

Abstract
Multiple sclerosis (MS) is an autoimmune demyelinating disease of the CNS resulting from a progressive loss of oligodendrocytes. Transaldolase (TAL) is expressed at selectively high levels in oligodendrocytes of the brain, and postmortem sections show concurrent loss of myelin basic protein and TAL from sites of demyelination. Infiltrating CD8+ CTLs are thought to play a key role in oligodendrocyte cell death. Cleavage by granzyme B (GrB) is predictive for autoantigenicity of self-proteins, thereby further implicating CTL-induced death in the initiation and propagation of autoimmunity. The precursor frequency and CTL activity of HLA-A2–restricted TAL 168–176–specific CD8+ T cells is increased in MS patients. In this paper, we show that TAL, but not myelin basic protein, is specifically cleaved by human GrB. The recognition site of GrB that resulted in the cleavage of a dominant TAL fragment was mapped to a VVAD motif at aa residue 27 by N-terminal sequencing and confirmed by site-directed mutagenesis. The major C-terminal GrB cleavage product, residues 28–337, had no enzymatic activity but retained the antigenicity of full-length TAL, effectively stimulating the proliferation and CTL activity of PBMCs and of CD8+ T cell lines from patients with MS. Sera of MS patients exhibited similar binding affinity to wild-type and GrB-cleaved TAL. Because GrB mediates the killing of target cells and cleavage by GrB is predictive of autoantigen status of self proteins, GrB-cleaved TAL-specific T cell-mediated cytotoxicity may contribute to the progressive destruction of oligodendrocytes in patients with MS.
Multiple sclerosis (MS) is a demyelinating disease of the CNS resulting from progressive loss of oligodendrocytes. In the acute stage of disease, lesions contain macrophages, CD4+ and CD8+ T cells, and immunoglobulin deposits, suggesting that the demyelination process is mediated by the immune system (1–3). Although the Ag or Ags driving this self-destructive process in MS have not been identified (4) the importance of myelin-derived Ags was demonstrated by their abilities to elicit an MS-like demyelinating disease, experimental allergic encephalomyelitis (EAE), in various animal models (5).
The major difficulties in applying the EAE model to MS stem from a lack of identification of relevant autoantigen(s). Studies on relapsing EAE have shown that different encephalitogenic molecules or epitopes within them that are compatible with the heterogeneity of the immune response in MS are selected, suggesting that disease initiation and relapse episodes are induced by different Ags (6, 7). Although cell-mediated mechanisms may have a primary role in EAE, augmentation of humoral immunity within the CNS is a well-recognized feature of MS (8). A breakdown of the blood–brain barrier would allow Abs to enter the CNS and cause demyelination by complement activation. In fact, complement may be directly involved in the death of oligodendrocytes (9). Most efforts have been focused on myelin basic protein (MBP) and proteolipid protein, which make up as much as 30 and 50% of CNS myelin, respectively (10, 11). T cell responses to MBP and proteolipid protein, or another oligodendrocyte-specific protein, myelin oligodendrocyte glycoprotein (MOG), did not differ considerably between MS patients and control donors (4). Although oral vaccination with a predefined inducing Ag may successfully prevent and treat disease in animal models (12), a similar approach with MBP in 30 patients with MS led to no significant clinical improvement (13).
Molecular mimicry (i.e., cross-reactivity between self-Ags and viral proteins) has been implicated in the initiation of autoimmunity and MS. Based on homology to retroviral sequences, a novel autoantigen, partially encoded by a retrotransposon and selectively expressed in oligodendrocytes at high levels (14), was identified as human transaldolase (TAL) (15). TAL is a key enzyme of the pentose phosphate pathway (PPP). Although glucose is largely metabolized through the glycolytic pathway and the tricarboxylic acid cycle, the significance of PPP in the brain has long been established. During brain development, PPP provides NADPH for the biosynthesis of lipids (16). The latter is particularly important at the period of active myelination (17, 18). The overall activity of PPP in the brain declines 5-fold from birth to maturity (19). Whereas under normal conditions only as little as 1% of the glucose enters the PPP (20), at times of rapid myelination, up to 60% of the glucose is metabolized via the PPP (21). Involvement of PPP in myelination provided a physiological explanation for the high level of TAL in oligodendrocytes (14, 22). PPP also plays an essential role in neutralization of oxygen radicals, and elevated levels of TAL expression increase susceptibility to apoptosis signals (23–25). The effector phase of the demyelination process in MS is thought to be mediated by reactive oxygen intermediates. Intralesional cytotoxic T cells produce TNF-β, which in turn induces apoptosis, an oxidative stress-mediated programmed cell death, of oligodendrocytes (26). The Fas receptor/ligand system has also been implicated in the death of oligodendrocytes (27, 28). Thus, oligodendrocyte-specific expression of TAL is possibly linked to production of large amounts of lipids, as a major component of myelin and vulnerability of the vast network of myelin sheaths to oxygen radicals. Immunohistochemical studies of postmortem brain sections revealed decreased staining by MBP- and TAL-specific Abs in MS plaques, indicating a concurrent loss of these Ags from sites of demyelination (22). Patients with MS have TAL Abs in their blood and cerebrospinal fluid (14, 22). TAL autoantibodies recognize immunoblotted and three-dimensional epitopes and inhibit enzymatic activity of TAL (29). By contrast, TAL Abs were absent in normal individuals and patients with other autoimmune and neurologic diseases (14) and, under identical conditions, no MBPAbs were found in the serum and cerebrospinal fluid of MS patients (22). The fact that TAL Abs were absent in controls, including patients with other neurologic diseases or systemic autoimmune diseases such as systemic lupus erythematosus and Sjögren’s syndrome, indicated that the autoimmune process targeting TAL may be specific for MS. In addition, TAL elicits proliferation, aggregate formation, and skewing of the TCR Vβ repertoire of peripheral blood T lymphocytes from patients with MS with respect to MBP as control Ag. The results suggested that TAL may be a more significant target than MBP of myelin-reactive T cells and of humoral autoreactivity in patients with MS (22).
Most reports attribute oligodendrocyte cell death, at least in part, to the cytotoxic effect of CD8+ T cells (30). Adoptively transferred MBP-specific (31) or MOG-specific CD8+ T cells induce severe CNS demyelination in animal models (32). In MS brain lesions, infiltrating CD8+ CTLs were reported to outnumber CD4+ T cells 10-fold (1). In addition, actively demyelinating lesions of MS-affected brains are enriched for clonally expanded CD8+ T cells, compared with CD4+ T cells (33, 34). These myelin-reactive CD8+ T cells may play a role in recruitment and retention of myelin-reactive CD4+ T cells by secreting proinflammatory soluble mediators to promote and mediate the inflammatory response in MS-affected brains. Full-length human rTAL or TAL peptide 168–176 (TALpep)–stimulated CTLs from HLA-A2+ MS patients kill TALpep-pulsed HLA-A2–transfected (Hmy A2.1), but not HLA-A3–transfected, control target cells. HLA-A2–transfected cells, but not control MO3.13 oligodendroglial cells, expressing high levels of endogenous TAL, are also killed by the CD8+ CTLs of MS patients without peptide-pulsing the targets, thus indicating that endogenously processed TAL was recognized by HLA-A2–restricted CTLs. Because granzyme B (GrB) mediates killing of target cells by CTLs (35) and cleavage by GrB is strongly predictive of autoantigen status of self-proteins (36), we tested the cleavage of TAL by GrB. In the current study, we show that the 38-kDa TAL Ag is specifically cleaved by GrB between aa 27 and 28. The resulting 310-aa C-terminal fragment shows a complete loss of enzymatic activity but fully retains the antigenicity of TAL and stimulates proliferation and cytotoxic activity of T cells from MS patients. Thus, stimulation of T cell-mediated cytotoxicity by GrB-cleaved TAL (TAL-GrB) may play a key role in the progressive destruction of oligodendrocytes in patients with MS.
