

Abstract
Friedreich’s Ataxia is the most common inherited ataxia in man. It is a mitochondrial disease caused by severely reduced expression of the iron binding protein, frataxin. A large GAA triplet expansion in the human FRDA gene encoding this protein inhibits expression of this gene. It is inherited in an autosomal recessive pattern and typically diagnosed in childhood. The primary symptoms include severe and progressive neuropathy, and a hypertrophic cardiomyopathy that may cause death. The cardiomyopathy is difficult to treat and is frequently associated with arrhythmias, heart failure, and intolerance of cardiovascular stress, such as surgeries. Innovative approaches to therapy, such as histone deacetylase inhibitors, and enzyme replacement with cell penetrant peptide fusion proteins, hold promise for this and other similar mitochondrial disorders. This review will focus on the basic findings of this disease, and the cardiomyopathy associated with its diagnosis.
Keywords: ataxia, heart, cardiomyopathy, mitochondria, genetic, neuropathy
INTRODUCTION
Friedreich’s Ataxia (FA) is the most common inherited ataxia in man. It is a relentlessly progressive cardio- and neurodegenerative disease typically beginning in childhood that leads to loss of motor skills and ultimately, inability to stand or walk within 10–15 years of onset [1]. It is caused by reduced expression of the protein frataxin, which is a small protein encoded in the nucleus and imported into the mitochondrial matrix. Patients develop a primary neurodegeneration of the dorsal root ganglia leading to the hallmark clinical findings of progressive ataxia [2–3] and debilitating scoliosis. Of particular note is that these patients also develop a severe cardiomyopathy leading to death in the 3rd to 5th decade of life [4]. There is no cure for this mitochondrial disease.
BACKGROUND
Friedreich’s Ataxia was originally described in 1863 by the German neurologist and pathologist, Nikolaus Friedreich, when he noted the onset of cardiac disturbance, ataxic gait, nystagmus, and loss of sensation in 6 patients across 2 families [5]. Physical diagnosis was later based on the clinical triad of hypoactive knee and ankle jerks, progressive cerebellar dysfunction, and pre-adolescent disease onset as described by McKusick [6]. Autopsies of patients with FA have shown degeneration of the spinocerebellar and lateral corticospinal tracts, and loss of cerebellum and dorsal root ganglia which explains the loss of proprioception and coordination in affected patients. Thus, muscle strength may remain good but there is loss of motor control. In heart, the left ventricular myocardium is most often moderately to severely hypertrophied in either a concentric or asymmetric distribution, and has a high degree of fibrosis and cardiomyocyte loss at time of death. FA is typically diagnosed around the age of puberty although it ranges from 5–15 years of age, and a late onset diagnosis has been described in some patients between 20–30 years of age. Diagnosis today is confirmed by PCR analysis of the triplet expansions within the FRDA gene. As is typical of mitochondrial diseases, those tissues with higher energy requirements and greater oxidant stress manifest symptoms earliest. Thus, neurologic symptoms consisting of loss of motor skills and cardiac dysfunction are the most common findings, although blindness, diabetes, and hearing loss are frequently diagnosed as well.
GENETICS
The incidence of FA is approximately 1 in 30,000 people (prevalence of 1:50,000) with equal frequency in males and females [7] and a carrier frequency of 1:60 to 1:120 [8–11]. The inheritance is autosomal recessive. It is highly unusual in that it is predominantly (>95%) caused by a GAA triplet expansion in the first intron of the human frataxin gene (FRDA) on chromosome 9q21.11 (reviewed in [4, 12]).
The triplet expansion found in FA, which often exceeds 800 repeats, causes the formation of a triple-stranded DNA helix as a result of the GAA repeats forming non-Watson-Crick pairings with the bases of the DNA double helix [13]. In contrast, most triplet expansions that cause disease are typically located in exons encoding the protein and thus, their expression causes protein mutations and inactivation. Triplex DNA can develop even more complicated structures, such as sticky DNA [14], leading to transcriptional inhibition and partial silencing of the FRDA locus with loss or severe reduction of frataxin protein (FXN). Additionally, GAA triplet expansions may also trigger chromatin condensation making the affected region of genomic DNA transcriptionally inactive [15]. Thus, in FA the protein expression is severely reduced but whatever protein is expressed is normally processed by mitochondria. This forms the basis for innovative therapy development today.
BASIC FINDINGS
Frataxin is an essential and highly conserved protein expressed in most eukaryotic organisms (reviewed by Puccio and Koenig [16]). It is expressed in virtually all tissues of the mammalian body and functions in mitochondrial iron homeostasis [17]. The 210 amino acid precursor FXN protein (23.1 kDa) contains a mitochondrial targeting sequence at the amino terminus that is processed in 2 steps by the mitochondrial matrix processing peptidase as it is imported into the matrix [18]. The mature form of FXN has been shown to be cleaved at amino acid 81 upon import into mitochondria yielding a 130 amino acid, 14.2 kDa protein [19–20].
Frataxin has been shown to bind iron along an acid ridge, although the binding affinity is quite low. Furthermore, human frataxin has been proposed to undergo iron-independent assembly into larger oligomeric subunits [21] capable of detoxifying iron by catalyzing the oxidation of Fe(II) to Fe(III) and storing the latter form as ferrihydrite mineral within the large oligomer that it forms [22–23]. The iron loading capacity of FXN is significant and may exceed 50–75 iron atoms per subunit in a 48-subunit oligomer [24].
