

Abstract
Importance of the field
PubChem is a public molecular information repository, a scientific showcase of the NIH Roadmap Initiative. The PubChem database holds over 27 million records of unique chemical structures of compounds (CID) derived from nearly 70 million substance depositions (SID), and contains more than 449,000 bioassay records with over thousands of in vitro biochemical and cell-based screening bioassays established, with targeting more than 7000 proteins and genes linking to over 1.8 million of substances.
Areas covered in this review
This review builds on recent PubChem-related computational chemistry research reported by other authors while providing readers with an overview of the PubChem database, focusing on its increasing role in cheminformatics, virtual screening and toxicity prediction modeling.
What the reader will gain
These publicly available datasets in PubChem provide great opportunities for scientists to perform cheminformatics and virtual screening research for computer-aided drug design. However, the high volume and complexity of the datasets, in particular the bioassay-associated false positives/negatives and highly imbalanced datasets in PubChem, also creates major challenges. Several approaches regarding the modeling of PubChem datasets and development of virtual screening models for bioactivity and toxicity predictions are also reviewed.
Take home message
Novel data-mining cheminformatics tools and virtual screening algorithms are being developed and used to retrieve, annotate and analyze the large-scale and highly complex PubChem biological screening data for drug design.
Keywords: PubChem, cheminformatics, data-mining, virtual screening, toxicity, polypharmacology
1. Introduction
PubChem is a scientific showcase of the Molecular Libraries Program (MLP), a US National Institutes of Health (NIH) Roadmap Initiative (http://mli.nih.gov/mli/) that aims to enhance chemical biology efforts through high-throughput screening (HTS) so as to identify small molecule probes effective at modulating a given biological process or disease. The PubChem database (http://PubChem.ncbi.nlm.nih.gov) was constructed in 2004 to facilitate information exchange and data sharing among the ten NIH-funded centers of the Molecular Libraries Screening Centers Network (MLSCN), which later was transformed to Molecular Libraries Probe Production Centers Network (MLPCN) in 2008. The MLPCN is composed of three different types of centers, i.e., Comprehensive Centers, Specialized Screening Centers and Specialized Chemistry Centers1.
Along with comprehensive online information on small molecule chemical structures and corresponding biological activity data, the PubChem database has now grown into a powerful public molecular information resource with online data analysis and subsetting tools to facilitate high-throughput/high-content screening (HTS/HCS) of small molecules that modulate the bioactivity of various targets. Maintained by the National Center for Biotechnological Information (NCBI)2, the PubChem database system consists of three primary relational databases: Substance (substance ID or SID), Compound (compound ID or CID) and Bioassay (assay ID or AID), which correspond to the three major query functions as shown in Figure 1. As of July 2010, the PubChem Substance database contains 69,170,468 entries of mixtures, extracts, complexes and uncharacterized substances; the PubChem Compound database contains 27,443,646 records of unique chemical structure compounds derived from the substance depositions; and the PubChem Bioassay database has 449,401 records. These data records were deposited by screening centers funded by the NIH Molecular Library Program, academic institutions, and industrial research organizations as illustrated in Figure 1A. The update list is referred to see the PubChem substance and bioassay data source information website 3.
Figure 1. Brief overview of the PubChem Database system.
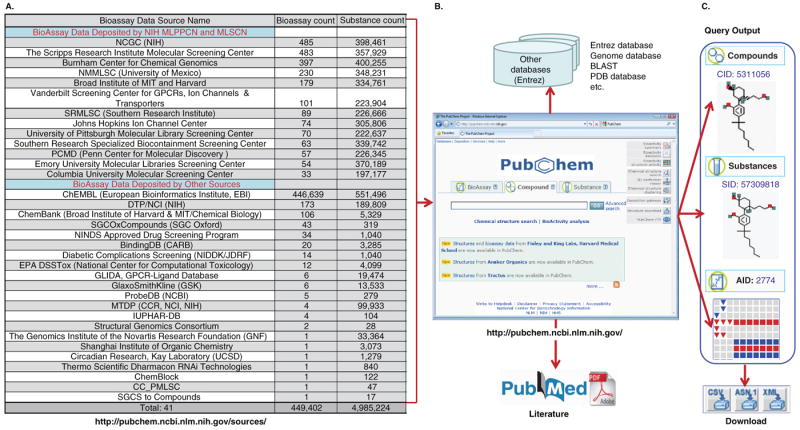
(A) The BioAssay data depositor list, including by NIH Molecular Libraries Probe Production Centers Network (MLPCN) and former Molecular Library Screening Centers Network (MLSCN) as well as other sources. (B) The PubChem database search window with online structure drawing/clustering, 3D view and data analysis tools, and the links to other databases as well as PubMed literature. (C) The query Output window (CID, SID and AID) and the files upload and download formats and tools
As PubChem continues to grow rapidly in the data collections as well as online data-mining analysis capability, research opportunities also emerge for the scientific community to exploit the available structural and biological data to enhance understanding and investigating the structure activity relationships (SAR), pharmacology, metabolism and toxicology profiles of target compounds, both in vitro and in silico. In the meantime, novel data-mining cheminformatics tools and virtual screening algorithms are being developed and used to retrieve, annotate and analyze the large-scale and highly complex PubChem biological screening data.
Since PubChem was launched, there has been rapidly increasing the number of research publications using PubChem chemical library and bioassay data for cheminformatics data-mining studies, virtual screening, SAR, in silico design, and on the like (Figure 2). The numbers of the PubChem-related research publications quickly increased from only a total of 32 PubChem-related articles in 2005 to 119 in 2009. A total of 459 PubChem-related articles have been published as of July 2010. Figure 2 provides a brief look at all 459 research publications (405 journal articles and 52 meeting abstracts) published where the word “PubChem” is used as a query. The associated research subjects were also used to survey the PubChem-related research fields. Until July 2010, there have been approximately 68 publications related to computational studies of PubChem data in the interested fields related to cheminformatics, data annotation, virtual screening and toxicity prediction research studies. There have also been a few PubChem review publications for which the reader is referred to the detailed reports on the PubChem database platform 4-6, chemical probe identities 7, 8, bioassay resources and bioactivity results 4, 9. The focus of this present review article is to give readers a most recent review of the PubChem database and the published PubChem library data-mining and virtual screening/in silico design research studies.
Figure 2. A literature survey of the research publications related to PubChem and the key research fields from 2005 to 2010.
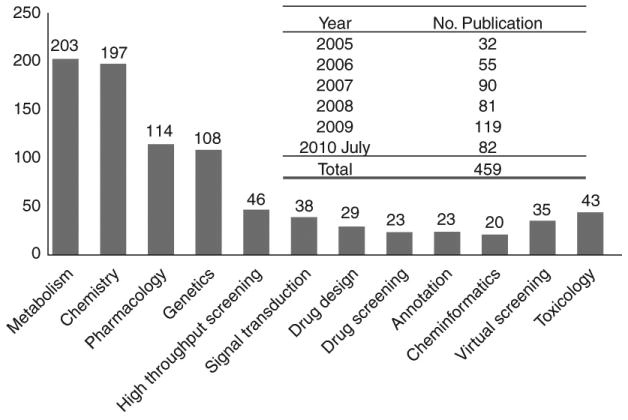
The insert table shows that the total number of the PubChem publication increases almost linearly from 2005 to now (The results were searched from SciFinder database).
2. PubChem dataset collections
Currently (July, 2010), the PubChem databases hold records for over 69 million substances (SID) containing 27 million unique chemical structures (or CID records) and 449,401 bioassays (AID). More than1.8 millions of these substances and 1.5 millions of compounds have bioactivity data in at least one of the thousands in vitro biochemical and cell-based screening assays, targeting more than 7,000 proteins and genes. The millions of compound records and bioassay data collections provide great opportunities for drug discovery research. They also, however, create a major challenge for scientists for the development of cheminformatics tools and modeling algorithms that are suitable to handle such high volumef of PubChem compound and bioactivity datasets for virtual screening and in silico drug design.