Materials and Methods
Patient and control cells
Fourteen patients with MS and eight healthy controls (HCs) were investigated. In the 14 patients, MS was diagnosed according to the criteria of Poser et al. (37). PBMCs were isolated from heparinized venous blood on Ficoll–Hypaque gradient and resuspended in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 mg/ml amphotericin B and cultured in a humidified atmosphere with 5% CO2 at 37°C. Cell-culture products were obtained from Cellgro (Mediatech, Herndon, VA). PBMCs were HLA class I Ag typed by the National Institutes of Health microcytotoxicity test (38). Viability and functional capacity of all PBMC samples have been monitored by trypan blue exclusion (>95%) and PHA-induced proliferative responses by thymidine [methyl-3H] (3H-TdR) incorporation, respectively (29).
Ags and Abs
Full-length recombinant human TAL (TAL-H) protein (clone 1425) was expressed as a fusion protein with GST by pGEX-2T plasmid vector (39), affinity purified through binding of GST to glutathione-coated agarose beads, cleaved from GST by 1 National Institutes of Health unit of thrombin (Sigma-Aldrich, St. Louis, MO) and separated from the agarose bead-bound GST by centrifugation (14). The purified TAL-H was quantified by the Bradford assay, analyzed by SDS-PAGE and Western blot, and tested for TAL enzyme activity, as earlier described (15). Synthetic TALpep was produced and purified to >99% homogeneity by Genemed (Genemed Synthesis, San Francisco, CA). MBP was purified from neurologically normal human brain according to the procedure of Deibler et al. (40). Purity of the MBP preparations was analyzed by SDS-PAGE and Western blot (22). BSA, Con A, and PHA were obtained from Sigma-Aldrich. Highly specific polyclonal rabbit Abs 169 and 170 directed to the 139-aa-long N-terminal segment of TAL were developed earlier (15).
Establishment of TAL-specific T cell lines
PBMCs were incubated in Petri dishes precoated with autologous serum for 1h at 37°C to remove monocytes (41). Nonadherent cells were enriched for CD8+ T cells by selective depletion of CD4+ cells using mAb-coated Dynabeads M-450 CD4 according to the manufacturer’s instructions (Cat. No. 111.08, Dynal, Lake Success, NY). Usually, a CD4 T cell depletion of >99% was achieved with Dynabeads M-450 CD4. The resultant effector cells (107 cells/donor) were stimulated with 5 μg/ml TAL or TALpep. On day 8, and subsequently at regular 7- to 10-d intervals, T cell lines (TCLs) were restimulated with autologous APCs derived from EBV-transformed B cells or PBMCs. APCs were generated by incubation with 5 μg/ml TAL or TALpep for 1 h, irradiated, and added to effector cells at a 1:1 ratio. In between restimulation with APCs, 10 U/ml human rIL-2 was also added to the effector cells.
Generation of B cell lines by EBV infection
EBV was harvested from the supernatant of B95-8 cells (42), filtered through a 0.45-μm mesh, and stored at −80°C. A total of 107 PBMCs were infected by EBV by incubation with 3 ml B95-8 supernatant plus 2 ml fresh complete RPMI medium at 37°C overnight. Next day, 4 ml fresh complete RPMI medium and cyclosporin A (Sigma-Aldrich) was added to a final concentration of 0.5 μg/ml, and cultures were left undisturbed for 10–14 d.
Proliferation assay
A total of 105 PBMCs per well were stimulated with and without 5 μg/ml full-length TAL (clone 1425), GrB-truncated TAL (clone 8427), negative control Ag BSA, and positive control Con A in 96-well plates for 72 h. The cultures were pulsed with 0.4 uCi [3H]-TdR for 12 h before cell harvesting. 3H-TdR incorporation was measured as counts per minute using a liquid scintillation counter and expressed as stimulation indexes (SIs) +/− SE of six parallel cultures.
T cell-mediated cytotoxicity assays
To assess cytotoxic potential and MHC class I restriction of the TAL peptide-reactive TCLs, HLA-A2–transfected (Hmy A2.1) and HLA-A3–transfected (Hmy A3.1) Hmy lymphoblastoma and HLA-A2–transfected (MO3.13/A2) and control MO3.13 oligodendroglial cells were used (43). MO3.13 (44) and Hmy cells spontaneously do not express any HLA class I or class II Ag (43, 44). Target cells were pulsed with 5 μg/ml TALpep for 2 h at 37°C. Then, 106 target cells were labeled with 200 μCi Na251CrO4 for 1 h, washed three times, and seeded at a concentration of 5 × 103 per well of U-bottom 96-well plates. Depending on availability, effectors were added at 2.5:1, 5:1, 10:1, 25:1, and 50:1 effector/target ratios. TALpep was added at 10 μg/ml during the cytotoxicity assay. Control cultures included peptide-pulsed and unpulsed target cells with peptide or medium alone. Maximal release was measured in the presence of 0.1 M HCl. After incubation for 4 h at 37°C, the supernatants were harvested, then counted in a γ counter. Percent cytotoxicity was calculated: 100 × [(test cpm − spontaneous cpm)/(maximal cpm − spontaneous cpm)].
Cytotoxic activity against adherent MO3.13 oligodendroglial cells (44) was measured by detachment of killed cells from the monolayer. A total of 2500 MO3.13 cells per well of flat-bottom 96-well plates were prelabeled with 3H-TdR (ICN Biomedicals, Irvine, CA), with or without 10 μg/ml TALpep, and allowed to form a nonconfluent monolayer for 24 h. Targets were washed three times, and effectors were added for 24 h. Subsequently, plates were washed six times to remove effectors and killed target cells, and cytotoxicity was determined based on the 3H-TdR content of remaining viable cells, as earlier described (41).
Western blot analysis
For Western blot analysis, 500 ng rTAL-H protein in 10 μl per well was separated by SDS-PAGE and electroblotted to nitrocellulose (45). Nitro-cellulose blots were incubated in 100 mM Tris, pH 7.5; 0.9% NaCl; 0.1% Tween 20; and 5% skim milk with TAL Ab 169 (15). The 70-kDa subunit of U1 snRNP was detected with a human Ab (46). Vinculin and β-actin were detected with mouse mAbs (47, 48). For detection of primary Abs, after washing, blots were incubated with HRP-conjugated secondary Abs (Jackson ImmunoResearch Laboratories, West Grove, PA). In between the incubations, the strips were vigorously washed in 0.1% Tween 20; 100 mM Tris, pH 7.5; and 0.9% NaCl. Protein bands were visualized by ECL (Western Lightning Chemiluminescence Reagent Plus, Perkin-Elmer, Boston, MA) using a Kodak Image Station 440CF and quantified by automated densitometry with the Kodak 1D Image Analysis Software (Eastman Kodak, Rochester, NY).
