

Abstract
Although membrane proteins constitute more than 20% of the total proteins, the structures of only a few are known in detail. An important group of integral membrane proteins are ion-transporting ATPases of the P-type family, which share the formation of an acid-stable phosphorylated intermediate as part of their reaction cycle. There are several crystal structures of the sarcoplasmic reticulum Ca2+ pump (SERCA) revealing different conformations, and recently, crystal structures of the H+-ATPase and the Na+/K+-ATPase were reported as well. However, there are no atomic resolution structures for other P-type ATPases including the plasma membrane calcium pump (PMCA), which is integral to cellular Ca2+ signaling. Crystallization of these proteins is challenging because there is often no natural source from which the protein can be obtained in large quantities, and the presence of multiple isoforms in the same tissue further complicates efforts to obtain homogeneous samples suitable for crystallization. Alternative techniques to study structural aspects and conformational transitions in the PMCAs (and other P-type ATPases) have therefore been developed. Specifically, information about the structure and assembly of the transmembrane domain of an integral membrane protein can be obtained from an analysis of the lipid–protein interactions. Here, we review recent efforts using different hydrophobic photo-labeling methods to study the non-covalent interactions between the PMCA and surrounding phospholipids under different experimental conditions, and discuss how the use of these lipid probes can reveal valuable information on the membrane organization and conformational state transitions in the PMCA, Na+/K+-ATPase, and other P-type ATPases.
Keywords: Ca2+-ATPase, hydrophobic photo-labeling, lipid-protein interaction, membrane protein, Na+/K+-ATPase, PMCA, P-type ATPase
INTRODUCTION
Although membrane proteins constitute more than 20% of all proteins, the structures of only a few are known in atomic detail. An important group of integral membrane proteins are ion-transporting ATPases of the P-type family, which are characterized by the formation of an acid-stable phosphorylated intermediate as part of their reaction cycle [1]. These proteins are essential for the establishment and maintenance of ion gradients across cellular membranes, and are thus indispensable for cell physiology [2]. Atomic resolution structural information is now available for a few members of this family. There are several crystal structures of the Ca2+ pump of sarcoplasmic reticulum (SERCA) corresponding to different conformations assumed during the reaction cycle [3-7], and recently, crystal structures of the H+-ATPase [8] and of the Na+/K+-ATPase were reported as well [9,10]. However, there are as yet no high-resolution structures for the majority of the P-type ATPases, including the family of plasma membrane Ca2+ ATPases (PMCAs). This underlines the continued need to develop new tools to probe the structure-function relationship in these proteins by alternative and complementary methods.
For many years, a primary focus in our laboratories has been to obtain structural information about the plasma membrane calcium pump (PMCA). This pump is an integral part of the Ca2+ signaling mechanism [11]. It is highly regulated by calmodulin, which activates the pump by binding to an auto-inhibitory region, thereby inducing a conformational change of the pump from an inhibited to an activated state [12,13]. Obtaining crystals of native PMCA is particularly challenging because there is no natural source from which this protein can be easily obtained in large quantities. In addition, the presence of several isoforms in the same tissue further complicates any effort to obtain a homogeneous sample suitable for crystallization. Although expression of recombinant PMCA in baculovirus-infected insect cells or yeast has been accomplished [13,14], attempts to isolate the pump in a homogeneous and functional state suitable for crystallization have met with limited success. These drawbacks led us to design alternative techniques for the study of structural aspects of the PMCA and potentially, of other P-type ATPases. Of particular interest for a better understanding of the pump mechanism are methods that yield information on the membrane-embedded part of the pump, which is the most difficult region to study by traditional methods of protein expression and analysis. Information about the structure and assembly of the transmembrane domain of an integral membrane protein can be obtained, however, from the analysis of the lipid–protein interactions. In this work, we describe an approach using different hydrophobic photo-labeling methods to study the non-covalent interactions between the PMCA and surrounding phospholipids under different experimental conditions that lead to known conformations.
HYDROPHOBIC PHOTO-LABELING METHOD TO STUDY LIPID-PROTEIN INTERACTIONS
Chemical labeling by photo-crosslinking has been used to identify proteins or portions of proteins exposed either to the membrane surface or to the hydrophobic membrane interior. The protein is covalently modified by a reagent that is confined to one side of the membrane or localized within the lipid bilayer. To locate portions of the protein in contact with lipids, highly non-polar reagents that partition into the hydrophobic core of the membrane must be used. The exclusive location within the membrane lipid bilayer is mandatory because a major source of artifacts in using these techniques arises when the label is not confined to the reactive space for which it has been designed. The reagents can be added to the membrane preparation and allowed to partition into the bilayer before being photo-activated (for a review, see [15,16]).
Valuable experimental information on the topology of membrane proteins can be obtained with the help of photoactivatable reagents designed to react within the hydrophobic milieu of the lipid bilayer. We will describe here in detail some specific examples from our own work on how these reagents can be used to obtain information about structural changes in the membrane region of P-type ATPases. A generalized scheme for the experimental procedure is given in Fig. (1). After pre-incubation of the photoactivatable reagent (R-F in Fig. 1) with the protein of interest (such as a naturally abundant or enriched and reconstituted membrane protein, M in Fig. 1), light of the appropriate wavelength is used to induce covalent cross-linking of the probe to the protein residue(s) in its vicinity. After washing and concentration (e.g., by TCA precipitation), the photo-labeled sample is separated by electrophoresis and the specific protein bands of interest are excised and evaluated for the specific incorporation of the probe. This can be conveniently done if the photoactivatable probe carries a radioactive label by determining the specific radioactivity of the excised protein (see Fig. 1). Further information on the location and stoichiometry of the specific incorporation of the photo-label can be obtained, e.g., after limited proteolysis of the protein and peptide sequencing/mass spectrometry.
Fig. (1). General scheme for membrane protein photo-labeling and analysis.

After pre-incubation of the photoactivatable reagent R-F (where F is the photoactivatable functional group) with the protein of interest (e.g., a naturally abundant or purified and reconstituted membrane protein M), light of the appropriate wavelength (hν) is used to induce covalent cross-linking of the probe to the protein residue(s) in its vicinity. After washing and concentration by TCA precipitation, the photo-labeled sample is separated by electrophoresis and the specific protein bands of interest are excised and evaluated for the specific incorporation of the probe. This can be conveniently done if the photoactivatable probe carries a radioactive label.
Several non-polar photoreactive reagents are used to specifically label the protein residues in contact with the bilayer interior. Most of the reagents made to date are based on nitrene or carbene chemistry. The particular interest in nitrenes and carbenes originates from their ability to attack even aliphatic C-H bonds. The photolabile precursors are azides, diazirines, and diazocompounds. Although nitrenes or azides are easier to introduce than carbenes in a photoactivatable probe, nitrenes do show selective reactivity with nucleophiles [17]. Moreover, carbenes are preferred because they generally lead to higher reaction yields. Carbenes and nitrenes are isoelectronic species and therefore display fundamentally the same chemistry. Although generally regarded as unselective, even for photoreagents based on carbene chemistry, a somewhat higher reactivity towards nucleophiles has been reported [15]. Therefore, an inspection of the number and distribution of amino acids bearing such functionalities, i.e., Trp, Cys, Lys, Met, Tyr, must be carried out on the protein of interest to allow valid comparisons of labeling extents. It turns out that these amino acid residues are similarly abundant and roughly evenly distributed along the predicted/identified transmembrane regions of the P-type AT-Pases. Thus, no significant bias in the extent of labeling would be expected due to a difference in amino acid composition among these proteins.