Although the exact function of FXN has not been defined, available data strongly suggests that FXN functions in the de novo biosynthesis of Fe-S clusters by presenting iron to IscU scaffold proteins [25–26]. It is also probable that FXN participates in heme production due to it’s interaction with ferrochelatase [27], as well as cytosolic Fe-S cluster proteins [28]. In its absence, free iron accumulates in mitochondria and is associated with loss of activity of Fe-S containing proteins [29], and loss of energy production [30–31] with probable cell death via apoptosis [32]. Given that there are a significant number of multi-subunit protein complexes in mitochondria with a Fe-S cluster and which also participate in electron transfer, it is highly likely that the decrease in FXN causes free radical generation and oxidant stress via electron loss and interaction with free iron [33].
ANIMAL MODELS
The development of animal models mimicking FA has been invaluable both for the study of the basic biology, as well as the clinical implications of this disease. Although highly useful, each of these models also has significant limitations. Two basic strategies have been used to recapitulate this disease in the mouse. In the first, a conditional ablation of the mouse FRDA gene was accomplished by the Puccio lab. Here, exon 4 of the FRDA gene was deleted using Cre mediated excision in a tissue specific fashion [34]. This causes complete absence of the FXN protein only in the target tissues. This was necessary because complete ablation of the FRDA gene in the mouse was embryonic lethal demonstrating the importance of early frataxin expression [35]. However, when the FRDA gene was ablated in heart and brain, or in heart alone (skeletal muscle expression was also ablated but has no phenotype), then significant phenotypes were generated that recapitulated many features of FA. For example, mice in which the FRDA gene was ablated in brain (neural tissues) and heart demonstrated a severe neurologic phenotype as well as cardiomyopathy (dilated) and very short lifespan [34]. These mice grow poorly (Figure 1A) when compared with their heterozygous littermates, and demonstrate ataxia and loss of proprioception on behavioral studies such as the rotarod. As with FA in the human heart, there is clear evidence of iron deposition in their hearts as shown by the Perls’ stain in Figure 1B. In contrast, mice in which the FRDA gene was ablated only in sarcomeric tissues (heart and skeletal muscle) predictably did not have ataxia but developed a cardiac phenotype of hypertrophic cardiomyopathy very reminiscent of the human heart disease [36]. Thus, this strategy mimics the phenotype of the human disease well, but not the correct genotype. They also are lacking any FXN protein in the affected tissues (“knocked out”) whereas humans have residual low level expression of FXN protein and thus, have a more variable presentation. These mice are especially useful for study of the histology of FA, as well as developing protein or enzyme replacement therapies, such as with cell penetrant peptide fusion proteins [37], e.g., TAT-Frataxin.
Figure 1.
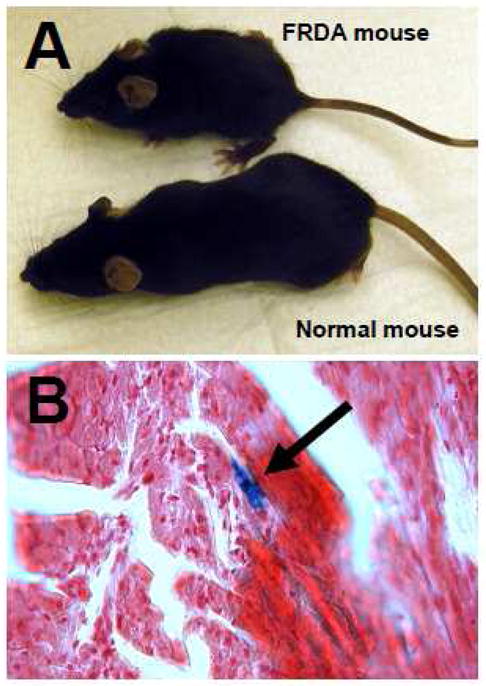
Panel A: Mouse model of Friedreich’s Ataxia. Mouse labeled ‘FRDA mouse’ has a conditional ablation of exon 4 in the FRDA gene in heart and brain. Below is an age matched, littermate heterozygous mouse (‘Normal mouse’). Panel B: Perls’ Stain for iron deposition in heart from a 28 day old conditional FRDA KO mouse. Arrow points to staining from iron deposition.
The cardiac findings in these conditional knockout mice are very similar to the hearts of patients with FA. In addition to iron deposition in the myocardium mentioned above, they also show a cardiomyopathy on echocardiography with evidence of diastolic dysfunction. For example, in Figure 2 the echocardiograms from a normal littermate mouse and a conditional FRDA knockout are shown. The M-mode of the normal mouse (Figure 2A) shows good left ventricular function and a normal mitral inflow pattern based on the E-A waveform. In panel B of the same figure, the M-mode demonstrates a dilated, poorly contractive left ventricle with slower heart rate and reversal of the E-A waveforms. Examination of the cardiomyocyte ultrastructure by electron microscopy is particularly informative in these mice. In Figure 3A and B, a normal mouse heart demonstrates normally shaped mitochondria tightly packed between rows of sarcomeres. Alignment of sarcomeres is highly regular and robust. In contrast, Figure 3C and D demonstrate the severe disorganization of the cardiomyocyte from an FRDA conditional knockout mouse heart. Here, there are very few sarcomeres remaining and mitochondria have proliferated extensively. There is little evidence of the tight relationship between energy generating mitochondria and the site of ATP utilization (sarcomere) seen in the normal mouse cardiomyocyte. This parallels exactly what is found in the human heart [38] and has strong clinical implications for patients with FA: left ventricular contractile function will ultimately decrease due to loss of contractile proteins and disruption of the normal mitochondria to sarcomere relationship.