Over the past few years, PubChem developer teams, cheminformatics research centers and many researchers from academic institutes and industries have developed a variety of online tools to access and analyze the PubChem Compound and Bioassay data records, including online search, FTP, and automated access to the data through the Entrez Utilities 10. Several review and research articles 4, 6, 11-14 have reported the current cheminformatics tools available to annotate and mine PubChem database via integrating PubChem with transcriptomic, proteomic and metabolomic datasets and ultimately translate chemical genomics screening data into information that will be benefit biomedical scientists and clinician who do not have extensive training in cheminformatics. The following sections cover the PubChem data sources and download information as well as how to construct high-quality structurally diverse compound libraries from PubChem database for virtual screening studies.
2.1. PubChem data sources and download formats
For computational chemistry, one of the important steps is to obtain reliable datasets for computer modeling studies, which requires an understanding of the biological targets within the data and knowledge of the data source. The PubChem Bioassay datasets available for cheminformatics modeling studies were deposited by NIH-funded screening centers with data records of 3.95 million substance counts (79.3% of the total substance counts) and 2315 bioassay counts (0.5% of total bioassay counts) 1, by European Bioinformatics Institute (ChEMBL) with data records of 551,496 substance counts (11% counts) and 446,639 bioassay counts (99.4%) 15, and also contributed by more than 40 of US agencies and various academic institutions and individual research laboratories (Figure 1).
Specifically, human tumor cell line screening data were deposited from the Developmental Therapeutic Program (DTP) 16 at the US National Cancer Institute (NCI), toxicology data from the DSSTox 17 program at the US Environmental Protection Agency (EPA), neurobiology and anticonvulsant data from the US National Institute of Neurological Disorders and Stroke (NINDS), Approved Drug Screening Program (ADSP) and the US National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP), high-throughput screening results from ChemBank 18, and target profiling and phenotypic assays from commercial vendors. In addition, a variety literature-extracted ligand-protein binding and bioactivity data are from the BindingDB 19, the IUPHAR 20, and the PDBBind 21 projects etc.
To date, PubChem BioAssay has biological activity data for more than millions of unique small molecule chemical structures and tens of thousands of siRNA probes annotated for several thousand different protein and gene targets from thousands of biochemical and cell-based bioassays. For a given PubChem Bioassay, all related data objects are stored in ASN.1 format (gzipped) and also available in XML and CSV file formats under Microsoft SQL relational database server with optimal database architecture for efficient storage, tracking and fast retrieval of large-scale biological test results. PubChem BioAssay datasets can be downloaded free via the PubChem FTP site 22. Assay data table and descriptions can also be retrieved and downloaded through a programmatic interface using the PubChem Power User Gateway (PUG/SOAP) facilities 4, 23. The various query functions are available for searches of compounds, substances, or bioactivity data information using either text or structure queries. Text searches include compound names, molecular formulas, keywords, descriptors or MeSH terms, etc. Structure searches include identity or similarity search as well as substructure or superstructure query with online structure-drawing tool, or using SMILES, SMARTS or InChI as a structure identifier (Figure 1). Users can search using any combination of Entrez and PubChem search tools, and then download data for further analyses and computational studies.
The various download export formats for chemical structures include SDF, SMILES, XML, InChI, images and ASN.1. Note that ASN.1 (Abstract Syntax Notation One) is a binary format. NCBI utilizes a textural description translated from ASN.1 as the PubChem native archive data format that is both computer and human readable 6. All other data formats (such as SDF) are converted from the original ASN.1. SDF file format is the industry standard 24 for conveyance of chemical structure information. SDF format, however, does not provide all aspects of the ASN.1 data and may not contain all archived information in ASN.1 data format. For the extensive description of PubChem data structure systems as well as annotation and analysis tools that link phenotypic outcome to the chemical structures of molecules screened, the reader is referred to the comprehensive description articles published by S. Bryant and NCBI scientists 4-6, 25. Furthermore, the quantitative assessment of the PubChem and other public databases as well as commercial databases of bioactive compounds was reported in a recent review 26.
In addition to the full records of PubChem compound library, several laboratories have developed cheminformatics tools to generated structurally diverse or 3D shape diverse sublibraries from the large PubChem database 27-29. These subset libraries are much smaller but representative to the parent library, showing minimum similarity and redundancy. Xie et al. at the Pittsburgh Molecular Library Screening Center (PMLSC) reported their studies on data-mined PubChem using a chemistry space based compound profiling algorithm to create a representative subset of compounds from PubChem 27. The total number of compounds was reduced to 540,000 from approximately 5.3 million to make in silico or in vitro screening of PubChem more manageable 27. In addition, 3D shape topology diversity 28, 29 and scaffold topology diversity 30 analyses were also applied to modeling PubChem chemical library, which are reviewed below.
2.2. PubChem representative subsets
PubChem as a valuable public molecular information resource repository has attracted many cheminformaticians and biologists to carry out in silico or in vitro high throughput screening (HTS) to identify novel hits with new chemical scaffolds and high affinity but low toxicity. One of the challenges for the scientists is to handle the multiple millions of compounds. It may be unrealistic to conduct direct HTS experiments to screen multi-millions of compounds in PubChem for each biological target on a weekly or monthly basis. For example, the NIH MLP only requires MLPCN comprehensive centers to perform 20 assays per year for screening about 300,000 compounds and then deposit the screening data into the PubChem database. Of course, modern HTS technologies can now screen millions of compounds more quickly and cheaply at costs much less than earlier HTS methods31 However, at screening rates of 300,000 compounds/week, it may still be a challenge for an academic institution to support on a weekly basis without the major funding support to cover the costs of HTS programs and resources as well as the assays development. Overcoming these limitations requires innovative approaches, either through development of fast and low-cost HTS experiments or by employing a cheminformatics approach to taper down the large compound library without losing its information of original molecular properties as well as structural diversity. A few of such representative sub libraries have been reported as discussed below.
2.2.1. Structurally-diverse representative subsets from PubChem
Xie et al. 27 have established a computational method of building representative sub-libraries from large PubChem compound database by combining a partitioning cell-based BCUT metric algorithm with pair-wise 2D fingerprint similarity search as illustrated in Figure 3. In their studies, they applied the established diversity analysis method to generate a representative sublibrary with ∼10% of the size of the parent compound library. Their results show the new subset has minimum similarity and redundancy, but greater structural diversity, and no loss of the molecular properties based upon distribution analyses of HB donor/acceptors, rotatable bonds, hydrophobes, MW, and logP, relative to the parent library PubChem 27. The generated representative subset (rePubChem) is made available to the cheminformatics community through former PMLSC websites.
Figure 3.
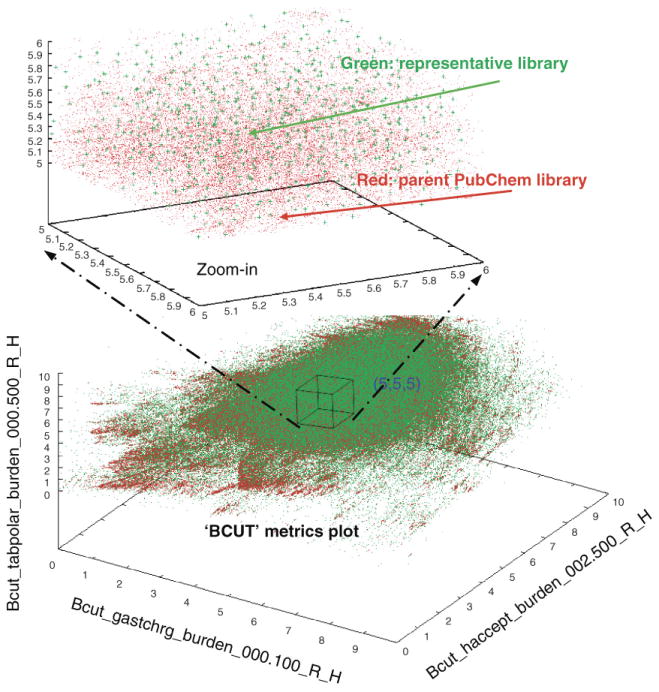
3D chemistry-space matrix plot of a representative sublibrary (green dots) created from the parent library PubChem database (red dots) by using the Diversity Analysis method based BCUT metrics calculation.