TAL enzyme activity
TAL enzyme activity was measured by the transfer of the dihydroxyacetone three-carbon unit from the donor D-fructose-6-phosphate to the acceptor D-erythrose-4-phosphate, as earlier described (15). Enzyme activity of 50 ng rTAL was assayed in the presence of 3.2 mM D-fructose 6-phosphate, 0.2 mM D-erythrose-4-phosphate, 0.1 mM NADH, 10 ug α-glycerophosphate dehydrogenase/triosephosphate isomerase at a 1:6 ratio in 1 ml PBS (pH 7.4) at room temperature by continuous absorbance reading at 340 nm for 20 min.
Cleavage of TAL by GrB and granzyme A
A total of 3 μg (4 μM) rTAL was incubated, with or without 50–150 nM GrB (GrB, Enzyme System Products, Dublin, CA), for 1 h at 37°C in 20 μl GrB reaction buffer (10 mM HEPES, pH 7.4; 2 mM EDTA; and 1% Nonidet P-40). TAL was also cleaved by GrB obtained as a gift from Nancy Thornberry (Merck, Rahway, NJ). For testing cleavage by granzyme A (GrA), 3 μg rTAL was incubated, with or without 125 nM or 250 nM GrA, for 1 h or 2 h in 20 μl 50 mM Tris-HCl (pH 7.5), 1 mM CaCl2, and 1 mM MgCl2 at 37°C. Cleavage products were analyzed by SDS-PAGE and Western blotting. For N-terminal sequencing, TAL-GrB fragments were separated by SDS-PAGE, electroblotted to polyvinylidene difluoride membrane, stained with Ponceau S, and excised for MALDI-TOF analysis at the Dana Farber/Harvard Cancer Center Proteomics Core Facility (Boston, MA).
Generation of TAL-GrB and GrB cleavage-resistant TAL by site-directed mutagenesis
To study enzymatic activity and antigenicity of TAL-GrB, the 34-kDa major cleavage product, comprising aa 28–337, was expressed as a fusion protein with GST. Clone 8427, capable of expressing aa 28–337 of TAL, was derived from clone 1425, using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The aa 1–27 of TAL were deleted by site-directed mutagenesis of clone 1425, using sense (5′-GATCTGGTTCCGCGTGGATCTACGGGCGACTTCCACGCCATC-3′) and antisense (5′-GATGGCGTGGAAGTCGCCCGTAGATCCACGCGGAACCAGATC-3′) primers. After digestion of template DNA with DpnI, the mutated expression vector, clone 8427, was transformed into XL I Blue Escherichia coli. Deletion of nucleotides encoding aa 1–27 in clone 8427 was confirmed by DNA sequencing. Cleavage of TAL by GrB at aa position 27 was investigated by site-directed mutagenesis of clone 1425, using sense (5′-CCACCGTGGTGGCCGCCACGGGCGACTTCC-3′) and antisense (5′-GGAAGTCGCCCGTGGCGGCCACCACGGTGG-3′) primers, replacing the aspartic residue with alanine (TAL-D27A). Expression of TAL-GrB(clone 8427)and GrB cleavage site-mutagenized TAL (TAL-D27A, clone 9182) was performed as described for wild-type TAL (TAL-WT).
ELISA
For ELISA, 96-well plates were precoated at 4°C with 1 μg peptide per well in 0.01 M NaHCO3 (pH 9.55). Uncoated sites were blocked with 10% goat serum/0.1% Tween 20 in PBS (pH 7.4) at room temperature for 1 h. Subsequently, sera were added to the wells in 10% goat serum/0.1% Tween 20 in PBS at a 1:1000 dilution. After incubation for 1 h, the plates were washed with 0.1% Tween 20 in PBS. The plates were then further incubated with HRP-conjugated secondary Abs, washed with 0.1% Tween 20 in PBS, and developed with 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid). Positive control rabbit sera 169 and 12484 were applied at a 1000-fold dilution, whereas human sera were tested at a 100-fold dilution. Relative optical densities of human sera were compared, with the mean of TAL-specific rabbit sera 169 and 12484 normalized to 1.0.
Statistics
Results were expressed as the mean ± SD or SE and analyzed by Student t test. The p values were calculated with GraphPad Software (San Diego, CA) and considered significant at <0.05.
Results
TAL-H is specifically cleaved by GrB
It was previously demonstrated that TAL-specific T cells effectively lyse TALpep–presenting oligodendroglioma cells in an HLA-A2–restricted manner in vitro (49). In vivo, oligodendrocytes are selectively destroyed in the brain of MS patients. In addition, TAL-H is expressed in oligodendrocytes at high levels in the brain (14, 22). Infiltrating CD8+ CTLs are thought to play a key role in oligodendrocyte cell death via GrB-initiated apoptosis (50). Cleavage by GrB is strongly predictive of the autoantigen status of self-proteins (36). Significantly, TAL-H contains predicted GrB cleavage sites. Therefore, we investigated whether GrB can cleave TAL. TAL and MBP were incubated with 100 nM GrB (Enzyme System Products, Dublin, CA) for 1–2 h at 37°C. GrB cleaved the 38-kDa rTAL-H Ag, resulting in a dominant 34-kDa fragment (Fig. 1A). This 34-kDa fragment was positively identified as TAL after immunoblotting with anti-TAL Ab 169 (Fig. 1B). In contrast, no GrB cleavage of MBP (4 μM) was observed by 100 nM GrB after 120 min incubation (Fig. 1A). TAL was resistant to cleavage by GrA (data not shown).
FIGURE 1.
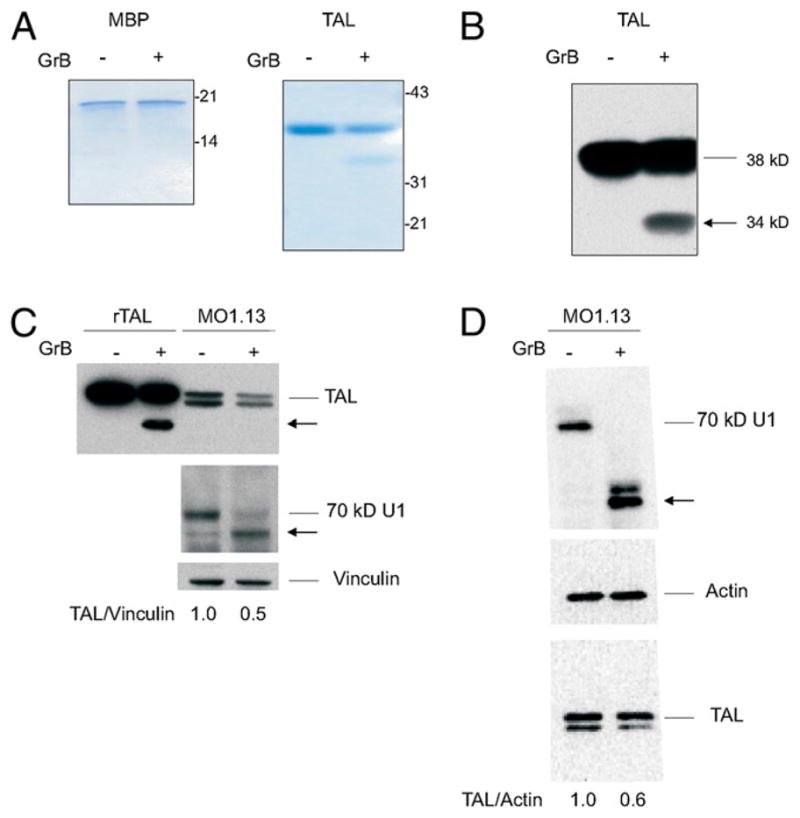
Cleavage of TAL by GrB. A, SDS-PAGE analysis of TAL and MBP exposed to GrB. TAL (38 kDa)and MBP (17.5 kDa) were incubated for 1 h at 37°C with 150 nM purified GrB. Reaction products were loaded into 12% Tris-glycine polyacrylamide gel, separated by SDS-PAGE, and stained with Coomassie brilliant blue. B, Western blot analysis of GrB-digested TAL electroblotted onto nitrocellulose and detected with anti-TAL Ab 169 (22). C, Western blot analysis of rTAL (rTAL, 100 ng/lane) and native TAL in lysates of MO3.13 cells (40 μg/lane) following incubation without (−) or with GrB (+) for 1 h at 37°C. The 70-kDa components of U1 small nuclear ribonucleoprotein and vinculin were detected as positive and negative controls, respectively, for monitoring GrB cleavage. D, Western blot analysis of native TAL in lysates of MO3.13 cells (40 μg/lane) following incubation without (−) or with (+) GrB for 1 h at 37°C. The 70-kDa components of U1 small nuclear ribonucleoprotein and actin were detected as positive and negative controls, respectively, for monitoring GrB cleavage. The TAL/actin ratio was determined with automated densitometry (48) and normalized to lysates incubated without GrB (1.0). Results reflect four independent digestions of MO3.13 lysates with GrB.