Most successful reagents of the general type mentioned above include the photo-labeling group TPD (trifluoromethyl phenyldiazirine), which generates a very reactive carbene species upon irradiation with UV light [15]. [3H]DIPETPD (Fig. 2A) was synthesized by Delfino and colleagues [18] as a photochemical bipolar phospholipidic probe designed to label in the middle plane of the lipid bilayer and employed in an attempt to probe the depth of membrane penetration of the fusion peptide of influenza virus hemagglutinin. The obvious difficulty with the use of such flexible bipolar molecules is that they can be integrated into membranes in different configurations. Indeed, when reconstituted into large unilamellar vesicles, only about half of the [3H]DIPETPD molecules were found to be in the desired transbilayer configuration. In addition, this probe is very difficult to incorporate into the membrane or lipid bilayer, and 3H is less convenient to detect than 125I.
Fig. (2). Photoactivatable hydrophobic labeling compounds.

A: [3H]bis-phosphatidyl ethanolamine (trifluoromethyl) phenyldiazirine ([3H]DIPETPD), B: 3-(trifluoromethyl)-3-(rn-[125I]iodophenyl)-diazirine ([125I]TID), C: 1-O-hexadecanoyl-2-O-[9-[[[2-[125I]iodo-4-(trifluoromethyl-3H-diazirin-3-yl)benzyl]oxy]carbonyl] nonanoyl]-sn-glycero-3-phosphocholine ([125I]TID-PC/16),. D: 8-(3′-[125I]iodo, 5′-azido-O-hexanoylsalicylamido)octanoic acid (AS86).
While the small hydrophobic [125I]TID (Fig. 2B) rapidly partitions into and randomly distributes within the entire membrane lipid phase, analogs of fatty acids and phospholipids integrate into membranes in an oriented manner. Moreover, [125I]TID can partition not only within the membrane bilayer but also into hydrophobic pockets of proteins.
[125I]TID-PC/16 (Fig. 2C) is an analogue of phosphatidylcholine endowed with a photoactivatable group at the end of one of the fatty acyl chains. Its physicochemical behavior is indistinguishable from that of phosphatidylcholine, i.e., it shows identical mobility on TLC (thin layer chromatography) plates using different solvent systems [19]. The fact that the probe is a PC analogue simplifies the quantitative analysis. Indeed, PC is generally chosen as the reference lipid for comparison of relative association constants of integral membrane proteins because the latter show no selectivity for this amphiphile.
With the purpose of examining the structural and kinetic details of the interaction between the PMCA enzyme and unsaturated fatty acids, we have synthesized the photoactivatable probe 8-(5′-azido-O-hexanoylsalicylamido)octanoic acid (AS86) (Fig. 2D). AS86 possesses a negative charge and a shape intended to mimic the bend of the double bond, which could represent essential features of the fatty acids interacting with the Ca2+ pump. This is a photoreactive probe that can be tagged with 125I, with a conformation resembling that of oleic acid. The azido function is positioned outwardly at the convex face of the molecule, thus improving the chance of productive reaction of the photo-generated nitrene with neighboring molecules [20].
STOICHIOMETRY OF LIPID–PROTEIN INTERACTION ASSESSED BY HYDROPHOBIC PHOTO-LABELING
The reagent [125I]TID-PC/16 (Fig. 2C) has been used to assess the number of lipid-associated sites in P-type AT-Pases. This reagent partitions in the phospholipid milieu and, upon photolysis, will react indiscriminately with its surrounding molecular cage [21]. The interaction between a membrane protein and the lipids in its immediate environment can therefore be examined directly with this probe. As proof of the general usefulness of this approach, the stoichiometry of the lipid annulus surrounding the three different P-type ion pumps PMCA, sarcoplasmic reticulum Ca2+-ATPase (SERCA), and Na+/K+-ATPase has been determined, yielding values of 17 ± 1, 18 ±3, 24 ±5, and 5.6 ±0.4 PC lipids per molecule of the PMCA, SERCA, Na+/K+-ATPase α-subunit, and Na+/K+-ATPase α-subunit, respectively [19]. These numbers agree reasonably well with the values measured by electron paramagnetic resonance (EPR) spectroscopy for SERCA (22 ± 2 and 24 ± 5 [22,23]) and Na+/K+-ATPase (30 and 34 [24,25]), and with the value predicted for the PMCA, considering that this protein resembles SERCA with ten transmembrane segments. For the Na+/K+-ATPase, the above approach allowed us to obtain values for the lipid–protein stoichiometry of the alpha and beta subunits independently. The data are consistent with a model in which the beta-subunit shows enhanced lipid exposure due to its peripheral arrangement in the non-covalent heterodimeric alpha/beta complex of the sodium pump [9,10]. A preferential interaction with lipids is also predicted for the beta segment based on its low amphipathicity and high hydrophobicity, suggesting a higher affinity for lipids.
Studying lipid–protein stoichiometry involves an analysis of the first shell of lipids in immediate contact with the membrane-embedded surface of the protein. Any suitable experimental approach must therefore distinguish lipids interacting with the protein from lipids belonging to the bulk phase. EPR is one of the most valuable tools in such investigations [26]. However, because EPR mainly analyzes the lipid behavior, its usefulness to study membrane proteins is limited when the membrane protein cannot be obtained in large amounts. The reason is that a sufficiently large population of lipids with restricted mobility should be attained to be able to get a measurable signal. On the other hand, no other (contaminant) membrane proteins should be present in the sample. The analytical development described above, which focuses on an analysis of the photo-labeled membrane protein, can overcome these drawbacks. The sensitivity of this method is determined by the specific radioactivity of the lipid probe and by the method used for protein quantification. Therefore, it can be applied to samples where the protein concentration is far too low for EPR, such as in the case of the PMCA. The method also allows the determination of lipid–protein stoichiometry in oligomeric systems, where different subunits of the same membrane protein can be simultaneously analyzed within the limits imposed by the SDS–PAGE separation.
INTERACTION OF UNSATURATED FATTY ACIDS WITH THE RED BLOOD CELL Ca2+-ATPASE
Knowledge of the location of specific sites in a particular protein can be of great help in characterizing its structure-function relationship. Early studies had shown that unsaturated fatty acids such as oleic acid, and acidic phospholipids such as phosphatidylserine and phosphatidylinositol, increase both the affinity for Ca2+ and the maximum activity of the Ca2+-ATPase of red blood cells devoid of endogenous calmodulin [27-29]. It was also proposed that, similarly to calmodulin and partial proteolysis by trypsin, those activating agents seem to act directly on the enzyme and not via membrane modifications [30]. Conversely, other authors postulated that the effect of phospholipids and fatty acids could occur as a consequence of modification of the lipid bilayer and not because of a direct action on the enzyme [31].
To examine the structural and kinetic details of the interaction between the enzyme and unsaturated fatty acids, we synthesized the photoactivatable probe 8-(5′-azido-O-hexanoylsalicylamido)octanoic acid (AS86) (Fig. 2D). As already mentioned, AS86 possesses a negative charge and a shape intended to mimic the kink introduced by a double bond in an unsaturated membrane lipid such as oleic acid (see Fig. 3).
Fig. (3). Superposition of AS86 and oleic acid structures.