Xie's laboratory has further developed the representative subset selection algorithm into a compound library profiling (CLP) method, and applied it to perform diversity analysis of newly synthesized cyclic ether compound library in comparison to the existing PMLSC compound collection and the PubChem database32, and also to characterize the diversity attributes of the synthesized bicyclic β-benzyloxy and β-hydroxy amide library in comparison to NIH small molecular repository (SMR) to which these new compounds are deposited 33. In these publications, the developed compound library profiling (CLP) algorithms provides valuable cheminformatics data mining tools to evaluate whether the newly synthesized compounds contribute to increase the diversity value of the existing NIH SMR compound libraries or commercial chemical library. It is anticipated that these methods and compound subsets will be valuable to a broad scientific community interested in acquiring/synthesizing structure-diverse compounds for efficient drug screening, and for more general applications requiring representative virtual compound libraries.
Additionally, Xie et al have applied the BCUT-based chemistry-space matrix calculation algorithm established above to build target-focused sub-libraries based on the few known active leads identified by PMLSC HTS experiments. Such a knowledge-based “cherrypicking” approach used a few known active compounds (or the corresponding chemistry-space matrices) to select additional compounds that have similar chemical-space matrices, and then cluster them to build target-focused sub-libraries. In these studies, the unbiased modeling of PMLSC datasets has demonstrated that the clustered libraries generated by this approach have hit rates remarkably better than classical HTS experiments as shown in Figure 4 (unpublished data). 3D diversity matrix plot shows the parent library (green dots: 65K compounds distributed to PMLSC from DPI) (Discovery Partners International, BioFocus Inc) and a generated sublibrary (targeting West Nile Virus NS2bNS3 Proteinase, NS2B) (blue dots: 220 compounds), computed by chemistry-space BCUT metrics diversity analysis approach 27. As shown in Figure 4, the focused library (220 compounds) (blue dots) were generated based on the two compounds known to bind NS2B (yellow dots) that were originally identified from a representative subset of 9013 compounds (above). There are 11 hits (red dots) in the focused library that match 11 of the 15 hits from HTS experiments on the full 65K compound library. The focused library thus gives a 5% (11/220) hit rate, which is 250 times higher than the 0.02% (15/65k) hit rate of the initial HTS experiments. In addition, the focused subset provides a concentrated subset for the secondary HTS experimental screening to further examine any possible false negative hits.
Figure 4. 3D matrix plot of the NS2B-focused sub-library (blue dots: 220 compounds) selected from the parent library (green dots: 65K compounds from PMLSC).
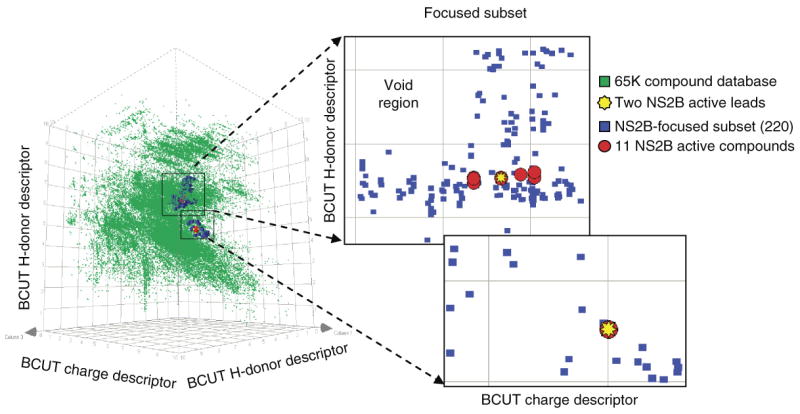
The focused subset was generated based on the two known NS2B-active leads (yellow dots) from the representative subsets (9013 compounds) by diversity analysis approach using cell-based chemistry-space BCUT metrics calculation. The focused library gives 5% hit rate (red dots: 11), which 250 times better than HTS experiments.
2.2.2. Other diverse sublibraries from PubChem
In addition to the chemistry space matrix compound profiling algorithm to mine PubChem library above, Fontaine et al. reported a 3D shape fingerprint similarity selection method using ROCS shape overlay comparison algorithm to mine PubChem database 28. In their studies, approximately 1.04 million PubChem compounds were obtained with filtering criteria: heavy atoms <28, rotatable bonds <6, and removing the structures with incomplete stereochemistry, ionized forms. Then, a set of a few thousands diverse structures was generated using 3D shape Tanimoto similarity (Tc) values of 0.75, and also analyzed under different 3D shape Tc values of 0.8 and 0.85. These calculated shape diverse subsets cover entirely the 3D shape space of the conformers of the 1.04 million PubChem compounds. Similar work 29 was also reported for assessment of conformational space of PubChem compounds by using conformation generation program Omega 34 and regression equation prediction as a function of RMSD.
The advantage of the above 3D shape overlap subset library selection methods is that it allows visualization of the superimposed compounds and a better understanding of the compound similarity. For millions or billions of conformers, however, 3D alignmentbased shape similarity searches in comparing with 3D shape fingerprint similarity method demands substantial computing capabilities and modeling resources in addition to the pre-computation requirements.
Furthermore, Wester and Oprea have reported their work using scaffold topologies to model various datasets from PubChem and DSSTox for toxicity studies 30. In their studies, they compared the results of different algorithms including the ring scaffold topology distribution in comparing to coarser-grained classification against several databases, including ChemNavigator (commercial chemicals), DNP (the Dictionary of Natural Products), WOMBAT (medicinal chemistry compounds with known bioactivity), and two subsets “active” from PubChem and DSSTox (toxic compounds) as well as GDB11 (General Database of Chemical Space of virtual small organic molecules with major atoms less than 11 or MW less than 160 Da)35. The topological results show that nearly ¾ of the scaffolds of toxic substances have two or less rings but 25% of DSStox and 4% of the PubChem actives do not contain rings (note: the DDSTOX chemical-index files have now been deposited into PubChem under “PubChem Substance”). The maximum topological diversity is observed in PubChem and 55% of PubChem's have less or equal to 8 ring size topologies.
In general, the subset chemical library will allow biologists and computational chemists to efficiently screen the sets of compounds that are small enough to be tractable, yet representative of the full set. Hits obtained from such a representative library can then be used to rationally parse the parent library, and generate a compound cluster or focus (similar compounds or analogs) for further bioassay validation, resulting in the rapid development of informative structure-activity relationships (SAR). The basic idea underlying the diverse subset selection strategy is based on a central premise of medicinal chemistry that structurally similar molecules have similar physicochemical properties and possibly similar biological activities, which premise has been enunciated in many computational chemistry and similarity-based virtual screening drug design studies.36-38 Of course, very similar molecules may in some cases possess very different activities, so called activity cliff that is defined as the ratio of the difference in activity of two compounds to their distance of separation in a given chemical space. The detailed discussions of outliers or activity cliffs are beyond this review but available in literature 39, 40.