Figure 2.
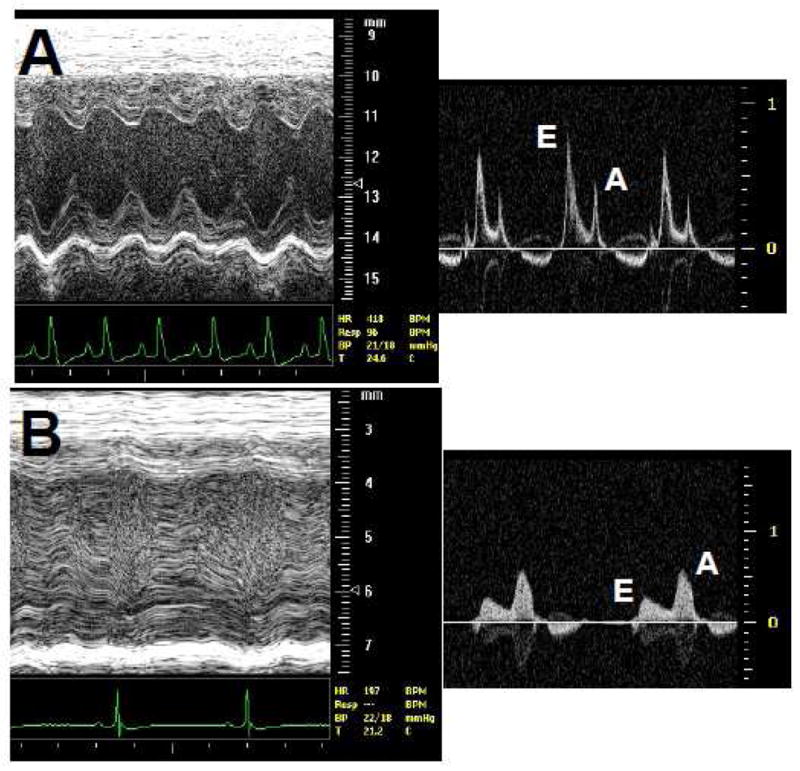
Echocardiogram from mice. Panel A shows excellent left ventricular function by M-mode echocardiography in a normal mouse. Distance in mm shown on right. Heart rate is shown below and is 407 beats per minute. The side panel shows mitral valve inflow with a normal E-A inflow pattern. Velocity in m/s shown on right. Panel B shows poor left ventricular function in a FRDA KO mouse. The left ventricular chamber is dilated compared with the normal heart. Heart rate is slower at 197 beats per minute. Side panel show mitral valve inflow with E-A reversal indicative of severe cardiomyopathy.
Figure 3.
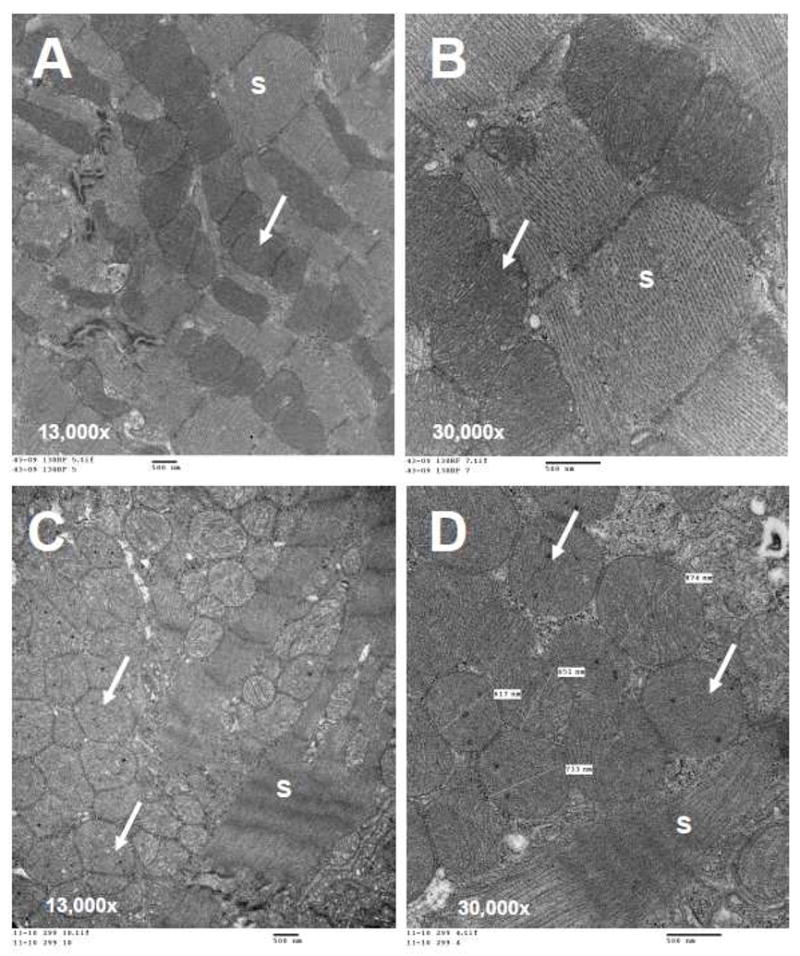
Electron microscopy of heart sections from mouse model of Friedreich’s Ataxia. Panels A and B are from a wild type mouse showing normal mitochondria (white arrows) in rows between abundant, well ordered sarcomeres (“S’). Panels C and D are from a conditional knockout mouse with ablation of the FRDA locus in heart and brain. Notice extreme proliferation of mitochondria (Panel C, white arrows) with intramitochondrial deposits (Panel D). There is a severe loss of sarcomeres (S). Bar = 500 nm in all 4 panels.