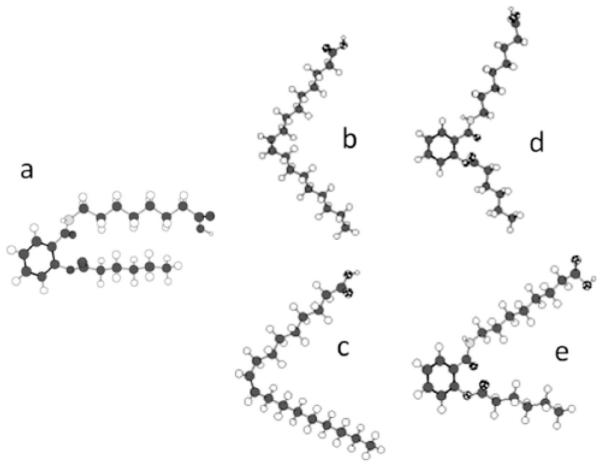
Global minimum conformers of AS86 (structure a) and oleic acid (structures b and c). Structures d and e result from least-squares flexible superimposition (allowing free rotation of torsional angles C-NH and OC-C) of conformer a over oleic acid conformers b and c, respectively. After the initial fit, each rotamer of conformer a was subjected to 1000 minimization cycles using the block diagonal Newton-Raphson algorithm to eliminate local tensions around the rotated dihedral angles. 17 atomic pairs along the chains were used for each comparison. The residual RMS deviations between b and d and between c and e were 0.805 and 0.878 Å, respectively. For details, see [20].
Experiments using AS86, which interacts non-covalently with the enzyme, revealed that it shares with oleic acid several properties with respect to its effects on the PMCA [20]: (i) it binds reversibly to the PMCA; (ii) in the absence of calmodulin, AS86 affects the Ca2+-ATPase activity in a biphasic behavior: at low concentrations it increases the affinity for Ca2+ and the maximum velocity of the enzyme, while at higher concentrations it decreases the maximum velocity of the Ca 2+-ATPase; (iii) in the presence o f calmodulin, AS86 moderately increases the affinity for Ca2+ but decreases the maximum velocity of the Ca2+-ATPase activity; and (iv) AS86 inhibits the activity of the Ca2+ pump lacking its calmodulin-binding domain following controlled proteolysis.
When AS86 was covalently bound to the native enzyme (following photo-activation), and the pump then activated by calmodulin, increasing amounts of AS86 decreased the maximum velocity of the Ca2+-ATPase activity without modifying the apparent affinity for Ca2+. These results could be explained by the existence of two different types of sites on the pump recognizing the reagent: one impacting the affinity for Ca2+ and the other inhibiting the effects of calmodulin. Importantly, when covalently bound, AS86 inhibits the enzyme lacking the calmodulin-binding domain, indicating that it acts through a site located outside the calmodulin-binding region.
125I-AS86 can be used to tag the purified PMCA. Competition with oleic acid for the interaction with the pump can then be easily demonstrated. Both the inhibitory effect on the calmodulin-dependent enzyme activity after covalent binding of AS86, and the photoadduct formation between the enzyme and 125I-AS86 are impaired by the presence of oleic acid in a concentration-dependent fashion (Fig. 4).
Fig. (4). Oleic acid competes with AS86 for binding to the PMCA.
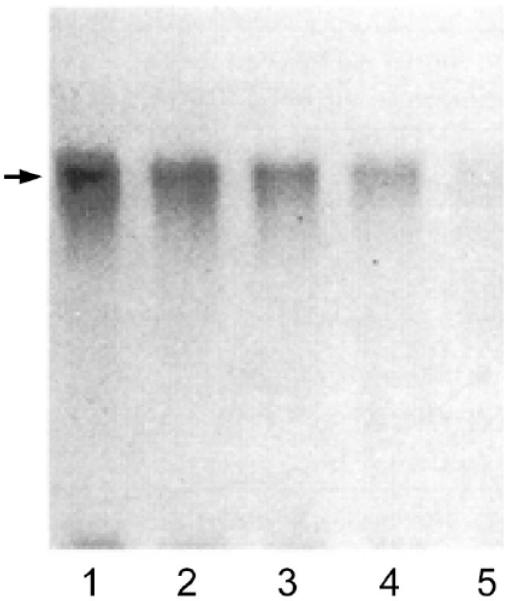
Autoradiogram of 0.46 μM purified Ca2+-ATPase (12 pmol) incubated with 1.6 μM 125I-AS86 (35 Ci/mmol, 40 pmol, 1.4 μCi) and increasing concentrations of oleic acid: 0 μM (lane 1), 25 μM (lane 2), 75 μM (lane 3), 225 μM (lane 4), and 600 μM (lane 5). Samples were irradiated for 5 minutes at 254 nm prior to SDS-PAGE. In all cases, the samples applied to gels contained 25 μl of the incubation mixture. The arrow indicates the position of the PMCA (138 kDa). The relative densities of the main labeled bands are 1.0, 0.53, 0.37, 0.25, and 0.09 in lanes 1-5, respectively. For details, see [20].
Although these data are not sufficient to pinpoint precisely where AS86 binds to the Ca2+ pump, it seems safe to infer the existence of a site(s) outside the (calmodulinbinding) C-terminal domain able to interact and establish a covalent link with the probe. The ability to tag the purified enzyme with 125I-AS86 could be useful to provide further insight on the location of the interaction sites.
HYDROPHOBIC PHOTO-LABELING OF THE Ca2+ PUMP REVEALS TOPOGRAPHIC FEATURES OF THE TRANSMEMBRANE DOMAIN AND DYNAMIC CHANGES UPON ACTIVATION BY CALMODULIN AND PHOSPHATIDIC ACID
[3H]DIPETPD (Fig. 2A) has been employed to explore the transmembrane organization of the Ca2+ pump [32], and demonstrated the existence of three hydrophobic clusters along the sequence of the pump. These clusters include the transmembrane regions of the pump, because labeling with [3H]DIPETPD is restricted mainly to the middle plane of the lipid bilayer [18,33].
Following identification of the major membrane-spanning hydrophobic clusters with [3H]DIPETPD, we took advantage of the higher specific activity provided by the radioactive iodine tag in [125I]TID (Fig. 2B), a generic hydrophobic probe, to undertake further labeling experiments. The efficient incorporation of radioactive label into peptides achievable with [125I]]TID significantly increased the resolution to determine the locale of labeling following enzymatic digestion of large labeled fragments of the pump. In this regard, it has been shown for other integral membrane proteins that the peptide labeling patterns do not differ much between phospholipid-based reagents bearing TPD [such as [3H]PTPC/1] [34] and [125I]TID, implying that the role of the chemistry of the reagent prevails over the position where the probe function is attached [34,35]. In agreement with this notion, [3H]DIPETPD and [125I]TID yielded similar profiles for the labeled peptides from the PMCA [32]. After controlled proteolysis of the Ca2+ pump with V8 protease, patterns of labeled peptides were compared in experiments in which the lipidic milieu of the pump was modified. The results were similar when the enzyme was labeled with [125I]TID, either in its natural environment (in situ) in red blood cell ghosts, or in lipid-detergent micelles. Hence, it can be concluded that the experimental procedures of solubilization, purification, and reconstitution of the Ca2+ pump do not introduce major changes in the transmembrane organization of this protein. In addition, the preservation of transport activity in reconstituted liposomes [32] and the maintenance of the kinetic properties and regulatory characteristics in the purified enzyme [36] support this view. These experiments validate hydrophobic photo-labeling as a method to address questions concerning the conformation of the membrane domain of the Ca2+ pump.