3. PubChem Benchmark Datasets and Virtual Screening Models
3.1. Opportunities and challenges of modeling PubChem library
There are many successful virtual screening methods reported for chemical probes or drug leads discovery, either based on ligand pharmacophores or receptor docking approaches 41-45. However, one of the basic preconditions to develop virtual screening approach for a drug target or conduct in silico validation studies is to ensure the availability of reliable bioassay datasets. The dataset is often randomly divided into training set and testing set, each may consist of known numbers of active and inactive data. The known datasets are used to assess the virtual screening methods according to their capability to separate the actives from inactive in the final ranking, or calculate the enrichment factor of the different in silico methods. Thus, dataset selection and model training/validation are another important step for virtual screening in addition to construct high-quality structurally diverse compound library as reviewed in the previous section.
As discussed above, the PubChem bioassay collection has rich data information. It provides great opportunities and also challenges for computational studies to reveal relationships between chemical structures and biological activities in order to assist virtual screening and computer-aided drug design. A common exercise for dataset extraction from PubChem is to first extract all bioassays that have defined the specific protein targets from PubChem and then extract the datasets that were screened by primary bioassays and also confirmed by secondary dose-response bioassays (Ki or EC50).
However, like other HTS/HCS screening experiments, the PubChem bioassay data may also suffer from the experimental noise and artifacts, such as false positives or false negatives. For example, bioassays are prone to a range of artifact caused by unspecific binding activity of the screened chemical compounds, such as off-target or cytotoxic effects in cell-based receptor bioassays 46. Thus, cautions should be taken when extracting bioactivity data from PubChem for computational studies. Usually, the secondary and confirmatory bioactivity data should be used in the computer modeling. In addition, the datasets extracted from PubChem or from the literature may also suffer from issues of compound analogues bias that are often prone to over-representation of certain scaffolds or chemical entities 47. The analog bias issue may cause overoptimistic estimates of virtual screening performance.
Thus, it is essential and important to have trust-worthy bioactivity datasets extracted from PubChem in order to develop reliable predictive models for virtual screening and method development in terms of performance assessment and algorithm validation.
While PubChem curators do check assay depositions, however there is no way to completely avoid erroneous data in PubChem as HTS data is prone to experimental noise. To deal with these complications of the large bioassay data in PubChem, several research laboratories have developed various cheminformatics tools and filtering methods to construct so called benchmark datasets by careful selection of the raw data sets from PubChem. These subsets will be great value for library design and virtual screening method development and validation. These reported studies include: developing predictive decision tree models from HTS data in PubChem 48, data mining PubChem using support vector machine (SVM) for inhibitor or ligand classifications 49-54 and aggregator identification 55, establishing GPU accelerated SVM for mining HTS data 56, and developing naïve Bayesian predictive models from PubChem Bioassay datasets 13. Figure 5 illustrates a Web-interfaced machine learning algorithm based ligand activity predictor and function classifier that were developed by Xie et al to predict active/inactive ligands and agonist/antagonist of 5HT1A using naïve Bayesian and SVM algorithms, respectively 57. The prediction models were constructed from over 1600 of known 5HT1A ligand datasets from the PubChem database (data source from GLIDA 58). These established data-mining algorithms provide valuable statistical approaches to mine large bioassay datasets for virtual screening. Ideally, with the high volume biological data from PubChem, cheminformaticians and computational chemists/biologists can use the unbiased datasets to develop and evaluate in silico drug design algorithms and methods for the best virtual screening performance across a range of datasets from PubChem. A few examples of the extracted PubChem datasets and cheminformatics studies are discussed below.
Figure 5. Web-interfaced 5HT1A ligand activity and function prediction server.
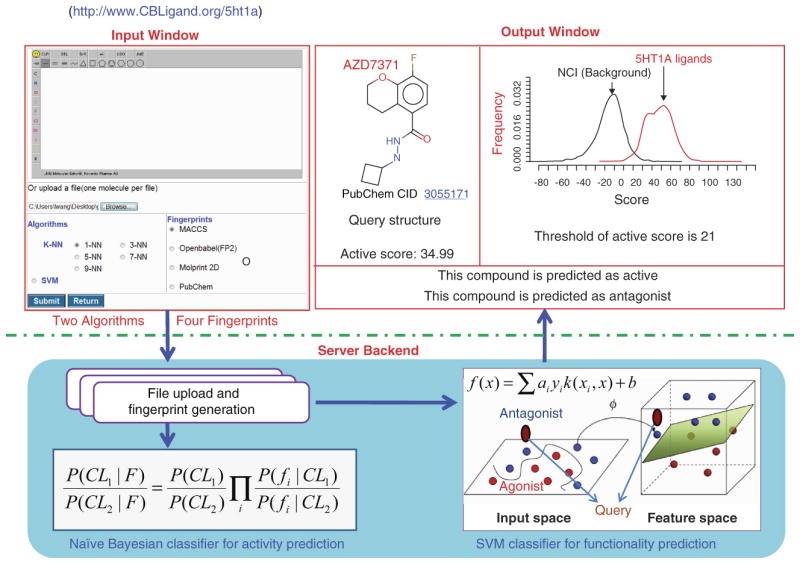
Input widow has online upload or structure drawing functions, four fingerprint generators and choice of prediction algorithms. Backend in server has built in file format conversion as well as naïve Bayecian classifer for ligand activity prediction and SVM classifier for ligand function prediction functions. Output window displays the result of query compound AZD7371 (PubChem CID 3055171), showing that the compound is predicted to be an active 5HT1A ligand with antagonistic function to the 5HT1A receptor. The prediction models were modeled from known 1600 of 5HT1A ligands from PubChem database (GLIDA depositor).
3.2. Benchmarking of the PubChem datasets and virtual screening models
Roherr and Baumann 59 have reported their cheminformatics research work on developing the maximum unbiased validation (MUV) datasets for virtual screening. The work was based on PubChem bioassay data by using refined K nearest neighbor (KNN) analysis algorithm. In their studies, all datasets and the chemical space samples were encoded by “simple” descriptors that are a vectorized form of the respective counts of all atoms in each molecule. The descriptors include the number of H-bonding acceptors and H-bond donors, the logP, the number of chiral centers and the number of ring systems 59. Subsequently, a workflow of topological optimization using MUV dataset design strategies, monitored by refined nearest neighbor analysis functions, was established to generate corresponding datasets of actives and inactives from PubChem. From the bioactivity data available in PubChem BioAssay, 18 subsets of bioactivity data, which were primarily screened and then confirmed by dose response assays, were extracted against 18 pharmaceutically relevant targets, and each dataset consists of 30 actives and 15000 inactives. The authors concluded that these benchmarked datasets are unbiased with regard to analogue bias and artificial enrichment, which can be used to maximize the unbiased validation of virtual screening methods. Their benchmarked unbiased datasets generated from PubChem and the associated statistics analysis tools are available for download at author's website 59.
Furthermore, Chen and Wild 13 have generated a Bayesian predictive models using 1133 bioassays in 2008 from the PubChem database. Using their workflow built by Pipeline Pilot package 14, the naïve Bayesian predictive model was built with the FCFP_6 circular substructural fingerprints 60 and the molecular descriptors encoded structural features and properties, including molecular weight, logP, number of H-bond acceptor and donors, the number of rotatable bonds. The developed models were accessed using Leave-One-Out validation (or internal validation) and the rational division of training and testing datasets validation (external validation) 61. Their studies showed that these predictive models are reasonably accurate by identifying high number of hits with the enrichment factor 3.6 and 5.7, which is much better than either similarity search or random screening. It is a good practice to build the predictive models using a rich and diverse compound datasets to ensure the development of a generalizable model with better accuracy. The variability in the accuracy (ROCV = 0.582–0.995, mean 0.881) is, however, still greater than that for models built using traditional QSAR data (ROCV = 0.985–0.998, mean 0.992)13. Thus, future work is necessary to improve the accuracy of predictive model by introducing a more diverse inactive set as baseline.