In the second strategy, mice have been generated in which there is a ‘knock in’ of the pathologic human FRDA gene with GAA repeats into the mouse by both the Pook lab [39] and the Pandolfo lab [40]. In these models, the native mouse FRDA genes have been ablated (KO) leaving expression of frataxin dependent only on the human FRDA gene, which also contains the triplet expansion region. The phenotype is less severe in these animals however; the genotype more accurately mimics the human disease. These animals have been especially useful for study of therapeutic approaches that increase transcription of the FRDA gene containing the triplet expansion, such as histone deacetylase inhibitors (HDAC inhibitors) [41].
CLINICAL FINDINGS
The clinical symptoms of FA are manifested as loss of coordination and motor control, diabetes, and a hypertrophic cardiomyopathy associated with death [2]. Other clinical findings, such as visual impairment, slurred speech, diabetes, and hearing loss are also frequently reported. There is a correlation between GAA repeat number and the onset and severity of clinical symptoms with higher repeat numbers typically associated with earlier onset, and a more severe cardiomyopathy [42–44]. The patient disease course typically reflects the length of the shorter allele, and compound heterozygous patients have been described in which one allele contains a point mutation and the other a triplet expansion.
The cardiomyopathy in FA is hypertrophic and typically maintains adequate systolic function [45] until shortly before death [44]. Cardiac MRI demonstrates the severe hypertrophy very well as shown in Figure 4A and B, but echocardiography is also excellent for long term evaluation of left ventricular wall thickness and function. Both ventricles are affected and arrhythmias are common, especially those of atrial origin. Although the primary cause of death in FA is heart failure [46], surprisingly little has been written about the natural history and pathology of the FA heart with most publications focusing instead on the dramatic neuropathology (Figure 5).
Figure 4.
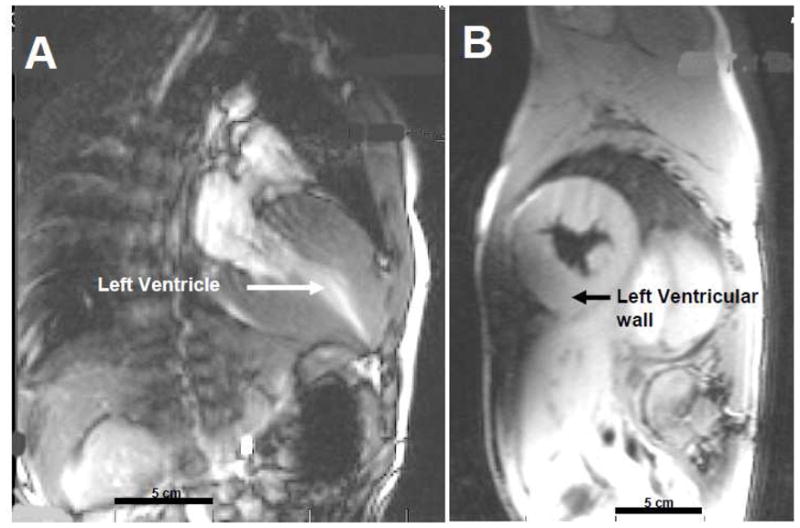
Cardiac MRI of a 10 year old male with Friedreich’s Ataxia. Panel A: Long axis view of hyperdynamic heart with contrast showing severe hypertrophy of left ventricular walls and mid-cavitary obliteration in systole. Ejection fraction = 81%. Panel B: Short axis of heart in systole showing hypertrophic cardiomyopathy. Bar = 5 cm in both panels.
Figure 5.
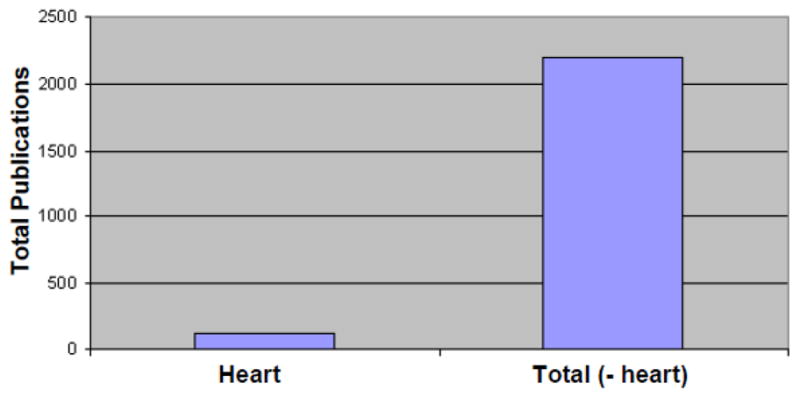
Graph of publications on Friedreich’s Ataxia from 1966–2010. Publications on the cardiomyopathy in this disease are significantly less in number when compared with those investigating the dramatic neurologic disease of Friedreich’s Ataxia.
Myocardial energy generation is significantly reduced in FA patients when compared with controls, and this reduction correlates with the degree of hypertrophy [47] or may even precede it’s development [31]. Use of the antioxidant and co-enzyme Q mimetic, Idebenone, may help improve myocardial energy deficiency and decrease hypertrophy [48–49]. In short term (1 year) studies, this has been true. Longer term studies using Idebenone have shown a reduction in cardiac mass but have not shown a decrease in arrhythmias or progression of heart failure [50–51] and there is no research on the clinical relevance of these changes. Antioxidant therapies have not shown any benefit or improvement in neurologic symptoms [52–53].
