purified and the ATP7A exon 15 specific primers noted above were used to amplify the region containing the patient’s mutation. Concurrently, a similar-sized region of ATP7A exon 8 without a BsmAI site was amplified (forward: 5′-GAAGCTTCTTTGGTCAAGAAGG-3′; reverse: 5′-TCAAAATCTGTTTTTTACTGACACTCC-3′ primer sequences), as well as a similar-sized region of exon 7 containing a BsmAI site (forward: 5′-AATGACGTGTGCCTCCTGCG-3′; reverse: 5′-GTGCTTTGTTGGTTGCCAGGG-3′ primer sequences). Quantitation of the exon 15 target was normalized to the exon 8 target for both digested and undigested samples using the expression 2CT(exon15)/ 2CT(exon8) where CT represents cycle threshold. The normalized quantity of exon 15 target in the digested sample (i.e., in which the mutant allele is removed by digestion) was divided by that of the undigested sample (in which both alleles are present) to determine the fraction of cells harboring the normal allele, as in the combined expression:
Analysis of exon 7 was used to assess efficiency of BsmAI digestion.
RESULTS
Clinical Outcome and Diagnostic Tests
At 20 months of age, the patient’s developmental skill levels, as assessed by the Denver Developmental Screening Test II [Frankenburg et al., 1992], included gross motor, 5 months; fine motor, 7 months; language, 7 months; and personal-social, 11 months. A brain MRI at 20 months of age showed atrophy and delayed myelination (Fig. 1). Clinical neurophysiology studies were normal, showing median nerve compound motor action potentials of 6.5 mV at the L wrist and 5.5 mV at the L elbow (normal value for 1–2 yr old infants: ≥3.5 mV), and median nerve conduction velocity of 50 m/s (normal value for 1–2 yr old infants: ≥35 m/s) (Kang, 2007). Median nerve sensory action potential was 36 μV (normal value for 1–2 yr old infants: ≥15 μV).
Figure 1.
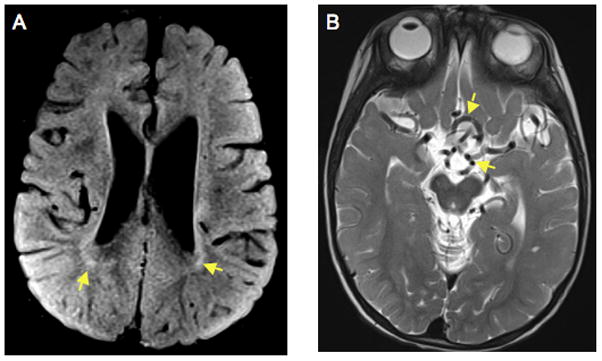
Brain MRI at 20 months of age shows cortical atrophy, delayed myelination and vascular tortuosity, consistent with classical severe Menkes disease. A. Axial FLAIR (Flow attenuated inversion recovery) image indicates dilated lateral venticles and hyperintense lesions in the white matter (arrows), consistent with delayed myelination. B. T2-weighted axial view demonstrating tortuous, kinked vessels in the Circle of Willis (arrows).
Biochemical Findings
Table I summarizes biochemical findings in the proband. His serum copper level was normal. Urine b2-microglobulin, a marker of renal tubular injury due to copper overload that is elevated in patients with Menkes disease, was also normal. Plasma catecholamine ratios, which reflect deficient activity of dopamine-b-hydroxylase, a copper-dependent enzyme [Kaler et al. 1993], were not consistent with Menkes disease. In contrast, cerebrospinal fluid copper and catecholamine ratios fell within the ranges established for classical Menkes disease (Table I).
Table I.