Additional machine learning algorithm was also applied to mine PubChem datasets. Weis et al. 49 established a support vector machine (SVM)-based classification algorithm to screen and identify the Factor XIa inhibitors from over 12 million compounds in PubChem database. In their studies, a support vector machine (SVM) classifier was trained to develop a predictive model using the Signature molecular descriptor on Factor XIa inhibitor HTS data. The resulting model had a 10-fold cross-validation accuracy. To further evaluate compounds identified as active by the SVM, docking studies were performed using AutoDock to generate a focused subset of compounds predicted to be active. It is anticipated that the established data mining technique for factor XIa inhibitor identification could also be applied to other bioassays in PubChem for identification of chemical probes.
As shown above, machine learning and statistical inference have provided alternative solution to model millions PubChem datasets and to develop predictive models for virtual screening. While these methods still need improvement, they do demonstrate robustness and predicting power to handle large amount of datasets from PubChem.
3.3. Virtual Screening based on the imbalanced bioassay data in PubChem
In addition to the given very large, high volume and complicated datasets mentioned above, another challenge to mining PubChem for virtual screening is the highly imbalanced nature of the PubChem data with only a small number of active compounds compared to inactive compounds. To deal with this issue, Bryant et al. reported a method for mining these highly imbalanced HTS data in PubChem51. In their work, the granular support vector machine (gSVM) repetitive under sampling method (gSVM-RUS) 62 and PubChem fingerprints 63 were used to build predictive models using the luciferase inhibition bioassay data that has the imbalanced ratio of active/inactive (1/377). The best predictive model developed showed hit rate of recognizing the active and inactive compounds at the accuracies of 86.6% and 88.9% with a total accuracy of 87.7% by cross-validation test and blind test, respectively. Their results demonstrated the robustness of the gSVM-RUS based predictive model in efficiently computing the highly imbalanced HTS data. It is anticipate that the developed gSVM-RUS algorithm may also help HTS assays such as luciferase-based HTS to develop a computational model to screen and identify potential interference compounds for the HTS assays.
4. PubChem Toxicology Prediction Modeling
4.1. Challenges for modeling of limited toxicity data from PubChem
Identification of hits or leads from HTS or virtual screening approaches is important first step. However, many screened compounds entering clinical studies do not survive through the numerous hurdles as a good pharmacological lead to be a drug on the market. Among many causes for attrition that have been studied, it has been noted that earlier attention to compound quality related to physical chemistry, drug metabolism and pharmacokinetics (DMPK), and toxicology/safety is necessary and important. In PubChem bioassay collection, cell viability studies as an indication of general cellular toxicity were also carried out using HTS cell proliferation experiments by measuring ATP concentration 64. The study attempts to correlate generic cell-proliferation to chemical features by analysis of cell proliferation results from different cell lines. Ideally, the development of predictive cellular toxicity models would be useful cheminformatics tools to mine the PubChem data in order to reliably predict compounds whether they are toxic or not. Here, the reviewer would like to point out that such generic cell proliferation assays do not address any specific mechanisms or targets involved in toxicity. It only gain some mechanistic insights by analysis of cell proliferation results from different cell lines in attempts to correlate generic cell proliferation to chemical features 65.
For modeling PubChem data for toxicology and pharmacology studies, there are several ADME/Tox databases available free or commercial on the web that can be used to facilitate computational toxicology model building and assistant drug design 66. A number of computational toxicology approaches has been developed and reported to predict whether a compounds that are toxic or have poor ADME properties, including linear regression models 67, neural networks models 68, Kohonen maps prediction model 69, Bayesian models 70, expert system models 71, QSAR models 72, target fishing technique 70 and Prediction of Activity Spectra for Substances (PASS) 73, and public sources for toxicity data review 17. These toxicology prediction methods are similar in nature but use a variety of molecular descriptors to derive predictive models either to predict LD50 values quantitatively or to perform classification of toxic or not toxic qualitatively. Here, the reviewer should point out that the PubChem toxicity data are still limited on certain species or specific classes of compounds; in particular it is still a challenge to identify toxic compounds in the absence of knowledge of toxicity mechanisms. Some of these related studies are reviewed below.
4.2. Cell toxicity predictive models from PubChem
Guha and Schuere investigated various aspects for developing computational models to predict cell toxicity based on cell proliferation screening data in PubChem 65. Based on the captured features in the datasets, several predictive models were generated to evaluate cell-based screening results and were used to identify and eliminate potentially undesired compounds. In addition, they explored the feasibility of utilizing cell proliferation data to predict animal toxicity using the datasets from PubChem and MDL databases. In their studies, human T cell (Jurkat) proliferation data were extracted from PubChem using Assay ID: AID's 364, 463 and 464, including over 60,000 data points with primary percent inhibition measured at 4 uM and about 800 IC50 data points. To investigate the generality of the workflow established, the cell proliferation quantitative HTS (qHTS) IC50 data points for 1334 compounds against 13 cell lines were extracted from various PubChem BioAssay collection (AID's). For a given cell line, a criteria was set that compounds with a pIC50 greater than the cutoff are labeled as toxic and those below as non-toxic.
To correlate the in vitro toxic prediction with the animal toxicity datasets, authors extracted the acute animal toxicity datasets from the Registry of Toxic Effects of Chemical Substances (RTECS) available through the MDL Toxicity Database (2006.2), including 103,041 chemical structures for 154,019 LD50 (mg/kg) data points (oral, intravenous, intraperitoneal, subcutaneous). A cutoff of LD50 was selected such that any molecule having a measured pLD50 of two standard deviations greater than the mean value was classified as toxic and the rest as non-toxic. To identify toxicity-indication structural pattern and derive the predictive models, Guha and Schurer used the BCI 1,052-bit structural descriptors as structural fingerprints, CATS2D descriptors as topological pharmacophoric fingerprints, and Molconn-Z real-value holistic descriptor for all three of the datasets mentioned above. Their work showed that the models generated exhibit reasonably good predictive performance on these highly imbalanced datasets from PubChem and MDL, with accuracy rates ranging from 56 to 80%. According to their data, simple structural descriptors, binary fingerprints or topological pharmacophores, do not appear to allow for a consistent discrimination between toxic and non-toxic classes. This is particularly true when comparing cell-based and animal toxicity.
Additional toxicity prediction models were also reported using PubChem datasets and DSSTox database. Edelstein et al. 74 extracted three datasets from the DSSTox database and integrated the information from PubChem and ChemBank for development of predictive toxicology models. Their studies showed that the correlated toxicology modeling can improve predictive performance over using chemical structure alone in a statistically significant way. Zhu et al. 75 also reported their work of modeling cell viability assay data to improve the prediction accuracy of conventional QSAR models of animal carcinogenicity. Their studies concluded that combining NTP-HTS profiles with conventional chemical descriptors could considerably improve the predictive power of computational approaches in toxicology.
Clearly, given no target information, modeling toxicity is complicated because of the multiple mechanisms and biological targets by which a compound may inhibit cell proliferation. Thus, it is recommended that specific mechanism related descriptors should be included, such as a variety of physicochemical descriptors (clogP, polar surface area, H-bonding, charge etc.) and bioactivity-based descriptors (biological descriptors76, target protein interaction descriptors derived from proteomics and gene expression 77, metabolism descriptors 78). These mechanism-related descriptors can help to understand specific mechanisms and possible interactions of a toxicant that may have with a biological systems.