Variability in the cardiac phenotype has been noted by multiple authors [45, 54]. Hypertrophy may progress to dilation with time giving the appearance by echocardiography that there is improvement of ventricular hypertrophy [54]. Earlier echocardiography and radionuclide studies have shown good systolic function of both ventricles but impaired diastolic filling [55–56]. These findings are consistent with the histological appearance of fibrosis in the left ventricular wall and suggest that there may be diastolic dysfunction in the FA heart. Virtually all hearts are affected in FA and subtle EKG findings are frequently among the earliest findings of cardiomyopathy. In Figure 6A and B, EKGs from two patients demonstrate the non-specific T-wave changes often seen in the lateral chest leads. EKG, however, is not diagnostic of FA and has not shown utility for prognosis in this disease.
Figure 6.

EKGs from 2 patients with Friedreich’s Ataxia showing non-specific T-wave abnormalities, e.g., flattened or inverted T-waves in the lateral chest leads. Panel B has a superior axis. Although EKG is abnormal in >90% of cases, it is not prognostic in this disease.
At the microscopic level, changes in the left ventricle include cardiomyocyte hypertrophy, focal necrosis, and diffuse fibrosis [57]. Electron microscopy shows mitochondrial proliferation, loss of contractile sarcomeres, and electron dense particles within mitochondria that may represent iron deposition [38]. This is strikingly similar to what is found in the mouse heart in which FXN is deficient (see Figure 3C and D). Recent studies of patients with FA using cardiac MRI and adenosine showed that the heart has significantly decreased myocardial perfusion reserve index, which paralleled the onset of metabolic syndrome [58]. Furthermore, the impaired perfusion reserve did not correlate with degree of hypertrophy or fibrosis suggesting that this may be an important tool to identify potential therapeutic targets to prevent heart failure development or progression.
The clinical implications of these findings are that the cardiac output of affected patients in FA may reflect the characteristics of a restrictive cardiomyopathy with a greater dependence on heart rate to maintain adequate output. Thus, there will predictably be an increase in left ventricular filling pressures with resultant atrial arrhythmias, which are commonly seen in advanced FA. Use of after load reducing agents, such as losartan or ACE inhibitors, may be beneficial in long term treatment of this heart disease. This may also decrease physiologic incentive for cardiac hypertrophy and energy utilization by ventricular cardiomyocytes. In contrast, those agents that slow heart rate and decrease oxygen utilization, such as carvedilol or other β-adrenergic blocking agents, may not be well tolerated. If diastolic filling is impaired due to a restrictive characteristic of this cardiomyopathy, then slowing the heart rate would predictably decrease cardiac output. For example, carvedilol is beneficial in adult onset ischemic cardiomyopathy, but had little effect on cardiomyopathies of childhood where the etiology of the heart failure is much different and variable [59].
TREATMENT
Treatment options at present logically include antioxidants, such as Idebenone, and iron chelation such as deferiprone [60–61]. Although early clinical trials have shown modest biochemical improvement [62], these therapies have not shown substantial sustained clinical improvement as they are designed to control downstream events resulting from the loss of FXN. It is also probable that significant and permanent damage has already occurred by the time the diagnosis of FA is made making these therapeutic approaches less effective. Recent studies have supported earlier intervention. For example, Boddaert et al, showed that iron chelation with a membrane permeant chelator that shuttles iron to transferrin reduced brain iron load and had greatest effect in the youngest patients [63]. If confirmed, this is a strong argument for beginning therapy for FA as early as possible and supports the drive towards newborn screening for FA (see commentary by G. Isaya [64]). Necessary expansion of newborn screening to include new diseases, such as FA, was advocated in a recent publication putting forth recommendations for uniform newborn screening [65], and work is already underway at Mayo Clinic to develop a protein-based assay for screening newborns for FA (supported by the Friedreich’s Ataxia Research Alliance). However, despite concerted clinical efforts with antioxidant and iron chelation strategies, no therapy has been substantially effective in this disease.
FUTURE DIRECTIONS
Friedreich’s Ataxia is a multisystem disorder that serves as a paradigm for mitochondrial disease. The gene defect is known, the affected organelle is known, and the inheritance is well understood and common. New discovery is rapidly advancing our understanding of the basic biology of FA and lessons learned here have high potential for application to other mitochondrial diseases. From a clinical research standpoint, there is a great need to understand the natural history and outcome of the heart disease in FA. Given the relatively less amount of literature examining the heart in FA compared with the neurologic and histological phenotype, greater emphasis on clinical studies and trials examining this mitochondrial cardiomyopathy are badly needed to advance our clinical understanding and speed development of therapies.
With regard to therapeutic development for FA, multiple lines of investigation are aggressively pursuing innovative strategies and will likely have application to other mitochondrial disorders as well. Two strategies are worthy of note and directly attack the problem in FA. In the first, the goal is to improve the ability of the DNA transcription machinery to read through the diseased FRDA gene by maintaining the DNA in an ‘uncompacted’ state. Thus, Joel Gottesfeld reported earlier that histone deacetylase inhibitors (HDAC inhibitors) would increase frataxin mRNA transcript and could increase FXN protein levels of cells in culture [15]. When HDAC inhibitors were applied to the Friedreich’s Ataxia mice with expression of the diseased human FRDA gene (the ‘knock in’ mouse above), there was an increase in FXN protein [41]. This strategy also has significant potential for application to other triplet expansion diseases.