Biochemical and Molecular Findings
| Source/reference ranges | Copper concentration (μg/dl) | DOPA/DHPG ratio | DOPAC/DHPG ratio | ?2-Microglobulin concentration (mg/L) | % Normal allele via PCR/densitometry | % Normal allele via Q-RT-PCR |
|---|---|---|---|---|---|---|
| Blood | 85.5 | 5 | 2.1 | 77.10 ± 5.48 | 68.90 ± 4.16 | |
| CSF | 0.249 ± 0.004 | 1.2 | 1.1 | |||
| Urine | 0.131 | |||||
| Fibroblast | 12.24 ± 5.24 | 12.34 ± 4.71 | ||||
| Normal values | 75–150 (B) 0.726 ± 0.31 (C)a | 1.7–3.3 (B)b 0.3–0.7 (C)b | 1.5–3.2 (B)b 0.1–0.4 (C)b | 0.000–0.300 | 100 | 100 |
| Untreated Menkes | 0–59 (B)c 0.239 ± 0.162 (C)d | 6.2–34.2(B)b 1.2–4.6 (C)b | 4.4–19.7 (B)b 0.8–5.4 (C)b | 2.527 ± 1.677e |
Blood copper concentration is mean of two determinations (89 and 82 μg/dl) at 15 and 20 months of age. The patient’s other biochemical results were measured at 20 months of age. Allele quantitation data reflect the mean ± SD from triplicate experiments.
B, blood; C, cerebrospinal fluid.
CSF copper levels (mean ± SD, n = 11) in normal pediatric subjects ranging in age from 1 to 36 months (mean age, 20.7 ± 13.9 months).
CSF copper levels (mean ± SD, n = 10) obtained as pretreatment baseline values in newborn infants with Menkes disease ranging in age from 5 to 30 days (mean age, 15 ± 8.7 days).
Urine β2-microglobulin concentration (mean ± SD, n = 9) obtained as pretreatment baseline values from newborn infants with Menkes disease ranging in age from 6 to 32 days (mean age = 15 ± 10.3 days).
Molecular Analyses
We confirmed mosaicism for the P1001L mutation in the proband’s genomic DNA (data not shown). The mutant allele was not detected in the mother. P1001L creates a novel BsmAI restriction site (Fig. 2A). PCR and BsmAI digestion indicated a different level of mosaicism in the patient’s blood compared to that in a primary fibroblast culture obtained from his skin biopsy. The wild type allele (294 bp fragment) was markedly less abundant in the latter tissue compared to the patient’s blood DNA and that from a normal control (Fig. 2B). We used densitometry to estimate the relative proportions of fragments associated with the wild type and mutant alleles in both tissues which indicated a wild type: mutant allele ratios of 77:23 in whole blood DNA and 12:88 in fibroblast DNA. We also used quantitative real-time PCR (Q-RT-PCR) experiments to compare mosaicism between tissues, which produced similar results, a wild type: mutant ratio of 69:31 in blood versus 12:88 in primary fibroblasts (Table I). The control for completeness of BsmA1 digestion (ATP7A exon 7) by Q-RT-PCR indicated 99% digestion in these experiments.
Figure 2.
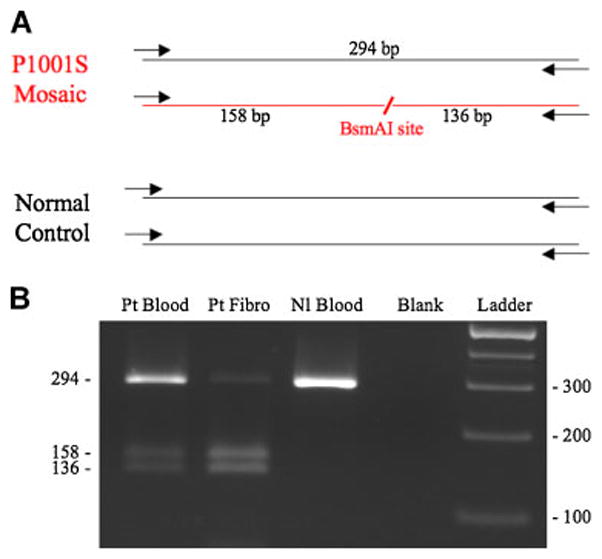
Analysis of mosaicism. A. Diagram of ATP7A exon 15 region that was amplified from genomic DNA from the mosaic Menkes disease patient and a normal control, showing position of a novel restriction site (BsmAI) created by the P1001L mutation. The 294 bp PCR products were digested with BsmAI, and analyzed on a 2% agarose gel. B. In the patient’s blood and fibroblasts (Fibro), presence of three restriction fragments denotes mosaicism for P1001L. Disparity between blood and fibroblasts in the proportions of normal and mutant alleles is reflected in the variable band intensities, indicating a higher percentage of the normal allele in the patient’s blood compared to fibroblasts. The relative proportion of normal and mutant alleles in each tissue was documented by densitometry (Table I). In contrast to the patient, a normal control DNA specimen (Nl Blood) shows only the expected 294 bp band.