4.3. Cardiac toxicity prediction models from PubChem
Another reported toxicology virtual screening study is the cardiac toxicity prediction model development. Li et al. have published their work on cardiac toxicity classification model using a combination of SVM and GRIND descriptors, and tested the model on a large set of hERG bioassay data from PubChem (AID:376) 50. Cardiac toxicity of drugs is a major concern in drug discovery. It often requires elimination or filtering out potential hERG channel inhibitors in an early stage of drug discovery process. In their studies, a large set of compounds (1948 compounds) with hERG activity was extracted from PubChem bioassay database (AID:376), containing 248 active and 1700 inactive compounds. Initially, docking studies were carried out using 561 molecules (495 from the training set and 66 from the testing set) on a constructed hERG homology model. Then, a binary classification prediction models were generated based on the 495 compounds using pharmacophore-based GRid-Independent Descriptors (GRIND) with a SVM classifier in order to discriminate between the hERG blockers and nonblockers. According to their studies, the models achieve an overall accuracy up to 94% with a Matthews coefficient correlation (MCC) of 0.86 (F-measure of 0.90 for blockers and 0.95 for nonblockers) 35. The models were also applied to a testing dataset of 66 compounds, showing 72% of the set was correctly predicted (F-measure of 0.86 and 0.34 for blockers and nonblockers, respectively). Finally, authors also evaluated the model on a large set of hERG bioassay data in PubChem, showing 73% accuracy (F-measure of 0.30 and 0.83 for blockers and nonblockers, respectively) and 10-20% improvement in the prediction of blockers compared to other methods. They concluded that the generated models based on GRIND descriptors and SVM classifier can be useful to filter potential hERG channel inhibitors.
In general, it is expected that the PubChem biological categorization of datasets is growing rapidly and it will become a valuable resources to advance the complicated pharmacology and toxicity prediction. In addition, more ADME data will be available from physical-chemical and ADMET in vitro assays either in the public and commercial databases or pharma companies' in-house databases. The current public toxicity databases are: DrugBank 79 (>4800 drug entries related to over 2500 non-redundant target proteins), Environmental Protection Agency's (EPA) Distributed Structure-Searchable Toxicity (DSSTox) Database 80 (over 10,000 unique chemicals), new EPA Aggregated Computational Toxicology Resource (ACToR) database 81 (over 300 chemicals pertaining to environmental toxicology). The commercial preclinical ADME/Tox databases include: Symyx database (metabolite and toxicity data) 82, Aureus AurSCOPE ® ADME/DDI database 83(drug-drug interaction and metabolic properties of drugs), PharmaPendium online resource 84 (FDA approved drug data and EMEA EPAR approval documents). These databases in combining with datasets from PubChem will provide rich molecular toxicology information for cheminformatics and virtual screening.
5. PubChem across-target polypharmacology network modeling
One of the important features of the PubChem data collection is that it reports the activity of compounds across multiple Bioassays, which allows to mining across-target bioactivity and study polypharmacological behaviors in the PubChem collection via cross-assay analysis studies. An example is given in Figure 6 to show that a PubChem Heatmap and clustering graphs displaying the cannabinoid ligand CP55940, a known analgesics and immunosuppressive agent, together with their biological test results that were obtained from HTS experiments against a group of related protein Targets. The compound CID104895, a stereoisomer of CP55940, is a a known potent GPCR ligand that has nanomolar binding affinity to cannabinoid receptors. As shown Figure 6, the compound CID104895 also shows across-target bioactivities to the other GPCRs proteins, including weak binding (green color: 10-100 uM range) to neuropeptide S receptor isoform A (target identification, GI#46395496: a G-protein coupled receptor for asthma susceptibility) and relative strong binding (yellow color 0.1-1 uM) to thyroid simulating hormone receptor (target identification, GI#38016895: another GPCR). The across-target multiple bioactivity data were measured by NIH NCGC using PubChem BioAssays AID1461 and AID926, respectively. More studies were reported on using PubChem data and online analysis tools to survey selectivity and across-target bioactivities of small molecules as discussed below.
Figure 6.
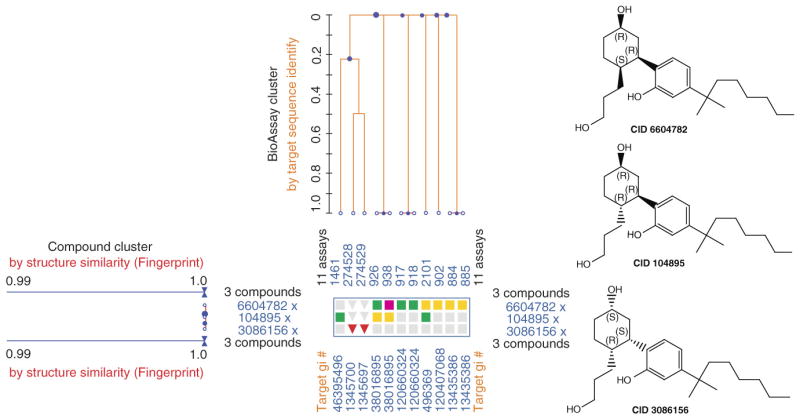
A graph of PubChem Heatmap and BioAssay Cluster showing three isomers of the compound CP55940 and the correspondent biological test results across multiple targets that were measured from HTS experiments against a group of related protein targets. The three stereoisomers are represented as PubChem Compound identifier ‘CID’ and the structures at the right sides of heatmap. The three compounds were searched based on 2D structure similarity showing a similarity value of 1.0 (displayed at the left side of the heatmap), indicating they are identical structure but stereoisomers. Bioassay Clusters of the 11 assays (represented as PubChem BioAssay identifier ‘AID’) were derived based on the sequence similarity of the tested protein targets, where the GenBank identifiers of the corresponding protein targets (gi#) are listed at the bottom of the heatmap view. Each cell in the Heatmap represents an individual activity outcome of a small molecule for the corresponding target, with ‘active’ results denoted by red, yellow or green color, and ‘inactive’ results denoted by blue color.
Chen et al. reported their modeling studies using PubChem as a data source to establish polypharmacology networks 85 in order to address the issue of high attrition rates arising from lack of efficacy and high toxicity. In their work, sets of data were extracted from PubChem bioassays collection, containing 602 bioassays for 506,190 distinct compounds, of which 90,290 compounds were active in at least one assays. The assays represented 258 unique protein targets, and each target was tested in multiple assays, whereas the assays that did not have an associated target were ignored. Then, authors derived a network representation of these assays collection and applied a bipartite mapping between the assays network and various biological pathways network as well as artificial drug-target network. The results demonstrated that mapping to a drug-target network can be allowed to prioritize new selective compounds, whereas mapping to other biological networks can be used to observe interesting target pairs detected in PubChem cross-assay analyses and the corresponding compounds in the context of biological systems.
Another similar cross-assay analysis study was reported by Han et al 9 regarding a datamining survey of across-target bioactivity results of small molecules in PubChem. In their report, two alternative target-grouping approaches were used to examine a compound's across-target bioactivity against 588,918 compounds under 660 bioassays. The established non-redundant target-based compound analysis methods revealed compounds that are selectively active against groups of protein targets that have identical or similar sequences. The target clustering analyses identified compounds that are bioactive across unrelated targets. One of such target compounds studied is myricetin (PubChem CID:5281672), or a flavonoid commonly found in natural food source. The compound was identified by PubChem across-target analysis as an inhibitor of multiple target proteins such as aldehyde dehydrogenase, Leishmania mexicana Pyruvate Kinase, Cytochrome P450, Stress-activated protein kinase and human RNase H, with a strong potency (IC50 <10 uM).
Clearly, PubChem provide bioactivity report via launching Entrez Bioassay Summary for scientists to examine and compare biological outcomes across multiple biological tests, which allows to view all active compounds across each BioAssay. The reviewer believe that data-mining analyses of bioactivity profile across a wide range of biological targets in PubChem provides promising statistical models to evaluate target specificity of promiscuous compounds for their selectivity and across-target properties. The systematic studies of polypharmacological behaviors in the PubChem collection via cross-assay analyseswill also offer better understanding of the biological mechanisms of ligand and protein interactions.