In the second approach, our laboratory has pursued an enzyme replacement strategy where the missing protein, FXN, is genetically fused to a cell penetrant peptide, TAT, to deliver the protein into the cell and mitochondria. We have reported earlier that TAT was capable of pulling exogenous proteins across cell and mitochondrial membranes to deliver a cargo to the matrix of mitochondria in both isolated cells and tissues of the intact mouse [37, 66–67]. TAT will even cross the placenta to deliver proteins to mitochondria of the fetus [68] thus offering the potential for initiating therapies for mitochondrial defects prior to birth. Studies are underway using the conditional ablation knock out mice from the Puccio lab to explore this approach. Early results are highly promising (data in preparation) and other investigators have also recently reported that TAT can deliver therapeutic proteins in mouse or human models of disease [69–71].
SUMMARY
In summary, our understanding of Friedreich’s Ataxia is advancing rapidly and will likely have application to other mitochondrial diseases. Novel therapeutic development is being aggressively pursued and takes advantage of recent discoveries at the bench that may be moved into patient care. There is a significant need to focus greater attention on the heart disease in FA to understand its long term outcome and to develop new therapeutic strategies using existing medications and approaches.
Acknowledgments
Support for this project was provided in part by the Friedreich’s Ataxia Research Alliance, and the National Institutes of Health 1P01HL 085098 and R21 NS052198. The funding sources had no role in the conduct or presentation of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- 2.Reddy PL, Grewal RP. Friedreich’s ataxia: a clinical and genetic analysis. Clinical neurology and neurosurgery. 2007;109:200–2. doi: 10.1016/j.clineuro.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 3.De Biase I, Rasmussen A, Endres D, Al-Mahdawi S, Monticelli A, Cocozza S, et al. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- 4.Patel PI, Isaya G. Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am J Hum Genet. 2001;69:15. doi: 10.1086/321283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedreich N. Ueber degenerative Atrophie der spinalen Hinterstränge. Virchows Archiv. 1863;26:391–419. [Google Scholar]
- 6.Boyer SH, Chisholm AW, McKusick VA. Cardiac aspects of Friedreich’s ataxia. Circulation. 1962;25:493–505. doi: 10.1161/01.cir.25.3.493. [DOI] [PubMed] [Google Scholar]
- 7.Schols L, Amoiridis G, Przuntek H, Frank G, Epplen JT, Epplen C. Friedreich’s ataxia: revision of the phenotype according to molecular genetics. Brain. 1997;120:2131–40. doi: 10.1093/brain/120.12.2131. [DOI] [PubMed] [Google Scholar]
- 8.Filla A, De Michele G, Marconi R, Bucci L, Carillo C, Castellano AE, et al. Prevalence of hereditary ataxias and spastic paraplegias in Molise, a region of Italy. J Neurol. 1992;239:351–3. doi: 10.1007/BF00867594. [DOI] [PubMed] [Google Scholar]
- 9.Romeo G, Menozzi P, Ferlini A, Fadda S, Di Donato S, Uziel G, et al. Incidence of Friedreich ataxia in Italy estimated from consanguineous marriages. Am J Hum Genet. 1983;35:523–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Skre H. Friedreich’s ataxia in Western Norway. Clin Genet. 1975;7:287–98. doi: 10.1111/j.1399-0004.1975.tb00331.x. [DOI] [PubMed] [Google Scholar]
- 11.Epplen C, Epplen JT, Frank G, Miterski B, Santos EJ, Schols L. Differential stability of the (GAA)n tract in the Friedreich ataxia (STM7) gene. Hum Genet. 1997;99:834–6. doi: 10.1007/s004390050458. [DOI] [PubMed] [Google Scholar]
- 12.Pandolfo M. Friedreich ataxia. Arch Neurol. 2008;65:1296–303. doi: 10.1001/archneur.65.10.1296. [DOI] [PubMed] [Google Scholar]
- 13.Gacy AM, Goellner GM, Spiro C, Chen X, Gupta G, Bradbury EM, et al. GAA instability in Friedreich’s Ataxia shares a common, DNA-directed and intraallelic mechanism with other trinucleotide diseases. Mol Cell. 1998;1:583–93. doi: 10.1016/s1097-2765(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. The Journal of biological chemistry. 2001;276:27171–7. doi: 10.1074/jbc.M101879200. [DOI] [PubMed] [Google Scholar]
- 15.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–8. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 16.Puccio H, Koenig M. Recent advances in the molecular pathogenesis of Friedreich ataxia. Hum Mol Genet. 2000;9:887. doi: 10.1093/hmg/9.6.887. [DOI] [PubMed] [Google Scholar]
- 17.Huang ML, Becker EM, Whitnall M, Rahmanto YS, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A. 2009;106:16381–6. doi: 10.1073/pnas.0906784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G. Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J Biol Chem. 2000;275:41469. doi: 10.1074/jbc.M006539200. [DOI] [PubMed] [Google Scholar]