DISCUSSION
We describe the first known example of somatic mosaicism for Menkes disease. Somatic mosaicism is known to affect clinical and biochemical phenotypes in numerous genetic disorders by selection in favor or against cells harboring the mutant allele [Kesari et al., 2009; Kvittingen, 1994; Leuer, 2001]. The proline 1001 residue is part of a highly conserved cysteine-proline-cysteine (CPC) transmembrane metal-biding site embedded in the sixth transmembrane domain of ATP7A, that is responsible for metal recognition and movement across the membrane permeability barrier [Argüello et al., 2007]. Therefore, it seems likely that P1001L would completely abrogate ATP7A function in cells that harbor it. Selection against mutant ATP7A alleles was suggested in tissue culture studies [Horn, 1983], and a higher percentage of normal cells might be expected on this basis for tissues that undergo turnover, e.g., blood, gastrointestinal epithelium, renal tubular epithelium.
The patient’s normal blood copper levels connote adequate copper transport at the level of gastrointestinal epithelial cells, which are derived from embryologic endoderm. The absence of elevated b2-microglobulin in the patient’s urine indicates that ATP7A function in renal tubular epithelial cells, derived from embryologic mesoderm [Horster 1999], is also adequate. Since the endothelial cells of brain capillaries that comprise the blood-brain barrier are, like all vasculature, formed from angioblasts of mesodermal lineage [Noden, 1989; Drake, 2000], we speculate that copper transport at the blood-brain barrier in this patient was not significantly impaired. However, he clearly manifests neurodevelopmental delays, as well as abnormal brain imaging (Fig. 1), and cerebrospinal fluid biochemical findings (Table I) that are consistent with severe Menkes disease and profound central nervous system copper deficiency.
The absence of chromosomal abnormalities or epigenetic alterations, the low likelihood of spontaneous reversion of an ATP7A mutation, and the absence of the P1001L allele in the patient’s mother all suggest that this mutation occurred after fertilization during an early stage of embryogenesis. We documented disparate levels of mosaicism in two tissues derived from different embryonic origins: the mutation was present at 23–31% frequency in whole blood (mesoderm-derived), compared with 88% in cultured fibroblasts from skin (ectoderm-derived). These molecular findings imply that the P1001L mutation in the patient preceded formation of the three primary embryonic lineages at gastrulation, with the ectoderm layer ultimately harboring a higher percentage of mutation-bearing cells than mesoderm or endoderm. Since the polarized epithelia of the choroid plexuses are clearly derived from ectoderm [Catala, 1998; Awatramani, 2003; Wolburg and Paulus, 2010], whereas brain capillary endothelial cells derive from mesodermal angioblasts, this infant’s clinical and biochemical findings strongly suggest that copper entry to the developing brain is primarily choroid plexus-mediated.