The first and second transmembrane segments M1 and M2 are labeled significantly and to a similar extent with reagents that probe the lip id-protein interface, such as [3H]DIPETPD and [125I]TID. On the other hand, the hydrophilic stretch connecting M1 and M2 includes a site susceptible to cleavage with V8 protease. This is consistent with the presence of an outer loop exposed to the aqueous solvent. This region is accessible to proteases under harsh conditions, and includes the epitope to antibody 1E4, as demonstrated by Feschenko et al. [37]. Thus, a consistent picture emerges for the topography of the N-terminal domain of the Ca2+-pump as determined by hydrophobic labeling and limited proteolysis (Fig. 5). Putting together these pieces of data, we proposed that a helical hairpin motif exists at the N-terminus of the pump. This hairpin may adopt a peripheral location in the transmembrane domain bundle, therefore exhibiting a substantial boundary region with surrounding membrane phospholipids. Interestingly, Stokes et al. [38] proposed a number of packing arrangements for the 10 helices belonging to the transmembrane domain of P-type ion pumps by model building on the basis of cryoelectron microscopy data at 14 Å resolution of the sarcoplasmic reticulum calcium pump [39]. All of these models position M1 and M2 side by side at the interface with membrane lipids.
Fig. (5). Scheme of proteolysis pattern of PMCA generated by V8 protease.

A: Cleavage in the cytosolic loops occurs at residues E322 and E661 as indicated by black arrows. The light and dark grey box represent the acidic phospholipid and the calmodulin binding site, respectively. Proteolysis by V8 protease results in three major fragments N (N-terminal), M (middle), and C (C-terminal). B: SDS gradient gel electrophoresis (4-20%) of PMCA after 30 (lane 1), 60 (lane 2) and 180 minutes (lane 3) of V8 proteolysis. The arrows show the fragments obtained. Molecular markers are shown in lane 4, with their masses indicated in kDa on the right.
More recently, we have used [125I]TID-PC/16 (Fig. 2C) incorporation to gain insight into the structural changes elicited by calmodulin and phosphatidic acid (PA) in the PMCA transmembrane region. The results showed that activation elicited by calmodulin involves structural rearrangements within the PMCA transmembrane domain markedly different from those taking place upon activation by acidic phospholipids. The effects of calmodulin and PA are additive as assessed by Ca2+-ATPase activity, suggesting that these ligands act through independent mechanisms (see Fig. 6). Neither calmodulin nor PA affected the apparent affinity constant of the other ligand, indicative of non-overlapping binding sites. Although calmodulin and PA could be expected to share a common structural effect because they both ultimately perturb a region connecting to transmembrane segment M3 in the PMCA, these ligands act through unrelated pathways. The overall exposure to surrounding lipids was found to be less in the presence of calmodulin than in the presence of PA and much less in the presence of either calmodulin or PA than in the presence of Ca2+ alone [40]. The level of compaction of the transmembrane region elicited by calmodulin appears to be maximal because, in the presence of this ligand, no further changes were observed in [125I]TID-PC/16 incorporation upon addition of PA. However, under the same experimental conditions, the effect of PA on the ATPase activity can be clearly demonstrated as an increased calculated value for activation (Fig. 6). This implies that the ATPase activation effect exerted by PA on calmodulin-bound PMCA does not have a corresponding compaction effect at the level of the transmembrane domain structure. Indeed, kinetic analysis of the activation by acidic phospholipids showed it to be the consequence of acceleration in the de-phosphorylation step (E2P to E2; see below for a brief explanation of the major conformational states of P-type ATPases). This is expected to occur mostly at the expense of structural changes in the cytoplasmic domain of the PMCA.
Fig. (6).

Ca2+-ATPase activation as a function of [calmodulin] in the presence or absence of 60 μM phosphatidic acid (A), and as a function of [phosphatidic acid] in the presence or absence of 0.2 μM calmodulin (B).
As mentioned above, by enzymatic digestion of photolabeled PMCA (Fig, 5), it is possible to dissect the domains within the transmembrane region that become specifically exposed to surrounding lipids. Calcium appears to elicit a general expansion of the transmembrane region in which every domain is about 50% more exposed to membrane lipids [41]. In the presence of calmodulin or PA, a striking decrease in [125I]TID-PC/16 incorporation into any of the hydrophobic domains is observed which, on average, is comparable to that observed for the undigested protein under each condition. However, the pattern of TID-PC incorporation between the different domains shows interesting variations. In the presence of calmodulin, the central domain included in fragment M (Fig. 5) becomes less exposed to phospholipids than the C-terminal domain included in fragment C, and much less than the N-terminal domain included in fragment N. Activation by calmodulin is due to its binding to an autoinhibitory domain located in the C-terminal tail of PMCA. Calmodulin binding dissociates this domain from a region located just upstream of transmembrane segment M3 of PMCA, thus relieving the auto-inhibition. It may therefore not be surprising that the most affected hydrophobic domains are those that include transmembrane segments directly connected with cytoplasmic targets of calmodulin binding, i.e., domain M which includes segment M3, and domain C which includes the transmembrane segment directly connected to the C-terminal autoinhibitory domain (Fig. 5).
The observed pattern is remarkably different in the presence of PA. Under this condition, it is the N-terminal domain (fragment N) that becomes less exposed. The central domain (fragment M) presents an intermediate exposure while the C-terminal domain shows the highest level of [125I]TID-PC/16 incorporation. The PA binding site is thought to be located within the cytoplasmic loop that connects transmembrane segments M2 and M3 (Fig. 5), partially overlapping with the C-terminal receptor region [42-44]. The most affected hydrophobic domains are again those including transmembrane segments directly connected with the postulated PA cytoplasmic binding region, i.e., domain N (which includes M2) and domain M (which includes M3). We emphasize, however, that the above analysis is extremely simplified. One should keep in mind that the “picture” of the transmembrane architecture of the PMCA is not only the consequence of cytoplasmic rearrangements that directly affect the connected transmembrane segments but also, and most probably mainly, the final outcome of long-range protein interactions. In this respect, and regarding cytoplasmic rearrangements, both calmodulin and PA were shown to increase the distance between the N- and C-terminal regions of the PMCA [13]. In contrast, at the transmembrane level, an overall compaction is the main effect elicited by both activators [40,41].
FOLLOWING CONFORMATIONAL CHANGES IN SERCA AND PMCA Ca2+ PUMPS WITH [125I]TID-PC/16
During the hydrolysis of ATP, P-type ATPases undergo phosphorylation and dephosphorylation reactions in which the terminal phosphoryl group of the nucleotide is first transferred to the beta-carboxyl of a conserved aspartic acid side chain in the enzyme, and subsequently to water [1]. Both the dephospho- and phospho-forms of the enzyme exist in at least two conformations, which are commonly designated E1 (or E1P) and E2 (or E2P). The changes in conformation alter the enzyme’s affinity for and reactivity to ATP, ADP, and Pi, as well as the affinities and accessibilities of sites that bind the transported cations [45]. In the E1 conformation, the pump is supposed to have high affinity for the exported sub-strate and low affinity for the imported one. This selectivity is reversfor the E2 conformation. The existence of E1 and E2 was first proposed on the basis of kinetic studies of phosphorylation [46], fluorescence [47], and analysis of tryptic digestion patterns [48], and more recently confirmed by crystallographic studies of several P-type pumps (see [49] for a recent review).