- 19.Condo I, Ventura N, Malisan F, Rufini A, Tomassini B, Testi R. In vivo maturation of human frataxin. Hum Mol Genet. 2007;16:1534–40. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 20.Schmucker S, Argentini M, Carelle-Calmels N, Martelli A, Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum Mol Genet. 2008;17:3521–31. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 21.O’Neill HA, Gakh O, Isaya G. Supramolecular assemblies of human frataxin are formed via subunit-subunit interactions mediated by a non-conserved amino-terminal region. J Mol Biol. 2005;345:433–9. doi: 10.1016/j.jmb.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill HA, Gakh O, Park S, Cui J, Mooney SM, Sampson M, et al. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry. 2005;44:537. doi: 10.1021/bi048459j. [DOI] [PubMed] [Google Scholar]
- 23.Schagerlof U, Elmlund H, Gakh O, Nordlund G, Hebert H, Lindahl M, et al. Structural basis of the iron storage function of frataxin from single-particle reconstruction of the iron-loaded oligomer. Biochemistry. 2008;47:4948–54. doi: 10.1021/bi800052m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nichol H, Gakh O, O’Neill HA, Pickering IJ, Isaya G, George GN. Structure of frataxin iron cores: an X-ray absorption spectroscopic study. Biochemistry. 2003;42:5971. doi: 10.1021/bi027021l. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Lyver ER, Knight SA, Pain D, Lesuisse E, Dancis A. Mrs3p, Mrs4p, and frataxin provide iron for Fe-S cluster synthesis in mitochondria. J Biol Chem. 2006;281:22493–502. doi: 10.1074/jbc.M604246200. [DOI] [PubMed] [Google Scholar]
- 26.Shan Y, Napoli E, Cortopassi G. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum Mol Genet. 2007;16:929–41. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- 27.Yoon T, Cowan JA. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem. 2004;279:25943–6. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- 28.Martelli A, Wattenhofer-Donze M, Schmucker S, Bouvet S, Reutenauer L, Puccio H. Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum Mol Genet. 2007;16:2651–8. doi: 10.1093/hmg/ddm163. [DOI] [PubMed] [Google Scholar]
- 29.Rotig A, de LP, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 30.Lodi R, Cooper JM, Bradley JL, Manners D, Styles P, Taylor DJ, et al. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc Natl Acad Sci USA. 1999;96:11492. doi: 10.1073/pnas.96.20.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lodi R, Rajagopalan B, Blamire AM, Cooper JM, Davies CH, Bradley JL, et al. Cardiac energetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: an in vivo 31P magnetic resonance spectroscopy study. Cardiovasc Res. 2001;52:111–9. doi: 10.1016/s0008-6363(01)00357-1. [DOI] [PubMed] [Google Scholar]
- 32.Pandolfo M. Iron and Friedreich ataxia. Journal of neural transmission. 2006:143–6. doi: 10.1007/978-3-211-45295-0_22. [DOI] [PubMed] [Google Scholar]
- 33.Schulz JB, Dehmer T, Schols L, Mende H, Hardt C, Vorgerd M, et al. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55:1719. doi: 10.1212/wnl.55.11.1719. [DOI] [PubMed] [Google Scholar]
- 34.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 35.Cossee M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, LeMeur M, et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9:1219. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- 36.Whitnall M, Rahmanto YS, Sutak R, Xu X, Becker EM, Mikhael MR, et al. The MCK mouse heart model of Friedreich’s ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc Natl Acad Sci U S A. 2008;105:9757–62. doi: 10.1073/pnas.0804261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rayapureddi JP, Tomamichel WJ, Walton ST, Payne RM. TAT Fusion Protein Transduction into Isolated Mitochondria Is Accelerated by Sodium Channel Inhibitors. Biochemistry. 2010 doi: 10.1021/bi101057v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michael S, Petrocine SV, Qian J, Lamarche JB, Knutson MD, Garrick MD, et al. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum (London, England) 2006;5:257–67. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 39.Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, et al. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- 40.Miranda CJ, Santos MM, Ohshima K, Smith J, Li L, Bunting M, et al. Frataxin knockin mouse. FEBS Lett. 2002;512:291. doi: 10.1016/s0014-5793(02)02251-2. [DOI] [PubMed] [Google Scholar]
- 41.Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montermini L, Richter A, Morgan K, Justice CM, Julien D, Castellotti B, et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41:675–82. doi: 10.1002/ana.410410518. [DOI] [PubMed] [Google Scholar]
- 43.Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G, et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet. 1996;59:554–60. [PMC free article] [PubMed] [Google Scholar]
- 44.Rajagopalan B, Francis JM, Cooke F, Korlipara LV, Blamire AM, Schapira AH, et al. Analysis of the factors influencing the cardiac phenotype in Friedreich’s ataxia. Mov Disord. 2010;25:846–52. doi: 10.1002/mds.22864. [DOI] [PubMed] [Google Scholar]
- 45.Kipps A, Alexander M, Colan SD, Gauvreau K, Smoot L, Crawford L, et al. The longitudinal course of cardiomyopathy in Friedreich’s ataxia during childhood. Pediatr Cardiol. 2009;30:306–10. doi: 10.1007/s00246-008-9305-1. [DOI] [PubMed] [Google Scholar]
- 46.Hanley A, Corrigan R, Mohammad S, MacMahon B. Friedreich’s ataxia cardiomyopathy: case based discussion and management issues. Ir Med J. 2010;103:117–8. [PubMed] [Google Scholar]