The polarized epithelia of choroid plexuses project their apical surfaces into the cerebrospinal fluid, with the basolateral sides facing the underlying capillaries. Although in most epithelial cells, ATP7A localizes to the basolateral surface following copper loading [Veldhuis et al, 2009; Kennerson et al., 2010], we have hypothesized that a reversal of protein sorting in choroid plexus epithelia results in ATP7A trafficking to the apical surface where it mediates brain copper delivery [Kaler, 2010]. Choroid plexuses clearly mediate brain entry of manganese, another member of the transition metal family [Aoki et al., 2004]. ATP7A could embed in the apical plasma membrane to pump copper across or, alternatively, may be associated with copper-laden vesicles that fuse with the apical surface and release copper into the cerebrospinal fluid. In support of this general concept, the Na+/K+ ATPase localizes to the apical rather than the basolateral membrane of choroid plexus epithelia [Alper et al. 1994], while all other polarized cells express Na+/K+ ATPase basolaterally.
Our biochemical and molecular findings provide new evidence that the blood-cerebrospinal fluid barrier is the primary route of copper delivery to the developing brain. If mesodermally derived tissues possess 69–77% levels of wild type ATP7A (Table I), brain capillary endothelial cells would not block copper entry. However, copper entry clearly was impaired throughout infancy in this patient and likely explains his Menkes disease phenotype. Thus, the ectodermally derived choroid plexus epithelia, presumably possessing only about 12% of the wild type allele, represents the location at which copper transport was likely defective.
In summary, we identified a Menkes disease patient with somatic mosaicism for a missense mutation, P1001L, in ATP7A that varied quantitatively between tissues from different embryonic lineages. The patient manifested a classic severe Menkes disease phenotype presumably due to the relatively higher frequency of cells harboring the disease-causing sequence variant in ectoderm, from which the choroid plexus epithelia are derived.
Acknowledgments
We gratefully acknowledge the patient and his family for participation in this study.
References
- Argüello JM, Eren E, Gonzàlez-Guerrero M. The structure and function of heavy metal transport P1B-ATPases. Biometals. 2007;20:233–248. doi: 10.1007/s10534-006-9055-6. [DOI] [PubMed] [Google Scholar]
- Alper SL, Stuart-Tilley A, Simmons CF, Brown D, Drenckhahn D. The fodrin-ankyrin cytoskeleton of choroid plexus preferentially colocalizes with apical Na+K(+)-ATPase rather than with basolateral anion exchanger AE2. J Clin Invest. 1994;93:1430–1438. doi: 10.1172/JCI117120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki I, Wu YJ, Silva AC, Lynch RM, Koretsky AP. In vivo detection of neuroarchitecture in the rodent brain using manganese-enhanced MRI. Neuroimage. 2004;22:1046–1059. doi: 10.1016/j.neuroimage.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Awatramani R, Soriano P, Rodriguez C, Mai JJ, Dymecki SM. Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat Genet. 2003;35:70–75. doi: 10.1038/ng1228. [DOI] [PubMed] [Google Scholar]
- Brill BR, Balciunas D, McCarra JA, Young ED, Xiong T, Spahn AM, Garcia-Lecea M, Korzh V, Ekker SC, Schimmenti LA. Development and Notch signaling requirements of the zebrafish choroid plexus. PLoS One. 2008;3(9):e3114. doi: 10.1371/journal.pone.0003114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Kaler SG, Yu PL, Meaney FJ. Metacarpophalangeal pattern profile analysis in Prader-Willi syndrome. Clin Genet. 1982;22:315–320. doi: 10.1111/j.1399-0004.1982.tb01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catala M. Embryonic and fetal development of structures associated with the cerebrospinal fluid in man and other species. Part I: The ventricular system, meninges and choroid plexuses. Arch Anat Cytol Pathol. 1998;46:153–169. [PubMed] [Google Scholar]