We also investigated whether TAL was cleaved in native lysate of the MO3.13 oligodendrocyte-like cell line. As shown in Fig. 1C and 1D, TAL was cleaved after the addition of GrB to MO3.13 lysates. The full-length form of TAL was reduced by 51 ± 6% in GrB-treated lysates relative to vinculin (Fig. 1C) or actin (Fig. 1D; p = 0.01). The 34-kDa cleavage product of TAL was not detected in native MO3.13 lysates, suggesting that the cleaved product may have been further degraded upon GrB proteolysis. Similar findings have been previously reported for cleavage of the native Bag1 protein by GrB (51).
Mapping of GrB cleavage sites in human TAL
Three potential GrB recognition motifs have been identified in TAL (Fig. 2). Upon digestion with GrB, only two cleavage products, a dominant 34-kDa fragment and a minor 25-kDa fragment (not shown), have been identified by Western blot analysis. Both fragments were gel purified and subjected to N-terminal sequencing. The peptide sequence obtained from the 34-kDa fragment, T-D FHAIDEY, indicated that TAL was cleaved after residue 27, contained within a typical VVAD motif (Fig. 2A). The minor 25-kDa GrB product was also sequenced, and its cleavage site was mapped after residue 108, corresponding to the recognition motif TEVD (Fig. 2A). There was no evidence for cleavage at the motif LLQD corresponding to residues 263–267. Digestion of TAL at aa position 27 by GrB was confirmed by site-directed mutagenesis of the VVAD motif. Replacement of the aspartic residue with alanine at aa position 27 blocked cleavage of the mutated protein TAL-D27A by GrB (Fig. 2B).
FIGURE 2.
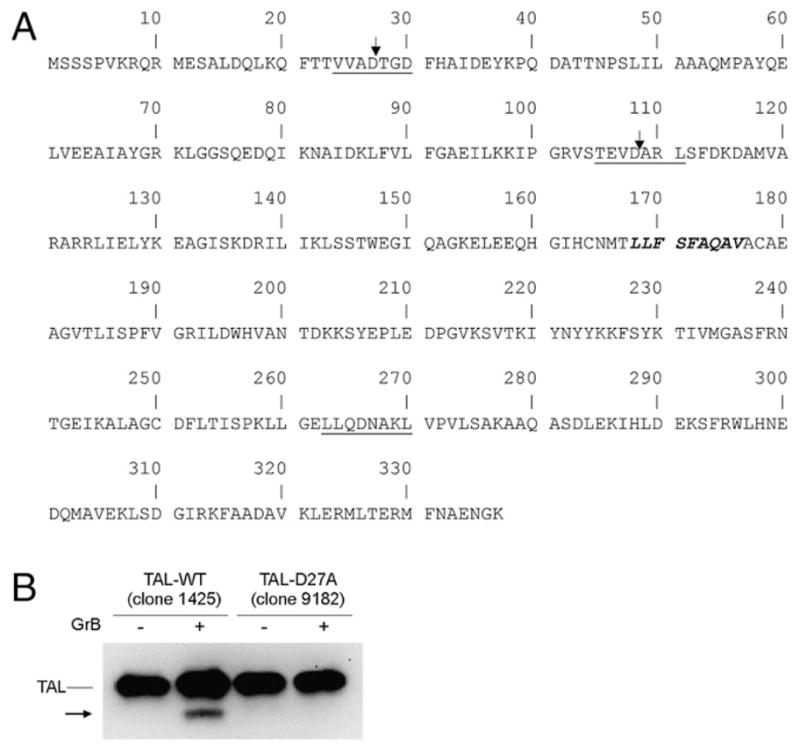
Mapping of GrB cleavage motifs in human TAL. A, Locations of GrB recognition motifs are underlined, and sites of cleavage are indicated by vertical arrows. CTL epitope residues 168–176 are italicized. B, Digestion of TAL-WT and mutated TAL carrying an aspartic-to-alanine substitution at aa position 27 (TAL-D27A). A total of 100 ng of recombinant protein was incubated without (−) or with (+) 150 nM GrB for 1 h at 37°C and detected by anti-TAL Ab 169, using Western blot.
The dominant 34-kDa TAL-GrB fragment exhibits no enzymatic activity but stimulates lymphocyte proliferation and CTL activity in patients with MS
The major 27–28 aa GrB cleavage site was selected for site-directed mutagenesis (Stratagene QuikChange) and subsequent protein expression of rTAL corresponding to the C-terminal 310-aa GrB cleavage product, TAL-GrB. The 34-kDa TAL-GrB peptide was expressed in the pGEX-2T vector following deletion of the first 27 aa of full-length TAL by site-directed mutagenesis, as described earlier (29). The 34-kDa TAL-GrB protein (TAL-GrB, clone 8427, missing the first 27 aa) was expressed as a fusion protein with GST, affinity purified, cleaved with thrombin, and analyzed by immunoblotting with anti-TAL Ab 169 and compared with the full-length 38-kDa WT rTAL-H protein (TAL-WT, clone 1425) as positive control (Fig. 3). Whereas the full-length TAL exhibited sp. act. of >16 U/mg protein, the 34-kDa TAL-GrB protein (aa 28–337) had no appreciable enzymatic activity (<0.1 U/mg).
FIGURE 3.