- 47.Bunse M, Bit-Avragim N, Riefflin A, Perrot A, Schmidt O, Kreuz FR, et al. Cardiac energetics correlates to myocardial hypertrophy in Friedreich’s ataxia. Ann Neurol. 2003;53:121–3. doi: 10.1002/ana.10419. [DOI] [PubMed] [Google Scholar]
- 48.Rustin P, Rotig A, Munnich A, Sidi D. Heart hypertrophy and function are improved by idebenone in Friedreich’s ataxia. Free Radic Res. 2002;36:467–9. doi: 10.1080/10715760290021333. [DOI] [PubMed] [Google Scholar]
- 49.Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di DS. Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology. 2003;60:1676. doi: 10.1212/01.wnl.0000055872.50364.fc. [DOI] [PubMed] [Google Scholar]
- 50.Pineda M, Arpa J, Montero R, Aracil A, Dominguez F, Galvan M, et al. Idebenone treatment in paediatric and adult patients with Friedreich ataxia: long-term follow-up. Eur J Paediatr Neurol. 2008;12:470–5. doi: 10.1016/j.ejpn.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 51.Velasco-Sanchez D, Aracil A, Montero R, Mas A, Jimenez L, O’Callaghan M, et al. Combined Therapy with Idebenone and Deferiprone in Patients with Friedreich’s Ataxia. Cerebellum (London, England) 2010 doi: 10.1007/s12311-010-0212-7. [DOI] [PubMed] [Google Scholar]
- 52.Kearney M, Orrell RW, Fahey M, Pandolfo M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database Syst Rev. 2009:CD007791. doi: 10.1002/14651858.CD007791.pub2. [DOI] [PubMed] [Google Scholar]
- 53.Lynch DR, Perlman SL, Meier T. A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch Neurol. 2010;67:941–7. doi: 10.1001/archneurol.2010.168. [DOI] [PubMed] [Google Scholar]
- 54.Hawley RJ, Gottdiener JS. Five-year follow-up of Friedreich’s ataxia cardiomyopathy. Arch Intern Med. 1986;146:483–8. [PubMed] [Google Scholar]
- 55.Palagi B, Picozzi R, Casazza F, Possa M, Magri G, Zoccarato O, et al. Biventricular function in Friedreich’s ataxia: a radionuclide angiographic study. Br Heart J. 1988;59:692–5. doi: 10.1136/hrt.59.6.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dutka DP, Donnelly JE, Palka P, Lange A, Nunez DJ, Nihoyannopoulos P. Echocardiographic characterization of cardiomyopathy in Friedreich’s ataxia with tissue Doppler echocardiographically derived myocardial velocity gradients. Circulation. 2000;102:1276–82. doi: 10.1161/01.cir.102.11.1276. [DOI] [PubMed] [Google Scholar]
- 57.Hewer R. The heart in Friedreich’s ataxia. Br Heart J. 1969;31:5–14. doi: 10.1136/hrt.31.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raman SV, Phatak K, Hoyle JC, Pennell ML, McCarthy B, Tran T, et al. Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: a mitochondrial cardiomyopathy with metabolic syndrome. Eur Heart J. 2010 doi: 10.1093/eurheartj/ehq443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, et al. Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA. 2007;298:1171–9. doi: 10.1001/jama.298.10.1171. [DOI] [PubMed] [Google Scholar]
- 60.Richardson DR. Friedreich’s ataxia: iron chelators that target the mitochondrion as a therapeutic strategy? Expert opinion on investigational drugs. 2003;12:235–45. doi: 10.1517/13543784.12.2.235. [DOI] [PubMed] [Google Scholar]
- 61.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. Faseb J. 2003;17:1972–4. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 62.Hart PE, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol. 2005;62:621–6. doi: 10.1001/archneur.62.4.621. [DOI] [PubMed] [Google Scholar]
- 63.Boddaert N, Le Quan Sang KH, Rotig A, Leroy-Willig A, Gallet S, Brunelle F, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–8. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- 64.Isaya G. Ironing out a therapy for Friedreich Ataxia. Blood. 2007;110:1–2. [Google Scholar]
- 65.Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P. Newborn Screening: Toward a uniform screening panel and system. Genetics in Medicine. 2006;8:1S–11S. doi: 10.1097/01.gim.0000223891.82390.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Del Gaizo Moore V, Payne RM. Transactivator of transcription fusion protein transduction causes membrane inversion. J Biol Chem. 2004;279:32541–4. doi: 10.1074/jbc.M405930200. [DOI] [PubMed] [Google Scholar]
- 67.Del Gaizo-Moore V, MacKenzie JA, Payne RM. Targeting proteins to mitochondria using TAT. Mol Genet Metab. 2003;80:170. doi: 10.1016/j.ymgme.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 68.Del Gaizo V, Payne RM. A novel TAT-Mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Molecular Therapy. 2003;7:720. doi: 10.1016/s1525-0016(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 69.Rapoport M, Saada A, Elpeleg O, Lorberboum-Galski H. TAT-mediated delivery of LAD restores pyruvate dehydrogenase complex activity in the mitochondria of patients with LAD deficiency. Mol Ther. 2008;16:691–7. doi: 10.1038/mt.2008.4. [DOI] [PubMed] [Google Scholar]
- 70.Gustafsson AB, Sayen MR, Williams SD, Crow MT, Gottlieb RA. TAT protein transduction into isolated perfused hearts: TAT-apoptosis repressor with caspase recruitment domain is cardioprotective. Circulation. 2002;106:735–9. doi: 10.1161/01.cir.0000023943.50821.f7. [DOI] [PubMed] [Google Scholar]
- 71.Toro A, Grunebaum E. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J Clin Invest. 2006 doi: 10.1172/JCI25052. [DOI] [PMC free article] [PubMed] [Google Scholar]