- Chelly J, Tumer Z, Tonnesen T, Petterson A, Ishikawa- Brush Y, Tommerup N, Horn N, Monaco AP. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- Choi BS, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009;1248:14–21. doi: 10.1016/j.brainres.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–1679. [PubMed] [Google Scholar]
- Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, Kopin IJ. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- Frankenburg WK, Dodds J, Archer P, Shapiro H, Bresnick B. The Denver II: a major revision and restandardization of the Denver Developmental Screening Test. Pediatrics. 1992;89:91–97. [PubMed] [Google Scholar]
- Godwin SC, Shawker T, Chang M, Kaler SG. Brachial artery aneurysms in Menkes disease. J Pediatr. 2006;149:412–415. doi: 10.1016/j.jpeds.2006.05.041. [DOI] [PubMed] [Google Scholar]
- Hall JG. Review and Hypotheses: Somatic Mosaicism: Observations Related to Clinical Genetics. Am J Hum Genet. 1988;43:355–363. [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40:721–728. doi: 10.1136/jmg.40.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn N. Menkes’ X-linked disease: prenatal diagnosis and carrier detection. J Inherit Metab Dis. 1983;6(Suppl 1):59–62. doi: 10.1007/BF01811325. [DOI] [PubMed] [Google Scholar]
- Horster MF, Braun GS, Huber SM. Embryonic renal epithelia: induction, nephrogenesis, and cell differentiation. Physiol Rev. 1999;79:1157–1191. doi: 10.1152/physrev.1999.79.4.1157. [DOI] [PubMed] [Google Scholar]
- Johanson CE. The Choroid Plexus-Arachnoid Membrane-Cerebrospinal Fluid System. In: Boulton Alan A, Baker Glen B, Walz Wolfgang., editors. The Neuronal Microenvironment. Neuromethods Series. Vol. 9. 1988. pp. 33–104. [Google Scholar]
- Kaler SG, Goldstein DS, Holmes C, Salerno JA, Gahl WA. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33:171–175. doi: 10.1002/ana.410330206. [DOI] [PubMed] [Google Scholar]
- Kaler SG. Menkes disease. In: Barness LA, editor. Advances in Pediatrics. Vol. 41. C.V. Mosby; 1994. pp. 263–304. [PubMed] [Google Scholar]
- Kaler SG. Diagnosis and therapy of Menkes disease, a genetic form of copper deficiency. Am J Clin Nutr. 1998;67:S1029–1034. doi: 10.1093/ajcn/67.5.1029S. [DOI] [PubMed] [Google Scholar]
- Kaler SG, Schwartz JP. Expression of the Menkes disease homolog in rodent neuroglial cells. Neurosci Res Commun. 1998;23:61–66. [Google Scholar]
- Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG. Small copper complexes for treatment of acquired and inherited copper deficiency syndromes. 2010. In: Thoene J, editor. Small Molecule Therapy for Genetic Disease. Cambridge University Press; 2010. [Google Scholar]
- Kang PB. Pediatric nerve conduction studies and EMG. In: Blum AS, Rutkove SB, editors. The Clinical Neurophysiology Primer. Humana; 2007. [Google Scholar]
- Kanumakala S, Boneh A, Zacharin M. Pamidronate treatment improves bone mineral density in children with Menkes disease. J Inherit Metab Dis. 2002;25:391–398. doi: 10.1023/a:1020103901969. [DOI] [PubMed] [Google Scholar]
- Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010;86:343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesari A, Neel R, Wagoner L, Harmon B, Spurney C, Hoffman EP. Somatic mosaicism for Duchenne dystrophy: evidence for genetic normalization mitigating muscle symptoms. Am J Med Genet A. 2009;149A:1499–1503. doi: 10.1002/ajmg.a.32891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Gitschier J, Packman S. Developmental expression of the mouse mottled and toxic milk genes suggests distinct functions for the Menkes and Wilson disease copper transporters. Hum Mol Genet. 1997;6:1043–1049. doi: 10.1093/hmg/6.7.1043. [DOI] [PubMed] [Google Scholar]