The photoactivatable probe [125I]TID-PC/16 (Fig. 2C) has been used in our laboratory to study the transmembrane region of the PMCA in different conformations [41]. By quantifying the amount of labeling by [125I]TID-PC/16, we were able to discern different PMCA conformers based on their differential interaction with surrounding phospholipids. The procedure was validated by performing [125I]TID-PC/16 incorporation studies on SERCA in different conditions (absence and presence of Ca2+, thapsigargin/EGTA) known to lead to specific conformations of the pump as determined by x-ray crystallography [6,7,50].
A major structural difference between SERCA and PMCA is the presence of an extended C-terminal autoinhibitory region in the PMCA that is not found in SERCA. PMCA activation by calmodulin involves binding of calmodulin to the C-terminal region, causing release of inhibitory interactions from the cytosolic core. This model for auto-inhibition (and its release) does not involve any changes in the transmembrane region. However, using the [125I]TID-PC/16 labeling method we could show that the auto-inhibited conformation is distinct (at least in the E1-Ca2+ state of the PMCA reaction cycle) and that the conformational changes induced by auto-inhibition expose additional surfaces to phospholipids [41]. Although the PMCA appears to have specific sites for interaction with acidic phospholipids [44], no such sites have been identified for neutral phospholipids such as PC. Thus, it is unlikely that higher affinity sites for the photoactivatable [125I]TID-PC/16 (which behaves as a PC analog) are exposed upon auto-inhibition. Rather, the observed change in [125I]TID-PC/16 incorporation reflects a major conformational difference in the membrane domain of the auto-inhibited and activated E1-Ca2+ states of the PMCA.
The usefulness of [125I]TID-PC/16 labeling to discriminate between distinct membrane domain conformations of membrane proteins was further demonstrated by studying the effect of the calmodulin-binding peptide C28 on SERCA. This peptide is known to inhibit SERCA [51]. Addition of C28 in conditions where the peptide inhibits SERCA (in the presence of Ca2+) induced an increase in [125I]TID-PC/16 labeling of this pump (see Table 1), suggesting that C28 induced an inhibitory conformation in SERCA similar to the one present in the PMCA in the absence of calmodulin [41]. The evidence obtained using [125I]TID-PC/16 to study the SERCA and PMCA under different activation/inhibition conditions allowed us to propose the existence of two different E1 conformations in these calcium pumps: E1I corresponds to the auto-inhibited PMCA and E1A to the activated one (in the presence of calmodulin or after removing the C-terminal tail). In SERCA, E1A is the conformation naturally assumed in the presence of Ca2+. Addition of C28 to SERCA mimics the E1I conformation of the auto-inhibited PMCA. It is quite remarkable that a short hydrophilic peptide like C28 drives such a profound change in the hydrophobic transmembrane region.
Table 1.
Relative Incorporation of [125I]TID-PC/16 into SERCA and PMCA Under Different Conditions
| Condition | SERCA | PMCA | ||
|---|---|---|---|---|
| Conformation | Specific Incorporation | Conformation | Specific Incorporation | |
| EGTA (no Ca2+) | E2 | 100 ± 3 | E2 | 100 ± 3 |
| EGTA + C28 | E2 | 98 ± 3 | E2 | 99 ± 3 |
| Ca2+ | E1A | 75 ± 4 | E1I | 155 ± 2 |
| Ca2+ + CaM | E1A | 70 ± 7 | E1A | 80 ± 2 |
| Ca2+ + C28 | E1I | 117 ± 3 | E1I | 150 ± 9 |
| Ca2+ + LaIII | E1 | ND | E1I | 156 ± 4 |
| Ca2+ + ATP + LaIII | E1P | 89 ± 4 | E1P | 155 ± 8 |
| Ca2+ + vanadate | E1, E2 | 92 ± 3 | E1, E2 | 125 ± 9 |
Results are the mean ± SEM of 3-7 independent experiments. ND; not determined. E1I and E1A are an inhibited and activated state of E1, respectively. For details, see [41].
The [125I]TID-PC/16 labeling method has also been used to analyze the effects of two inhibitors of PMCA, LaIII and vanadate, on the conformational state of the membrane domain [41]. LaIII is known to stabilize the E1Ca state, and in the presence of ATP it blocks the reaction cycle in E1P [52]. LaIII and ATP produced the same level of [125I]TID-PC/16 incorporation into the PMCA as Ca2+ alone (Table 1), suggesting that the transmembrane arrangement of E1P is similar to that of E1Ca (E1I). Earlier experiments on the kinetics of vanadate inhibition showed antagonism by Ca2+ [53,54], consistent with Ca2+ and vanadate binding to alternate conformations. Vanadate binds to the E2 conformation of SERCA, hence the apparent vanadate affinity also depends on the El to E2 equilibrium. The finding (Table 1) of an intermediate level of [125I]TID-PC/16 reagent incorporation in the presence of vanadate and Ca2+ is indeed indicative of the coexistence of E1 and E2 states in these conditions.
DETERMINATION OF THE DISSOCIATION CONSTANTS FOR Ca2+ AND CALMODULIN FROM THE PLASMA MEMBRANE CA2+ PUMP
The photoactivatable phosphatidylcholine analog [125I]TID-PC/16 has also been used to measure equilibrium constants for the dissociation of ligands from PMCA complexes and to draw structural conclusions about the regulation of the transport of Ca2+ in the presence of different modulators. This exploits the fact that the interaction of the PMCA with regulators such as Ca2+, calmodulin and acidic phospholipids is accompanied by conformational changes in the membrane region [40]. To perform these experiments, the labeling of PMCA with [125I]TID-PC/16 was quantified as a function of the added regulator, thus measuring the shift of conformation E2 (in the absence of Ca2+) to the auto-inhibited conformation E1I and of conformation E1I to the activated E1A state, and titrating the effect of Ca2+ under different conditions.
Fig. (7) shows an example where the transition of PMCA from the E2 to E1I conformation was followed by [125I]TIDPC/16 incorporation as a function of increasing [Ca2+]. The results from this and similar experiments in the presence of different modulators indicate that the PMCA possesses a high affinity site for Ca2+ regardless of the presence or absence of activators. The studies suggest that PMCA activity is modulated through the C-terminal domain, which induces an auto-inhibited conformation for Ca2+ transport but does not modify the affinity for Ca2+ at the transmembrane domain. Calmodulin and calmodulin-like treatments affect the C-terminal domain to drive the auto-inhibited conformation E1I to the activated E1A conformation, thereby modulating the transport of Ca2+.
Fig. (7). [Ca2+] dependence of incorporation of [125I]TID-PC/16 to PMCA.
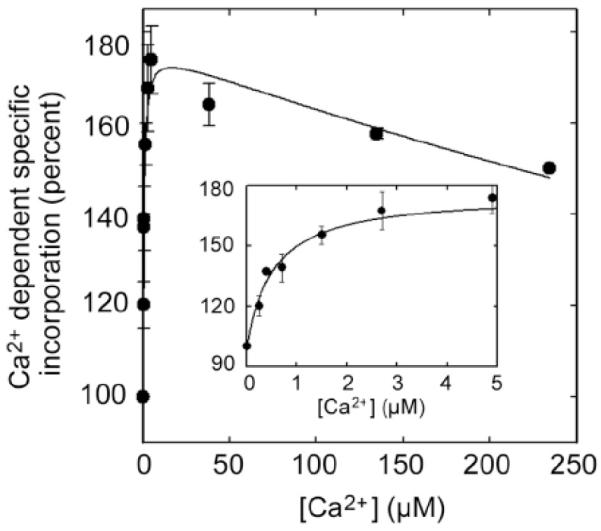
Purified PMCA devoid of calmodulin was incubated at 37°C in the presence of different amounts of Ca2+, and after 3 min [125I]TIDPC/16 was irradiated with UV light (λ ~ 360 nm) and incorporation determined as described [40]. The inset shows the incorporation of [125I]TID-PC/16 at low concentration of Ca2+. From [40] with permission.