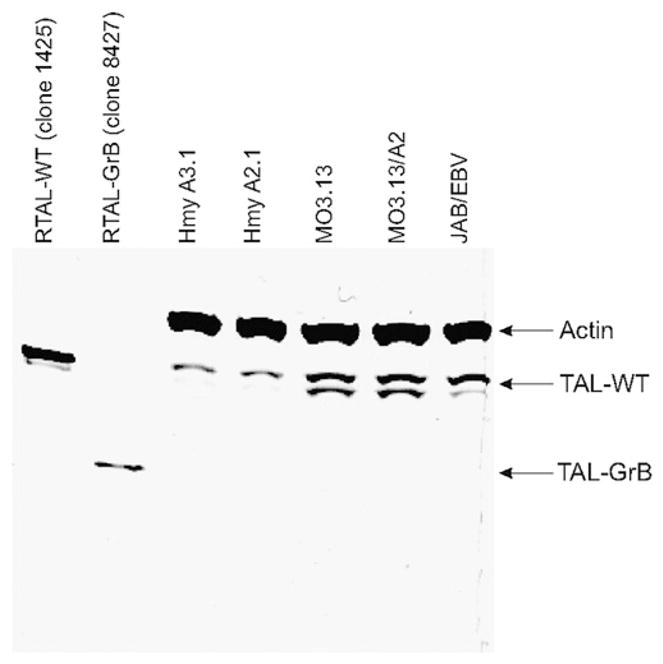
Western blot analysis of TAL-WT (recombinant clone 1425) and the 310-aa C-terminal GrB cleavage product (TAL-GrB, recombinant clone 8427) and TAL protein expression in Hmy A3.1, Hmy A2.1, MO3.13, MO3.13/A2, and EBV-transformed B cells of MS patient JAB (JAB/EBV). A total of 100 ng of TAL-WT or TAL-GrB recombinant protein and 40 ug of whole-cell lysates was analyzed per lane and developed by simultaneous incubation with Abs to TAL and human β-actin, as earlier described (15).
The antigenicity recombinant proteins corresponding to the mutant TAL-GrB and TAL-WT were compared using PBMCs from HLA-A2+ MS patients (JAB, PET, RYD, SOL), HLA-A2+ MS patients (PCA, DET, GIR), and HLA-A2+ HCs (RUS, NIL). TAL-GrB and TAL-WT elicited increased proliferative responses by HLA-A2+ MS PBMCs, SIs of 3.2 ± 0.14 and 3.4 ± 0.18, respectively, exceeding the SI of control Ag BSA (SI = 1.2; p < 0.0001; Fig. 4). Both TAL-WT (p = 0.021) and TAL-GrB elicited a higher SI with HLA-A2+ MS than did HLA-A2+ control PBMCs (p = 0.010).
FIGURE 4.

Proliferative responses to TAL-WT (clone 1425) and TAL-GrB (clone 8427) by PBMCs from HLA-A2+ MS patient JAB and HLA-A2+ control donor BN. A total of 105 cells per well were stimulated for 72 h with 5 μg/ml TAL-WT, TAL-GrB, negative control Ag BSA (left y-axis) or polyclonal stimulator Con A (right y-axis). Data show mean ± SD of SIs of six parallel cultures. *p < 0.0001 reflects comparison of TAL-WT–stimulated or TAL-GrB–stimulated cells with BSA-stimulated cells of MS patient JAB. **p = 0.021; ***p = 0.010.
Next, we investigated the ability of TAL-GrB and TAL-WT to activate HLA-A2–restricted CTLs in MS patients. We used HLA-A2–transfected MO3.13 oligodendroglioma and Hmy lymphoblastoma cells as targets. TAL-GrB and TAL-WT elicited comparable CTL activities by PBMCs of HLA-A2+ MS patients, but not in HLA-A2+ MS patients or HLA-A2+ HCs. Similar to earlier findings (49), MO3.13 oligodendroglioma, but not Hmy cells, were killed by CTLs with prior pulsing with TALpep (Fig. 5). This suggested that MO3.13 oligodendroglioma cells may more effectively present TAL owing to higher levels of endogenous expression. Indeed, expression of TAL relative to β-actin was 4-fold higher in MO3.13 oligodendroglioma, compared with Hmy cells (Fig. 3). Moreover, TAL-GrB fully retained the capacity of TAL-WT to stimulate CTL activity of TCL from HLA-A2+ MS patients JAB (JTE) and KEF (KTE) (Fig. 6).
FIGURE 5.

Cytotoxic activity of TAL-WT–stimulated and TAL-GrB–stimulated PBMCs from HLA-A2+ MS patients (JAB, PET, RYD, SOL), HLA-A2− MS patients (PCA, DET, GIR), and HLA-A2+ HCs (RUS, NIL) against MO3.13/A2 (HLA-A2–transfected) and control MO3.13, as well as Hmy-A2.1 (HLA-A2–transfected) and control Hmy-A3.1 (HLA-A3–transfected) target cells. Target cells were pulse labeled with or without TALpep. A total of 106/ml PBMCs were stimulated with and without 5 μg/ml TAL-WT, TAL-GrB, or BSA for 7 d and added to target cells at 10:1 effector/target ratio. Values significantly exceeding control levels are indicated by brackets. *p < 0.005; **p < 0.0005; ***p < 0.0001.
FIGURE 6.

Cytotoxicity by TAL-specific TCLs from MS patients was assessed against MO3.13/A2 (HLA-A2–transfected) and control MO3.13, as well as Hmy-A2.1 (HLA-A2–transfected) and control Hmy-A3.1 (HLA-A3–transfected) target cells. JTE and KTE TCLs were generated from HLA-A*0201–positive MS patients and maintained by weekly stimulation with autologous EBV-transformed B cells pulsed with 5 μg/ml rTAL (49). Values significantly exceeding control levels are indicated by brackets. *p < 0.005; **p < 0.0005; ***p < 0.0001.
Autoantibodies in sera of MS patients bind TAL-WT and TAL-GrB with similar affinity
Patients with MS have TAL Abs in their blood and cerebrospinal fluid (14, 22). By contrast, TAL Abs were absent in normal individuals and patients with other autoimmune and neurological diseases (14) and, under identical conditions, no MBP Abs were found in serum and cerebrospinal fluid of MS patients (22). As shown in Fig. 7, TAL-WT and TAL-GrB were recognized by autoantibodies in the sera of 12 patients with similar affinity. Rabbit anti-TAL-H Abs 169 (15) and 12484 (29) at 1:1000 dilution and MS sera at 1:100 dilution had similar binding affinity to rTAL-WT and TAL-GrB (data not shown). Binding by MS sera to TAL-WT or TAL-GrB markedly exceeded binding by sera of HCs (Fig. 7).
FIGURE 7.
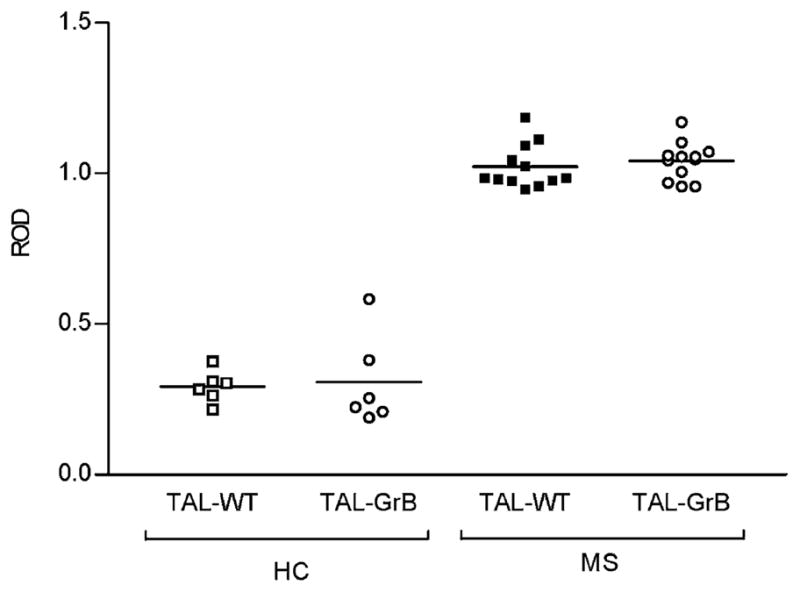
Testing of binding affinity to TAL-WT and TAL-GrB by sera of 12 MS patients and 6 HCs, using ELISA. Binding affinity of MS sera exceeded the binding affinity of control sera to both TAL-WT (p = 2.7 × 1013) and TAL-GrB (p = 9.4 × 1011), using a two-tailed t test.