- Kuo YM, Zhou B, Cosco D, Gitschier J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc Natl Acad Sci U S A. 2001;98:6836–6841. doi: 10.1073/pnas.111057298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvittingen EA, Rootwelt H, Berger R, Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type I. J Clin Invest. 1994;94:1657–1661. doi: 10.1172/JCI117509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuer M, Oldenburg J, Lavergne JM, Ludwig M, Fregin A, Eigel A, Ljung R, Goodeve A, Peake I, Olek K. Somatic mosaicism in hemophilia A: a fairly common event. Am J Hum Genet. 2001;69:75–87. doi: 10.1086/321285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PC, Chen YW, Centeno JA, Quezado M, Lem K, Kaler SG. Downregulation of myelination, energy, and translational genes in Menkes disease brain. Mol Genet Metab. 2005;85:291–300. doi: 10.1016/j.ymgme.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Lutsenko S, Petris MJ. Function and regulation of the mammalian copper-transporting ATPases: insights from biochemical and cell biological approaches. J Membr Biol. 2003;191:1–12. doi: 10.1007/s00232-002-1040-6. [DOI] [PubMed] [Google Scholar]
- Menkes JHM, Alter M, Steigleder GK, Weakley DR, Sung JH. A sex-linked recessive disorder with retardation of growth, peculiar hair and focal cerebellar degeneration. Pediatrics. 1962;29:764–769. [PubMed] [Google Scholar]
- Mercer JF, Livingston J, Hall B, Paynter JA, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, Glover TW. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- Nischwitz V, Berthele A, Michalke B. Speciation analysis of selected metals and determination of their total contents in paired serum and cerebrospinal fluid samples: An approach to investigate the permeability of the human blood-cerebrospinal fluid-barrier. Anal Chim Acta. 2008;627:258–269. doi: 10.1016/j.aca.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Noden DM. Embryonic origins and assembly of blood vessels. Am Rev Respir Dis. 1989;140:1097–1103. doi: 10.1164/ajrccm/140.4.1097. [DOI] [PubMed] [Google Scholar]
- Phillips M, Camakaris J, Danks DM. Comparisons of copper deficiency states in the murine mutants blotchy and brindled. Changes in copper-dependent enzyme activity in 13-day-old mice. Biochem J. 1986;238:177–183. doi: 10.1042/bj2380177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D, Ravindranath T, Kaler SG. Internal Jugular Phlebectasia in Menkes Disease. Int J Pediatr Otorhinolarygol. 2007;71:1145–1148. doi: 10.1016/j.ijporl.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W, Wolburg H. Development of the blood-brain barrier. Trends Neurosci. 1990;13:174–178. doi: 10.1016/0166-2236(90)90043-a. [DOI] [PubMed] [Google Scholar]
- Rouault TA, Zhang DL, Jeong SY. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab Brain Dis. 2009;24:673–684. doi: 10.1007/s11011-009-9169-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal MB. The choroid plexuses and the barriers between the blood and the cerebrospinal fluid. Cell Mol Neurobiol. 2000;20:183–196. doi: 10.1023/a:1007045605751. [DOI] [PubMed] [Google Scholar]
- Scheiber IF, Mercer JF, Dringen R. Copper accumulation by cultured astrocytes. Neurochem Int. 2010;56:451–460. doi: 10.1016/j.neuint.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Veldhuis NA, Gaeth AP, Pearson RB, Gabriel K, Camakaris J. The multi-layered regulation of copper translocating P-type ATPases. Biometals. 2009;22:177–190. doi: 10.1007/s10534-008-9183-2. [DOI] [PubMed] [Google Scholar]
- Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- White SR, Reese K, Sato S, Kaler SG. Spectrum of EEG findings in Menkes disease. Electroenceph Clin Neurophysiol. 1993;87:57–61. doi: 10.1016/0013-4694(93)90175-u. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;10:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]