[125I]TID-PC/16 incorporation was also used to follow the transition from E1I to E1A in the PMCA as a function of added calmodulin (Fig. 8) [40]. This allowed calculation of the dissociation constant for binding of native CaM to the PMCA in equilibrium, yielding a value (9.6 ± 0.8 nM) very close to that obtained in steady state conditions by evaluating the half-maximal activation of Ca2+-ATPase (7.2 ± 1.4 nM). Importantly, these experiments showed that following conformational state transitions in the PMCA via changes in [125I]TID-PC/16 incorporation allow an independent calculation of the dissociation constant of non-labeled calmodulin (and likely, of other modulators of activity) from the PMCA.
Fig. (8). Calmodulin (CaM) dependence of the incorporation of [125I]TID-PC/16 to PMCA in the presence of saturating concentration of Ca2+.
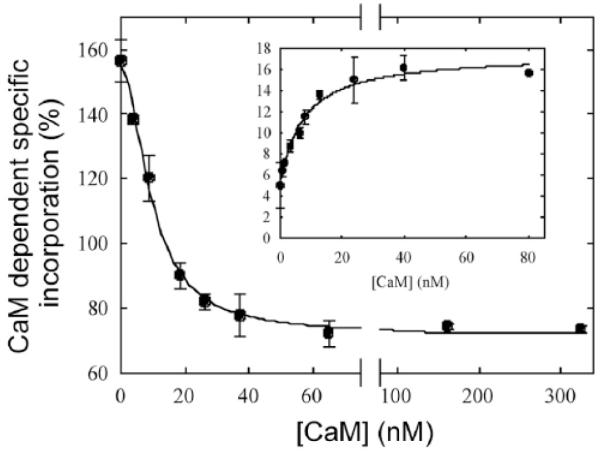
Incorporation of [125I]TID-PC/16 was determined in a medium containing 100 μM Ca2+ at 37 °C. Inset: Ca2+-ATPase activity as a function of the CaM concentration. From [40] with permission.
These studies are the first to determine the equilibrium constants for the dissociation of Ca2+ and calmodulin ligands from PMCA complexes through the change of transmembrane conformations of the pump. The data further suggest that the transmembrane domain of the PMCA undergoes major rearrangements resulting in altered lipid accessibility upon Ca2+ binding and activation.
CONFORMATIONAL CHANGES AND DISTRIBUTION OF TRANSMEMBRANE SEGMENTS IN THE NA+/K+-ATPase
Through a series of careful experiments, Blanton and McCardy [55] described the use of [125I]TID and [125I]TIDPC/16 to provide a definition of the lipid-protein interface of the Na+/K+-ATPase alpha-subunit and to detect movements of the transmembrane segments that occur during ion transport. Taking advantage of the different capacities of [125I]TID and [125I]TIDPC/16 to distribute in the membrane, Na+/K+-ATPase was photo-labeled under conditions that stabilize the two different conformations E1 and E2, and the sites of labeling were then characterized. The results showed that [125I]TIDPC/16 labeled the two conformers to a similar extent, whereas there was an enhanced photoincorporation of [125I]TID in the E2 conformation. Analysis of the amino-terminal sequence of proteolytic fragments revealed that the increased photoincorporation in the E2 conformation was mainly confined to the transmembrane segments M5 and M6. According to the spatial arrangement determined from the crystal structure of the SERCA (which at the time was the only high-resolution P-type ATPase structure available), these segments are not in direct contact with the phospholipid bilayer (see Fig. 9). The increased labeling of M5 and M6 in the E2 conformation may reflect conformational changes in these segments associated with the ion transport pathway, allowing the relatively small [125I]TID molecule access to specific residues in M5/M6 [55]. The accuracy of the location of M5/M6 in the central core of the pump was recently confirmed in the crystal structures of the Na+/K+-ATPase in the E2 conformation [9,10,56].
Fig. (9). Model of the spatial organization of the membrane segments in the Na+/K+-ATPase proposed by Blanton and McCardy [55].
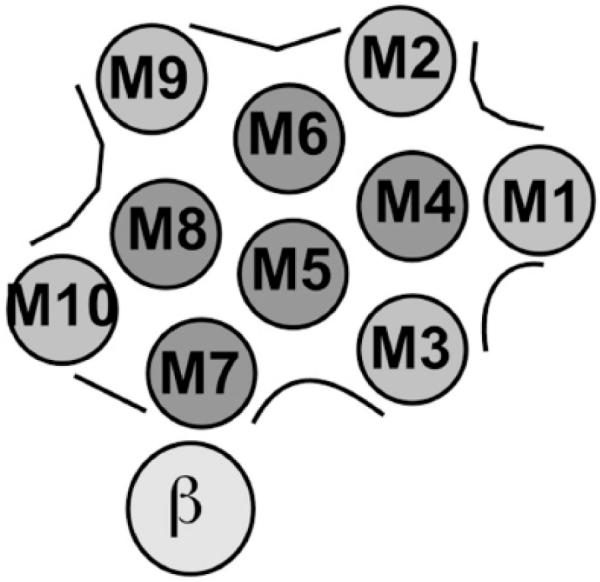
The 10 predicted transmembrane helices of the α-subunit are labeled M1 – M10. The transmembrane segments likely to be exposed to the lipid bilayer are shaded in light gray, those shielded from the bilayer are in dark gray. The single transmembrane domain of the β-subunit is also shown.
Using the same strategy in experiments measuring photo-labeling with [125I]TIDPC/16, Blanton and McCardy [55] found that the bulk of the lipid-protein interface of the Na+/K+-ATPase alpha-subunit is confined to the transmembrane segments M1, M3, M9, and M10 (but interestingly, not M2 as would be predicted from the model in Fig. 9). The extent of [125I]TID and [125I]TIDPC/16 incorporation into proteolytic fragments containing these segments was the same whether the fragments were isolated from ATPase labeled in the E1 or E2 conformation. The authors concluded that the transmembrane segments of the Na+/K+-ATPase alpha-subunit that lie at the lipid-protein interface do not undergo major conformational movements during the E1-E2 transition. The recent structural studies on the Na+ /K+-ATPase and comparisons with E1 and E2 structures of the SERCA pump have largely validated this conclusion and the positions of the transmembrane segments in the model depicted in Fig. (9) (see [49] for a recent review). On the other hand, the observed insensitivity of M2 to [125I]TIDPC/16 photo-labeling may be explained by shielding from bulk lipid caused by the tighter packing of M2 against M3 and M4 than depicted in the original model (Fig. 9), as well as by contacts with M1 and the core of the membrane domain involved in ion transport [9,10].
In contradiction with the lack of differential photoincorporation of [125I]TIDPC/16 found by Blanton and McCardy, we recently measured a small but consistent enhancement of photo-labeling in the E2 conformation of the Na+/K+-ATPase using this reagent (JPFC Rossi and RC Rossi, unpublished observation), which may reflect a slight but significant increase in lipid accessibility of some of the transmembrane segments during the E1-E2 transition [10]. On the other hand, the lack of labeling of M7 led Blanton and McCardy to correctly postulate that this segment was responsible for the interaction with the beta subunit (see Fig. 9). It is remarkable that this conclusion was posed when no sufficiently precise information on the three-dimensional structure of the Na+/K+-ATPase was yet available.