Discussion
Autoimmunity has been attributed to molecular mimicry between viral Ags and self-proteins, resulting in cross-reactivity of MHC-restricted T cells and Abs (52, 53). In MS, cross-reactivity between microbial and myelin Ags has been implicated in immune-mediated destruction of oligodendrocytes and subsequent demyelination (54, 55). In addition to molecular mimicry, cleavage by GrB is an indication of autoantigen status (36). The majority of autoantigens across several human autoimmune diseases are efficiently cleaved by GrB in vitro, producing unique fragments (36). Cleavage by GrB can expose cryptic epitopes of peptides from a wide spectrum of apoptotic cells (47), including neurons, which in turn can trigger self-reactive T cells (56).
GrB is produced by CTLs, and it enters target cells through perforin-generated pores where it triggers apoptosis through cleavage and activation of caspase-3 (35). In MS brain lesions, infiltrating CD8+ CTLs outnumber CD4+ T cells (1), and oligodendrocyte cell death is mostly attributed to the cytotoxic effect of CD8+ T cells (30). Adoptively transferred MBP-specific (31) or MOG-specific CD8+ T cells induce severe CNS demyelination in animal models (32). Although MBP-specific CD8 T cells were detected in human PBL, there was no difference in cytotoxicity of MBP 110–118 peptide-stimulated CD8 T cells between healthy donors and MS patients (57, 58). Recently, increased precursor frequency of MBP 111–119–reactive CD8 T cells was found in a minority of MS patients (59). Cytosolic proteins, like highly soluble TAL, constitute a major source of peptides presented to CD8+ T cells by HLA class I molecules (60). Among 14 peptides with predicted HLA-A2 binding stabilities of > 100 min (t1/2) at 37°C, TALpep has the highest binding affinity for HLA-A2, and it is specifically recognized by CD8+ CTLs in HLA-A2+ MS patients. The precursor frequency, cytotoxic activity, and IFN-γ production of TALpep-specific CD8 T cells were increased in each of seven HLA-A2+ MS patients, compared with seven HLA-A2− MS patients, four HLA-A2+ patients with other neurological diseases, four HLA-A2− patients with other neurological diseases, and four HLA-A2+ and two HLA-A2− healthy donors (49). TAL is expressed at selective high levels in oligodendrocytes (14), and immunohistochemical studies of postmortem brain sections revealed decreased staining by MBP- and TAL-specific Abs in MS plaques, indicating a concurrent loss of these Ags from sites of demyelination (22). In this study, we demonstrate that TAL is cleaved by GrB, which may play a significant role in autoimmunization during the oligodendrocyte-specific cytotoxic process in MS. Apoptotic oligodendrocytes may release TAL-GrB fragments, exposing potentially autoantigenic cryptic epitopes that autoreactive CTLs could respond to, and in turn perpetuate the oligodendrocyte-cytotoxic process. Therefore, GrB-mediated cleavage, in addition to molecular mimicry with viral proteins and high level of expression in oligodendrocytes, may contribute to the antigenicity of TAL. In the context of TAL, not only the retention of autoantigenicity but also the loss of enzymatic activity, which was recently found to influence apoptosis susceptibility in cell type and pathway-specific manners (48, 61), may be relevant for the pathogenesis of MS. Cleavage by GrB, subsequent proteolysis, and inactivation of TAL could enhance the susceptibility of oligodendrocytes to apoptosis and therefore promote the release of antigenic peptides from TAL as well as from other potential autoantigens. Although it may be too speculative at this point, the degradation of TAL could result in its trimming into the 168–176 dominant epitope (49) and its loading into HLA-A2/MHC class I complexes (62). This process may stimulate the presentation of TALpep to CD8+ T cells directly by dying oligodendrocytes or indirectly by APCs, such as microglia, following the uptake of apoptotic oligodendrocytes in the brain. The latter mechanism (i.e., Ag uptake from dying tumor cells) was found to enhance Ag presentation and antitumor immunity (63).
The three-dimensional structure of human TAL has been determined by x-ray crystallography, and surface exposure of aa residues 1–115 and 265–290 was confirmed by binding of poly-clonal rabbit Abs raised against enzymatically active full-length rTAL and autoantibodies of MS patients to a set of 33 peptides overlapping TAL (64). Remarkably, the most exposed and antigenic peptide, residues 21–35, contain the major GrB cleavage site between aa 27 and 28 (64). The minor fragment was generated by cleavage after aa 108 comprised within an exposed recognition motif. By contrast, the LLQD motif corresponding to aa 263–267 is not exposed on the surface of TAL, which may explain a lack of cleavage by GrB at this site.
The major 34-kDa TAL-GrB peptide had no enzymatic activity; however, it elicited proliferative responses and CTL activities similar to those by full-length TAL-WT. The HLA-A2–binding CTL recognition motif 168–176, which is retained in TAL-GrB, is located inside the α-β barrel and therefore is not exposed on the surface of TAL (64). Recognition of TAL 168–176 by CTLs of MS patients is consistent with the notion that the autoimmune T cell repertoire is primarily directed against cryptic self-determinants (56). Both the TAL-WT and the TAL-GrB were recognized by HLA-A*0201–restricted CD8+ CTLs (Figs, 5, 6). We previously showed that all TAL-specific CD8+ TCLs expressed TCRVβ14 49. A recent study demonstrated that oligoclonally expanded CTL lines from MS patients preferentially use TCRVβ14, CDR3, and BJ segments matching those of TAL-specific TCLs (65). Importantly, CD8+ T cells expressing these TCRs derived exclusively from HLA-A2–positive individuals, further supporting the notion that these expanded T cell clones are actually targeting the TAL protein (65).
On the basis of binding to an array of 33 peptides, each 15 aa long, overlapping TAL by 5 aa, four immunodominant B cell epitopes have been identified in MS patients (64): 9/13 TAL-reactive sera recognized peptide 24 (residues 231–245), 10/13 sera recognized peptide 28 (residues 271–285), and peptides 11 (residues 101–115) and 32 (residues 311–325) were recognized by 5/13 sera. Ab 12484 raised against enzymatically active full-length TAL showed high-affinity binding to peptides 11 and 32 but failed to recognize peptides 24 and 28, suggesting that the two latter peptides may be cryptic or nonimmunogenic in the rabbit. Because TAL is released from sites of demyelination in the brain of MS patients (22), it is plausible that these cryptic epitopes are exposed by GrB-mediated cleavage. The prevalence of autoantibodies to TAL is increased in MS patients (14, 22), and TAL-reactive MS sera exhibited similar binding affinity to WT or GrB-cleaved Ag (data not shown). Along the same line, autoantibodies directed against GrB-cleaved SS-B/La Ag were found in sera from patients with primary Sjögren’s syndrome (66).
In summary, the current study shows that TAL is specifically cleaved by GrB between aa 27 and 28. The resulting 310-aa C-terminal fragment has no enzymatic activity but fully retains the antigenicity of TAL, stimulates proliferation and cytotoxic activity of T cells, and is recognized by autoantibodies from MS patients. Thus, the cleavage by GrB may play a key role in the autoantigenicity of TAL and contribute to the destruction of oligodendrocytes in MS.