6. Expert Opinion
In the reviewer's opinion, PubChem is a powerful small molecule information repository and has valuable features and multiple functions with advantages over other available public or commercial databases of bioactivity data. First, all compound structures, bioassay conditions and experimental readouts are publicly accessible online. Second, most of the compound collections tested in each bioassay were already pre-selected for maximizing structural diversity and acquisition of the “druglike” properties. Third, all tested compounds, including both actives and inactives for each bioassay are archived in the database and available for structure-activity analyses. Most importantly, millions of unique small molecule chemical structures and tens of thousands of siRNA probes were biologically annotated by millions of biological activity outcomes. In PubChem, these compounds were measured using thousands biochemical and cell-based bioassays for different protein and gene targets. In addition, PubChem Substance, Compound, and BioAssay databases are fully integrated within NCBI's Entrez data retrieval system 86. With the PubChem Power User Gateway (PUG) programmatic interface 23 and the Entrez Programming Utilities (eUtils) 10, one can perform automated chemogenomics analysis of the tested compounds and their bioactivities by correlating with the target proteins or DNA information87 as well as other database resources25.
However, PubChem data also suffers from the typical tendency of HTS/HCS assays for false positives and negatives as well as many highly imbalanced datasets. Cautions should be taken in utilizing these datasets as it may affect the virtual screening baseline noise and deviate the virtual screening prediction accuracy. It is also noticeable that PubChem toxicity HTS data is still limited to certain species or specific classes of compounds although more in vitro cellular toxicity data are becoming available in PubChem. Thus, it is still challenging to identifying toxic compounds in the absence of knowledge of toxicity mechanisms. It should be also noted that the in contrast to animal toxicity studies, the generic cell proliferation assays do not address any specific mechanisms or targets involved in toxicity. The cell-based toxicity studies, however, can be very cost-effective and suitable for high-throughput screening.
Taken all together, PubChem datasets can be used as a rich source to aid developing predictive models for cheminformatics and virtual screening for in silico drug design studies as reviewed above. Of course, one will not expect the accuracy to be as high as for high-quality experimental sets. On the other hand, it is always recommended to make sure that models are built using a rich enough diversity of compounds for a generalizable model to be derived. It is also notable that the reported machine-learning derived models, as reviewed above, would be appropriate for virtual screening (or pre-screening) in order to reduce the number of compounds that need to be experimentally screened from large datasets like PubChem, but not for prediction on small sets that require very high levels of accuracy. Overall, the public accessibility to such HTS/HCS assay data is particularly valuable to the scientific community, since this kind of critical information needed by drug discovery research is typically held by pharmaceutical companies. The open-access and information-rich repository will make PubChem an even more valuable and powerful public resource for cheminformatics data-mining and virtual screening as well as biomedical and drug discovery research in the future.
Article highlights box.
PubChem is a database of chemical molecules, a component of NIH's Molecular Libraries Roadmap Initiative.
PubChem provides information on the biological activities of small molecules from a multitude of depositors. Currently, it has 69,170,468 entries of substances (SID), 27,443,646 records of unique chemical structure compounds (CID), and 449,401 records of Bioassays (AID).
Multiple million records of PubChem datasets provide great opportunities and also challenges for developing cheminformatics tools and modeling algorithms that are suitable to handle high volumes of PubChem compound and bioactivity records for virtual screening and in silico drug design.
As other HTS/HCS, PubChem bioactivity data may also suffer from the experimental noise and artifacts, such as false positives or false negatives. It is important to have confirmatory bioactivity datasets extracted from PubChem in order to develop reliable predictive models for virtual screening method development and validation
Novel data-mining cheminformatics tools and virtual screening algorithms are being developed and used to retrieve, annotate and analyze the large-scale and highly complex PubChem bioactivity data in order to facilitate computer-aided drug design.
Acknowledgments
The author would like to thank Dr. Billy Day for reading this manuscript.
Declaration of Interest: XQ Xie has received funding support for his laboratory from the NIH (R01DA025612 and P50 GM067082).
7. Annotated bibliography
Papers of special note have been highlighted as either of interests (*) or of considerable interest (**) to readers.
- 1.MLPCN. Molecular Libraries Probe Production Centers Network (MLPCN), NIH Molecular Libraries Program (MLP) cited; Available from: http://mli.nih.gov/mli/mlpcn/mlpcn/
- 2.NCBI. NIH, National Center for Biotechnological Information (NCBI) cited; Available from: http://www.ncbi.nlm.nih.gov/
- 3.PubChem-Sources. PubChem Substance and Bioassay data source information website. cited; Available from: http://pubchem.ncbi.nlm.nih.gov///sources/
- 4**.Wang Y, Bolton E, Dracheva S, Karapetyan K, Shoemaker BA, Suzek TO, et al. An overview of the PubChem BioAssay resource. Nucleic Acids Res. 2010 Jan;38(Database issue):D255–66. doi: 10.1093/nar/gkp965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5**.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009 Jul 1;37(Web Server issue):W623–33. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: Integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–41. [Google Scholar]
- 7.Huryn DM, Cosford NDP. The molecular libraries screening center network (MLSCN): identifying chemical probes of biological systems. Annu Rep Med Chem. 2007;42:401–16. doi: 10.1016/S0065-7743(07)42026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oprea TI, Bologa CG, Boyer S, Curpan RF, Glen RC, Hopkins AL, et al. A crowdsourcing evaluation of the NIH chemical probes. Nat Chem Biol. 2009 Jul;5(7):441–7. doi: 10.1038/nchembio0709-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Han L, Wang Y, Bryant SH. A survey of across-target bioactivity results of small molecules in PubChem. Bioinformatics. 2009 Sep 1;25(17):2251–5. doi: 10.1093/bioinformatics/btp380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Entrez. NIH NCBI Entrez Programming Utilities (eUtils) cited; Available from: www.ncbi.nlm.nih.gov/entrez/query/static/eutils_help.html.
- 11.Rosania GR, Crippen G, Woolf P, States D, Shedden K. A cheminformatic toolkit for mining biomedical knowledge. Pharm Res. 2007 Oct;24(10):1791–802. doi: 10.1007/s11095-007-9285-5. [DOI] [PubMed] [Google Scholar]
- 12.Guha R, Gilbert K, Fox G, Pierce M, Wild D, Yuan H. Advances in cheminformatics methodologies and infrastructure to support the data mining of large, heterogeneous chemical datasets. Curr Comput-Aided Drug Des. 2010;6(1):50–67. doi: 10.2174/157340910790980115. [DOI] [PubMed] [Google Scholar]
- 13**.Chen B, Wild DJ. PubChem BioAssays as a data source for predictive models. Journal of Molecular Graphics and Modelling. 2010;28(5):420. doi: 10.1016/j.jmgm.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Hassan M, Brown RD, Varma-O'Brien S, Rogers D. Cheminformatics analysis and learning in a data pipelining environment. Molecular Diversity. 2006;10(3):283–99. doi: 10.1007/s11030-006-9041-5. [DOI] [PubMed] [Google Scholar]
- 15.ChEMBL. ChEMBL is a database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data). European Bioinformatics Institute (EBI), European Molecular Biology Laboratory (EMBL) http://www.ebi.ac.uk/chembldb/ [cited; Available from: http://pubchem.ncbi.nlm.nih.gov//sources/sources.cgi?mode=contact&dsn=ChEMBL.