CONCLUSION
The above examples illustrate the usefulness of photoactivatable hydrophobic probes as tools to investigate the structural and dynamic properties of the membrane domain of complex membrane proteins for which high-resolution structural information from x-ray crystallography or NMR spectroscopy is still challenging to obtain. As a complementary technique, such photo-labeling studies will continue to provide important insights into mechanistic aspects and overall architectural principles of membrane proteins such as the P-type ion pumps.
ACKNOWLEDGEMENTS
The present work was supported by NIH grant RO1 NS51769 (to EES), NIH Fogarty International Center Grant R03 TW006837 (to EES, RCR and JPFCR), and by ANPCYT, CONICET and UBACYT from Argentina. We are greatly indebted to Dr. J. Brunner (Department of Biochemistry, Swiss Federal Institute of Technology (ETH), Zurich, Switzerland) for the kind gift of TTD-PC/16 (tin precursor).
REFERENCES
- [1].Pedersen PL, Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci. 1987;12:146–50. [Google Scholar]
- [2].Thever MD, Saier MH. Bioinformatic characterization of P-type ATPases encoded within the fully sequenced genomes of 26 eukaryotes. J Membr Biol. 2009;229:115–30. doi: 10.1007/s00232-009-9176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 2000;405:647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- [4].Toyoshima C, Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418:605–11. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- [5].Toyoshima C, Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004;430:529–35. doi: 10.1038/nature02680. [DOI] [PubMed] [Google Scholar]
- [6].Takahashi M, Kondou Y, Toyoshima C. Interdomain communication in calcium pump as revealed in the crystal structures with transmembrane inhibitors. Proc Nat Acad Sci USA. 2007;104:5800–5. doi: 10.1073/pnas.0700979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Olesen C, Picard M, Winther AM Lund, et al. The structural basis of calcium transport by the calcium pump. Nature. 2007;450:1036–42. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- [8].Pedersen BP, Buch-Pederson MJ, Morth JP, Palmgren MG, Nissen P. Crystal structure of the plasma membrane proton pump. Nature. 2007;450:1111–4. doi: 10.1038/nature06417. [DOI] [PubMed] [Google Scholar]
- [9].Morth JP, Pedersen BP, Toustrup-Jensen MS, et al. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–9. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- [10].Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4Å resolution. Nature. 2009;459:446–50. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- [11].Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT, Enyedi Á . Plasma membrane Ca2+ ATPases as dynamic regulators of cellular calcium handling. Ann NY Acad Sci. 2007;1099:226–36. doi: 10.1196/annals.1387.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sarkadi B, Enyedi A, Foldes-Papp Z, Gardos G. Molecular characterization of the in situ red cell membrane calcium pump by limited proteolysis. J Biol Chem. 1986;26:9552–7. [PubMed] [Google Scholar]
- [13].Corradi GR, Adamo HP. Intramolecular fluorescence resonance energy transfer between fused autofluorescent proteins reveals rearrangements of the N- and C-terminal segments of the plasma membrane Ca2+ pump involved in the activation. J Biol Chem. 2007;282:35440–8. doi: 10.1074/jbc.M703377200. [DOI] [PubMed] [Google Scholar]
- [14].Heim R, Iwata T, Zvaritch E, et al. Expression, purification, and properties of the plasma membrane Ca2+ pump and of its N-terminally truncated 105-kDa fragment. J Biol Chem. 1992;267:24476–84. [PubMed] [Google Scholar]
- [15].Brunner J. New photolabeling and crosslinking methods. Annu Rev Biochem. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- [16].Brunner J. Use of photocrosslinkers in cell biology. Trends Cell Biol. 1996;6:154–7. doi: 10.1016/0962-8924(96)40001-0. [DOI] [PubMed] [Google Scholar]
- [17].Staros JV. Aryl azide photolabels in biochemistry. Trends Biochem Sci. 1980;5:320–2. [Google Scholar]
- [18].Delfino JM, Schreiber SL, Richards FM. Design, synthesis, and properties of a photoactivatable membrane-spanning phospholipidic probe. J Am Chem Soc. 1993;115:3458–74. [Google Scholar]
- [19].Giraldo AM Villamil, Castello PR, González-Flecha FL, Moeller JV, Delfino JM, Rossi JPFC. Stoichiometry of lipid-protein interaction assessed by hydrophobic photolabeling. FEBS Lett. 2006;580:607–12. doi: 10.1016/j.febslet.2005.12.078. [DOI] [PubMed] [Google Scholar]
- [20].Rossi JP, Delfino JM, Caride AJ, Fernández HN. Interaction of unsaturated fatty acids with the red blood cell Ca2+-ATPase. Studies with a novel photoactivatable probe. Biochemistry. 1995;34:3802–12. doi: 10.1021/bi00011a038. [DOI] [PubMed] [Google Scholar]
- [21].Brunner J, Semenza G. Selective labeling of the hydrophobic core of membranes with 3-(trifluoromethyl)-3-(m-[125I]iodophenyl-)diazirine, a carbene-generating reagent. Biochemistry. 1981;20:7174–82. doi: 10.1021/bi00528a019. [DOI] [PubMed] [Google Scholar]
- [22].Thomas DD, Bigelow DJ, Squier TC, Hidalgo C. Rotational dynamics of protein and boundary lipid in sarcoplasmic reticulum membrane. Biophys J. 1982;37:217–25. doi: 10.1016/S0006-3495(82)84671-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Silvius JR, McMillen DA, Saley ND, Jost PC, Griffith OH. Competition between cholesterol and phosphatidylcholine for the hydrophobic surface of sarcoplasmic reticulum Ca2+-ATPase. Biochemistry. 1984;23:538–47. doi: 10.1021/bi00298a022. [DOI] [PubMed] [Google Scholar]
- [24].Brotherus JR, Griffith OH, Brotherus MO, Jost PC, Silvius JR, Hokin LE. Lipid-protein multiple binding equilibria in membranes. Biochemistry. 1981;20:5261–67. doi: 10.1021/bi00521a026. [DOI] [PubMed] [Google Scholar]
- [25].Esmann M, Watts A, Marsh D. Spin-label studies of lipid-protein interactions in (Na+,K+)-ATPase membranes from rectal glands of Squalus acanthias. Biochemistry. 1985;24:1386–93. doi: 10.1021/bi00327a016. [DOI] [PubMed] [Google Scholar]
- [26].Marsh D, Pali T. The protein-lipid interface: perspectives from magnetic resonance and crystal structures. Biochim Biophys Acta. 2004;1666:118–41. doi: 10.1016/j.bbamem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- [27].Ronner P, Gazzotti P, Carafoli E. A lipid requirement for the (Ca2+ + Mg2+)-activated ATPase of erythrocyte membranes. Arch Biochem Biophys. 1977;179:578–83. doi: 10.1016/0003-9861(77)90146-1. [DOI] [PubMed] [Google Scholar]
- [28].Niggli V, Adunyah ES, Penniston JT, Carafoli E. Purified (Ca2+Mg2+)-ATPase of the erythrocyte membrane: reconstitution and effect of calmodulin and phospholipids. J Biol Chem. 1981;256:395–401. [PubMed] [Google Scholar]