Acknowledgments
This work was supported in part by Grant RO1 DK 49221 from the National Institutes of Health and Grant RG 2466 from the National Multiple Sclerosis Society.
Abbreviations used in this paper
- EAE
experimental allergic encephalomyelitis
- GrA
granzyme A
- GrB
granzyme B
- 3H-TdR
thymidine [methyl-3H]
- HC
healthy control
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- PPP
pentose phosphate pathway
- SI
stimulation index
- TAL
transaldolase
- TAL-GrB
human recombinant GrB-cleaved TAL
- TAL-H
human transaldolase
- TALpep
TAL peptide 168–176
- TAL-WT
human recombinant wild-type TAL
- TCL
T cell line
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- 2.Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- 3.Traugott U, Scheinberg LC, Raine CS. On the presence of Ia-positive endothelial cells and astrocytes in multiple sclerosis lesions and its relevance to antigen presentation. J Neuroimmunol. 1985;8:1–14. doi: 10.1016/s0165-5728(85)80043-6. [DOI] [PubMed] [Google Scholar]
- 4.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. [Review] Annu Rev Immunol. 1992;10:153–187. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 5.Roder J, Hickey WF. Mouse models, immunology, multiple sclerosis and myelination. [news] Nat Genet. 1996;12:6–8. doi: 10.1038/ng0196-6. [DOI] [PubMed] [Google Scholar]
- 6.McCarron RM, Fallis RJ, McFarlin DE. Alterations in T cell antigen specificity and class II restriction during the course of chronic relapsing experimental allergic encephalomyelitis. J Neuroimmunol. 1990;29:73–79. doi: 10.1016/0165-5728(90)90149-h. [DOI] [PubMed] [Google Scholar]
- 7.Whitham RH, Jones RE, Hashim GA, Hoy CM, Wang RY, Vandenbark AA, Offner H. Location of a new encephalitogenic epitope (residues 43 to 64) in proteolipid protein that induces relapsing experimental autoimmune encephalomyelitis in PL/J and (SJL x PL)F1 mice. J Immunol. 1991;147:3803–3808. [PubMed] [Google Scholar]
- 8.Genain CP, Cannella B, Hauser SL, Raine CS. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med. 1999;5:170–175. doi: 10.1038/5532. [DOI] [PubMed] [Google Scholar]
- 9.Zajicek JP, Wing M, Scolding NJ, Compston DA. Interactions between oligodendrocytes and microglia. A major role for complement and tumour necrosis factor in oligodendrocyte adherence and killing. Brain. 1992;115:1611–1631. [PubMed] [Google Scholar]
- 10.Fuller KG, Olson JK, Howard LM, Croxford JL, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Med. 2004;102:339–361. doi: 10.1385/1-59259-805-6:339. [DOI] [PubMed] [Google Scholar]
- 11.Lees MB, Brostoff SW. Proteins of myelin. In: Morrell P, editor. Myelin. Plenum Publishing Corporation; New York: 1984. pp. 197–224. [Google Scholar]
- 12.Gaur A, Fathman CG. Immunotherapeutic strategies directed at the trimolecular complex. [Review] Adv Immunol. 1994;56:219–265. doi: 10.1016/s0065-2776(08)60453-8. [DOI] [PubMed] [Google Scholar]
- 13.Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, Hafler DA. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259:1321–1324. doi: 10.1126/science.7680493. [DOI] [PubMed] [Google Scholar]
- 14.Banki K, Colombo E, Sia F, Halladay D, Mattson DH, Tatum AH, Massa PT, Phillips PE, Perl A. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J Exp Med. 1994;180:1649–1663. doi: 10.1084/jem.180.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banki K, Halladay D, Perl A. Cloning and expression of the human gene for transaldolase. A novel highly repetitive element constitutes an integral part of the coding sequence. J Biol Chem. 1994;269:2847–2851. [PubMed] [Google Scholar]
- 16.Mayes PA. The pentose phosphate pathway and other pathways of hexose metabolism. In: Murray RK, Granner DK, Mayes PA, Rodwell VW, editors. Harper’s Biochemistry. Appleton & Lange; Norwalk, CT: 1993. pp. 201–211. [Google Scholar]
- 17.Jacobson S. Sequence of myelination in the brain of the albino rat. A. Cerebral cortex, thalamus and related structures. J Comp Neurol. 1963;121:5–29. doi: 10.1002/cne.901210103. [DOI] [PubMed] [Google Scholar]
- 18.McDougal DB, Jr, Schulz DW, Passonneau JV, Clark JR, Reynolds MA, Lowry OH. Quantitative Studies of White Matter : I. Enzymes involved in glucose-6-phosphate metabolism. J Gen Physiol. 1961;44:487–498. doi: 10.1085/jgp.44.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baquer NZ, Hothersall JS, McLean P, Greenbaum AL. Aspects of carbohydrate metabolism in developing brain. Dev Med Child Neurol. 1977;19:81–104. doi: 10.1111/j.1469-8749.1977.tb08027.x. [DOI] [PubMed] [Google Scholar]
- 20.Balazs R. Carbohydrate metabolism. In: Lajtha A, editor. Handbook of Neurochemistry. Plenum Press; New York: 1970. pp. 1–36. [Google Scholar]
- 21.Hotta SS. Glucose metabolism in brain tissue: the hexosemonophosphate shunt and its role in glutathione reduction. J Neurochem. 1962;9:43–51. doi: 10.1111/j.1471-4159.1962.tb07491.x. [DOI] [PubMed] [Google Scholar]
- 22.Colombo E, Banki K, Tatum AH, Daucher J, Ferrante P, Murray RS, Phillips PE, Perl A. Comparative analysis of antibody and cell-mediated autoimmunity to transaldolase and myelin basic protein in patients with multiple sclerosis. J Clin Invest. 1997;99:1238–1250. doi: 10.1172/JCI119281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banki K, Hutter E, Colombo E, Gonchoroff NJ, Perl A. Glutathione levels and sensitivity to apoptosis are regulated by changes in trans-aldolase expression. J Biol Chem. 1996;271:32994–33001. doi: 10.1074/jbc.271.51.32994. [DOI] [PubMed] [Google Scholar]
- 24.Banki K, Hutter E, Gonchoroff NJ, Perl A. Molecular ordering in HIV-induced apoptosis. Oxidative stress, activation of caspases, and cell survival are regulated by transaldolase. J Biol Chem. 1998;273:11944–11953. doi: 10.1074/jbc.273.19.11944. [DOI] [PubMed] [Google Scholar]
- 25.Banki K, Hutter E, Gonchoroff NJ, Perl A. Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signaling. J Immunol. 1999;162:1466–1479. [PMC free article] [PubMed] [Google Scholar]
- 26.Selmaj K, Raine CS, Farooq M, Norton WT, Brosnan CF. Cytokine cytotoxicity against oligodendrocytes. Apoptosis induced by lymphotoxin. J Immunol. 1991;147:1522–1529. [PubMed] [Google Scholar]
- 27.D’Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP. Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med. 1996;184:2361–2370. doi: 10.1084/jem.184.6.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowling P, Shang G, Raval S, Menonna J, Cook S, Husar W. Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J Exp Med. 1996;184:1513–1518. doi: 10.1084/jem.184.4.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banki K, Perl A. Inhibition of the catalytic activity of human transaldolase by antibodies and site-directed mutagenesis. FEBS Lett. 1996;378:161–165. doi: 10.1016/0014-5793(95)01446-2. [DOI] [PubMed] [Google Scholar]
- 30.Lassmann H. Neuropathology in multiple sclerosis: new concepts. [Review] [74 refs] Mult Scler. 1998;4:93–98. doi: 10.1177/135245859800400301. [DOI] [PubMed] [Google Scholar]
- 31.Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. [see comment] J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- 33.Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, Wekerle H, Hohlfeld R, Goebels N. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA. 2004;101:2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schröder R, Deckert M, Schmidt S, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8:451–460. doi: 10.1016/s1074-7613(00)80550-6. [DOI] [PubMed] [Google Scholar]
- 36.Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–826. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, Johnson KP, Sibley WA, Silberberg DH, Tourtellotte WW. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol. 1983;13:227–231. doi: 10.1002/ana.410130302. [DOI] [PubMed] [Google Scholar]
- 38.Oudshoorn M, I, Doxiadis I, van den Berg-Loonen PM, Voorter CE, Verduyn W, Claas FH. Functional versus structural matching: can the CTLp test be replaced by HLA allele typing? Hum Immunol. 2002;63:176–184. doi: 10.1016/s0198-8859(01)00384-6. [DOI] [PubMed] [Google Scholar]
- 39.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 40.Deibler GE, Martenson RE, Kies MW. Large scale preparation of myelin basic protein of several mammalian species. Prep Biochem. 1972;2:139–165. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 41.Perl A, Gonzalez-Cabello R, Láng I, Gergely P. Effector activity of OKT4+ and OKT8+ T-cell subsets in lectin-dependent cell-mediated cytotoxicity against adherent HEp-2 cells. Cell Immunol. 1984;84:185–193. doi: 10.1016/0008-8749(84)90089-3. [DOI] [PubMed] [Google Scholar]
- 42.Chan MA, Stein LD, Dosch HM, Sigal NH. Heterogeneity of EBV-transformable human B lymphocyte populations. J Immunol. 1986;136:106–112. [PubMed] [Google Scholar]
- 43.Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci USA. 1994;91:10859–10863. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLaurin J, Trudel GC, Shaw IT, Antel JP, Cashman NR. A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J Neurobiol. 1995;26:283–293. doi: 10.1002/neu.480260212. [DOI] [PubMed] [Google Scholar]
- 45.Towbin HH, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Casciola-Rosen LA, Miller DK, Anhalt GJ, Rosen A. Specific cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J Biol Chem. 1994;269:30757–30760. [PubMed] [Google Scholar]
- 47.Roberts KM, Rosen A, Casciola-Rosen LA. Methods for inducing apoptosis. In: Perl A, editor. Autoimmunity:Methods and Protocols. Humana; Totowa, N.J: 2004. pp. 115–128. [DOI] [PubMed] [Google Scholar]
- 48.Qian Y, Banerjee S, Grossman CE, Amidon W, Nagy G, Barcza M, Niland B, Karp DR, Middleton FA, Banki K, Perl A. Trans-aldolase deficiency influences the pentose phosphate pathway, mitochondrial homoeostasis and apoptosis signal processing. Biochem J. 2008;415:123–134. doi: 10.1042/BJ20080722. [DOI] [PubMed] [Google Scholar]
- 49.Niland B, Banki K, Biddison WE, Perl A. CD8+ T cell-mediated HLA-A*0201-restricted cytotoxicity to transaldolase peptide 168–176 in patients with multiple sclerosis. J Immunol. 2005;175:8365–8378. doi: 10.4049/jimmunol.175.12.8365. [DOI] [PubMed] [Google Scholar]
- 50.Pouly S, Antel JP. Multiple sclerosis and central nervous system demyelination. [Review] [132 refs] J Autoimmun. 1999;13:297–306. doi: 10.1006/jaut.1999.0321. [DOI] [PubMed] [Google Scholar]
- 51.Hostetter DR, Loeb CRK, Chu F, Craik CS. Hip is a pro-survival substrate of granzyme B. J Biol Chem. 2007;282:27865–27874. doi: 10.1074/jbc.M704312200. [DOI] [PubMed] [Google Scholar]
- 52.Oldstone MBA. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- 53.Perl A. Pathogenesis and spectrum of autoimmunity. In: Perl A, editor. Autoimmunity Methods and Protocols. Humana Press; Totowa, N.J: 2004. pp. 1–8. [Google Scholar]
- 54.Boccaccio GL, Steinman L. Multiple sclerosis: from a myelin point of view. [Review] [87 refs] J Neurosci Res. 1996;45:647–654. doi: 10.1002/(SICI)1097-4547(19960915)45:6<647::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 56.Moudgil KD, Sercarz EE. The T cell repertoire against cryptic self determinants and its involvement in autoimmunity and cancer. Clin Immunol Immunopathol. 1994;73:283–289. doi: 10.1006/clin.1994.1200. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe M, Muramatsu M, Hirai H, Suzuki T, Fujisawa J, Yoshida M, Arai K, Arai N. HTLV-I encoded Tax in association with NF-kappa B precursor p105 enhances nuclear localization of NF-kappa B p50 and p65 in transfected cells. Oncogene. 1993;8:2949–2958. [PubMed] [Google Scholar]
- 58.Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J Immunol. 1998;160:3056–3059. [PubMed] [Google Scholar]
- 59.Zang YCQ, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, Killian J, Zhang JZ. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. 2004;172:5120–5127. doi: 10.4049/jimmunol.172.8.5120. [DOI] [PubMed] [Google Scholar]
- 60.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–299. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 61.Hanczko R, Fernandez D, Doherty E, Qian Y, Vas G, Niland B, Telarico T, Garba A, Banerjee S, Middleton FA, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009;119:1546–1557. doi: 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yewdell JW, Bennink JR. Cut and trim: generating MHC class I peptide ligands. [Review] [33 refs] Curr Opin Immunol. 2001;13:13–18. doi: 10.1016/s0952-7915(00)00175-8. [DOI] [PubMed] [Google Scholar]
- 63.Calderwood SK, Theriault JR, Gong J. Message in a bottle: role of the 70-kDa heat shock protein family in anti-tumor immunity. [Review] [105 refs] Eur J Immunol. 2005;35:2518–2527. doi: 10.1002/eji.200535002. [DOI] [PubMed] [Google Scholar]
- 64.Esposito M, Venkatesh V, Otvos L, Weng Z, Vajda S, Banki K, Perl A. Human transaldolase and cross-reactive viral epitopes identified by autoantibodies of multiple sclerosis patients. J Immunol. 1999;163:4027–4032. [PubMed] [Google Scholar]
- 65.Somma P, Ristori G, Battistini L, Cannoni S, Borsellino G, Diamantini A, Salvetti M, Sorrentino R, Fiorillo MT. Characterization of CD8+ T cell repertoire in identical twins discordant and concordant for multiple sclerosis. J Leukoc Biol. 2007;81:696–710. doi: 10.1189/jlb.0906584. [DOI] [PubMed] [Google Scholar]
- 66.Huang M, Ida H, Kamachi M, Iwanaga N, Izumi Y, Tanaka F, Aratake K, Arima K, Tamai M, Hida A, et al. Detection of apoptosis-specific autoantibodies directed against granzyme B-induced cleavage fragments of the SS-B (La) autoantigen in sera from patients with primary Sjögren’s syndrome. Clin Exp Immunol. 2005;142:148–154. doi: 10.1111/j.1365-2249.2005.02888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]