- [29].Sarkadi B, Enyedi A, Nyers A, Gárdos G. The function and regulation of the calcium pump in the erythrocyte membrane. Ann NY Acad Sci. 1982;402:329–48. doi: 10.1111/j.1749-6632.1982.tb25753.x. [DOI] [PubMed] [Google Scholar]
- [30].Niggli V, Adunyah ES, Carafoli E. Acidic phospholipids, unsaturated fatty acids and proteolysis mimic the effect of calmodulin on the purified erythrocyte Ca2+-ATPase. J Biol Chem. 1981;256:8588–92. [PubMed] [Google Scholar]
- [31].Nelson DR, Hanahan DJ. Phospholipid and detergent effects on (Ca2+-Mg2+) ATPase purified from human erythrocytes. Arch Biochem Biophys. 1985;236:720–30. doi: 10.1016/0003-9861(85)90678-2. [DOI] [PubMed] [Google Scholar]
- [32].Castello PR, Caride AJ, Flecha FLG, Fernández HN, Rossi JPFC, Defino JM. Identification of transmembrane domains of the red cell calcium pump with a new photoactivatable phosopholipidic probe. Biochem Biophys Res Commun. 1994;201:194–200. doi: 10.1006/bbrc.1994.1688. [DOI] [PubMed] [Google Scholar]
- [33].Stegmann T, Delfino JM, Richards FM, Helenius A. The HA2 subunit of influenza hemagglutinin inserts into the target membrane prior to fusion. J Biol Chem. 1991;266:18404–10. [PubMed] [Google Scholar]
- [34].Harter C, James P, Bächi T, Semenza G, Brunner J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide”. J Biol Chem. 1989;264:6459–64. [PubMed] [Google Scholar]
- [35].Meister H, Bachofen R, Semenza G, Brunner J. Membrane topology of light-harvesting protein B870-alpha of Rhodospirillum rubrum G-9+. Amino acid residues in contact with the lipid bilayer as inferred from labeling with photogenerated carbenes. J Biol Chem. 1985;260:16326–31. [PubMed] [Google Scholar]
- [36].Kosk-Kosicka D. Comparison of the red blood cell Ca2+-ATPase in ghost membranes and after purification. Mol Cell Biochem. 1990;99:75–81. doi: 10.1007/BF00230336. [DOI] [PubMed] [Google Scholar]
- [37].Feschenko MS, Zvaritch EI, Hofmann F, et al. A monoclonal antibody recognizes an epitope in the first extracellular loop of the plasma membrane Ca2+ pump. J Biol Chem. 1992;267:4097–101. [PubMed] [Google Scholar]
- [38].Stokes DL, Taylor WR, Green NM. Structure, transmembrane topology and helix packing of P-type ion pumps. FEBS Lett. 1994;346:32–8. doi: 10.1016/0014-5793(94)00297-5. [DOI] [PubMed] [Google Scholar]
- [39].Toyoshima C, Sasabe H, Stokes DL. Three-dimensional cryoelectron microscopy of the calcium ion pump in the sarcoplasmic reticulum membrane. Nature. 1993;362:469–71. doi: 10.1038/362469a0. [DOI] [PubMed] [Google Scholar]
- [40].Mangialavori I, Gomes M Ferreira, Pignataro MF, Strehler EE, Rossi JPFC. Determination of the dissociation constants for Ca2+ and calmodulin from the plasma membrane Ca2+ pump by a lipid probe that senses membrane domain changes. J Biol Chem. 2010;285:123–30. doi: 10.1074/jbc.M109.076679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mangialavori I, Giraldo AM Villamil, Buslje C Marino, Gomes M Ferreira, Caride AJ, Rossi JPFC. A new conformation in sarcoplasmic reticulum calcium pump and plasma membrane Ca2+ pumps revealed by a photoactivatable phospholipidic probe. J Biol Chem. 2009;284:4823–28. doi: 10.1074/jbc.M806912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zvaritch E, James P, Vorherr T, Falchetto R, Modyanov N, Carafoli E. Mapping of functional domains in the plasma membrane Ca2+ pump using trypsin proteolysis. Biochemistry. 1990;29:8070–6. doi: 10.1021/bi00487a012. [DOI] [PubMed] [Google Scholar]
- [43].Brodin P, Falchetto R, Vorherr T, Carafoli E. Identification of two domains which mediate the binding of activating phospholipids to the plasma-membrane Ca2+ pump. Eur J Biochem. 1992;204:939–46. doi: 10.1111/j.1432-1033.1992.tb16715.x. [DOI] [PubMed] [Google Scholar]
- [44].Filoteo AG, Enyedi A, Penniston JT. The lipid-binding peptide from the plasma membrane Ca2+ pump binds calmodulin and the primary calmodulin-binding domain interacts with lipid. J Biol Chem. 1992;267:11800–5. [PubMed] [Google Scholar]
- [45].Glynn IM, Karlish SJD. Occluded cations in active transport. Annu Rev Biochem. 1990;59:171–205. doi: 10.1146/annurev.bi.59.070190.001131. [DOI] [PubMed] [Google Scholar]
- [46].Post RL, Kume S, Tobin T, Orcutt B, Sen AK. Flexibility of an active center in sodium-plus-potassium adenosine triphosphatase. J Gen Physio. 1969;54:306–26. doi: 10.1085/jgp.54.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Karlish SJ, Yates DW. Tryptophan fluorescence of (Na++K+)-ATPase as a tool for study of the enzyme mechanism. Biochim Biophys Acta. 1978;527:115–30. doi: 10.1016/0005-2744(78)90261-9. [DOI] [PubMed] [Google Scholar]
- [48].Jørgensen PL, Petersen J. High-affinity 86Rb-binding and structural changes in the alpha-subunit of Na+,K+-ATPase as detected by tryptic digestion and fluorescence analysis. Biochim Biophys Acta. 1982;705:38–47. doi: 10.1016/0167-4838(82)90333-8. [DOI] [PubMed] [Google Scholar]
- [49].Bublitz M, Poulsen H, Morth JP, Nissen P. In and out of the cation pumps: P-type ATPase structure revisited. Curr Op Struct Biol. 2010;20:431–9. doi: 10.1016/j.sbi.2010.06.007. [DOI] [PubMed] [Google Scholar]
- [50].Toyoshima C, Nomura H, Sugita Y. Structural basis of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. FEBS Lett. 2003;555:106–10. doi: 10.1016/s0014-5793(03)01086-x. [DOI] [PubMed] [Google Scholar]
- [51].Enyedi A, Penniston JT. Autoinhibitory domains of various Ca2+ transporters cross-react. J Biol Chem. 1993;268:17120–5. [PubMed] [Google Scholar]
- [52].Luterbacher S, Schatzmann HJ. The site of action of La3+ in the reaction cycle of the human red cell membrane Ca2+-pump ATPase. Experimentia. 1983;39:311–2. doi: 10.1007/BF01955322. [DOI] [PubMed] [Google Scholar]
- [53].Rossi JP, Garrahan PJ, Rega AF. Vanadate inhibition of active Ca2+ transport across human red cell membranes. Biochim Biophys Acta. 1981;648:145–50. doi: 10.1016/0005-2736(81)90029-8. [DOI] [PubMed] [Google Scholar]
- [54].Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem. 1992;267:14483–9. [PubMed] [Google Scholar]
- [55].Blanton MP, McCardy EA. Identifying the lipid-protein interface and transmembrane structural transitions of the Torpedo Na,K-ATPase using hydrophobic photoreactive probes. Biochemistry. 2000;39:13534–44. doi: 10.1021/bi0015461. [DOI] [PubMed] [Google Scholar]
- [56].Ogawa H, Shinoda T, Cornelius F, Toyoshima C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc Nat Acad Sci.USA. 2009;105:13742–7. doi: 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]