- 16.Driscoll JS. The preclinical new drug research program of the National Cancer Institute. Cancer treatment reports. 1984;68(1):63–76. [PubMed] [Google Scholar]
- 17*.Richard AM, Gold LS, Nicklaus MC. Chemical structure indexing of toxicity data on the internet: moving toward a flat world. Curr Opin Drug Discov Devel. 2006 May;9(3):314–25. [PubMed] [Google Scholar]
- 18.Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, et al. ChemBank: a small-molecule screening and chemoinformatics resource database. Nucleic Acids Res. 2008;36(Database Iss):D351–D59. doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35(Database Iss):D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harmar AJ, Hills RA, Rosser EM, Jones M, Buneman OP, Dunbar DR, et al. IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Res. 2009;37(Database Iss):D680–D85. doi: 10.1093/nar/gkn728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R, Fang X, Lu Y, Wang S. The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. Journal of Medicinal Chemistry. 2004;47(12):2977–80. doi: 10.1021/jm030580l. [DOI] [PubMed] [Google Scholar]
- 22.PubChem-FTP. NIH PubChem FTP site. cited; Available from: ftp://ftp.ncbi.nlm.nih.gov/pubchem/Bioassay.
- 23.PubChem-PUG. Pubchem Power User Gateway (PUG) cited; Available from: ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_pug.pdf.
- 24.SDF. SDF file format is the industry standard (http://www.symyx.com/downloads/public/ctfile/ctfile.jsp) for conveyance of chemical structure information.
- 25*.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010 Jan;38(Database issue):D5–16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Southan C, Varkonyi P, Muresan S. Quantitative assessment of the expanding complementarity between public and commercial databases of bioactive compounds. Journal of Cheminformatics. 2009;1(1):10. doi: 10.1186/1758-2946-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Xie XQ, Chen JZ. Data mining a small molecule drug screening representative subset from NIH PubChem. J Chem Inf Model. 2008 Mar;48(3):465–75. doi: 10.1021/ci700193u. [DOI] [PubMed] [Google Scholar]
- 28*.Fontaine F, Bolton E, Borodina Y, Bryant S. Fast 3D shape screening of large chemical databases through alignment-recycling. Chemistry Central Journal. 2007;1(1):12. doi: 10.1186/1752-153X-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borodina YV, Bolton E, Fontaine F, Bryant SH. Assessment of Conformational Ensemble Sizes Necessary for Specific Resolutions of Coverage of Conformational Space. J Chem Inf Model. 2007;47(4):1428–37. doi: 10.1021/ci7000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wester MJ, Pollock SN, Coutsias EA, Allu TK, Muresan S, Oprea TI. Scaffold topologies. 2. Analysis of chemical databases. J Chem Inf Model. 2008 Jul;48(7):1311–24. doi: 10.1021/ci700342h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armstrong JW. A review of high-throughput screening approaches for drug discovery. American Biotechnology Laboratory; 1999. Apr, pp. 26–27. [Google Scholar]
- 32.Mao S, Probst D, Werner S, Chen J, Xie X, Brummond KM. Diverging Rh(I)-catalyzed carbocylization strategy to prepare a library of unique cyclic ethers. J Comb Chem. 2008 Mar-Apr;10(2):235–46. doi: 10.1021/cc7001843. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Xiao Q, Ma C, Xie XQ, Floreancig PE. Construction of a Bicyclic beta -Benzyloxy and beta -Hydroxy Amide Library through a Multicomponent Cyclization Reaction. Journal of Combinatorial Chemistry. 2009;11:640–44. doi: 10.1021/cc800200h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.OMEGA. Omega is a conformation geneartion program. cited; Available from: http://www.eyesopen.com/products/applications/omega.html.
- 35.Fink T, Bruggesser H, Reymond JL. Abstracts of Papers, 229th ACS National Meeting, San Diego, CA, United States, March 13-17, 2005. 2005. Virtual exploration of the small molecule chemical universe below 160 daltons. COMP-344. [DOI] [PubMed] [Google Scholar]
- 36.Pozzan A. Molecular descriptors and methods for ligand based virtual high throughput screening in drug discovery. Current Pharmaceutical Design. 2006;12(17):2099–110. doi: 10.2174/138161206777585247. [DOI] [PubMed] [Google Scholar]
- 37.Johnson MA, Maggiora GM, Lajiness MS, Moon JB, Petke JD, Rohrer DC. Molecular similarity analysis: applications in drug discovery. Methods and Principles in Medicinal Chemistry (Advanced Computer-Assisted Techniques in Drug Discovery) 1995;3:89–110. [Google Scholar]
- 38.Willett P. Similarity-based virtual screening using 2D fingerprints. Drug Discovery Today. 2006;11(23 & 24):1046–53. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Maggiora GM. On Outliers and Activity CliffsWhy QSAR Often Disappoints. Journal of Chemical Information and Modeling. 2006;46(4):1535–35. doi: 10.1021/ci060117s. [DOI] [PubMed] [Google Scholar]
- 40.Guha R, Van Drie JH. Structure–Activity Landscape Index: Identifying and Quantifying Activity Cliffs. Journal of Chemical Information and Modeling. 2008;48(3):646–58. doi: 10.1021/ci7004093. [DOI] [PubMed] [Google Scholar]
- 41.Shoichet BK. Virtual screening of chemical libraries. Nature (London, United Kingdom) 2004;432(7019):862–65. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004;3(11):935–49. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 43.Jain AN. Ligand-Based Structural Hypotheses for Virtual Screening. Journal of Medicinal Chemistry. 2004;47(4):947–61. doi: 10.1021/jm030520f. [DOI] [PubMed] [Google Scholar]
- 44.Chen JZ, Xie XQ. GPCR Structure-Based Virtual Screening Approach for the CB2 Antagonist Search. J Comput Info Modeling. 2007;47:1626–37. doi: 10.1021/ci7000814. [DOI] [PubMed] [Google Scholar]
- 45.Chen JZ, Han XW, Liu Q, Makriyannis A, Wang J, Xie XQ. 3D-QSAR Studies of Arylpyrazole Antagonists of Cannabinoid Receptor Subtypes CB1 and CB2. A Combined NMR and CoMFA Approach. Journal of Medicinal Chemistry. 2006;49(2):625–36. doi: 10.1021/jm050655g. [DOI] [PubMed] [Google Scholar]
- 46.Crisman TJ, Parker CN, Jenkins JL, Scheiber J, Thoma M, Kang ZB, et al. Understanding False Positives in Reporter Gene Assays: in Silico Chemogenomics Approaches To Prioritize Cell-Based HTS Data. J Chem Inf Model. 2007;47(4):1319–27. doi: 10.1021/ci6005504. [DOI] [PubMed] [Google Scholar]
- 47.Good AC, Oprea TI. Optimization of CAMD techniques 3. Virtual screening enrichment studies: a help or hindrance in tool selection? Journal of Computer-Aided Molecular Design. 2008;22(3-4):169–78. doi: 10.1007/s10822-007-9167-2. [DOI] [PubMed] [Google Scholar]
- 48.Han L, Wang Y, Bryant SH. Developing and validating predictive decision tree models from mining chemical structural fingerprints and high-throughput screening data in PubChem. BMC Bioinformatics. 2008;9:401. doi: 10.1186/1471-2105-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weis DC, Visco DP, Jr, Faulon JL. Data mining PubChem using a support vector machine with the Signature molecular descriptor: Classification of factor XIa inhibitors. Journal of Molecular Graphics and Modelling. 2008;27(4):466. doi: 10.1016/j.jmgm.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 50.Li Q, Jorgensen FS, Oprea T, Brunak S, Taboureau O. hERG classification model based on a combination of support vector machine method and GRIND descriptors. Mol Pharm. 2008 Jan-Feb;5(1):117–27. doi: 10.1021/mp700124e. [DOI] [PubMed] [Google Scholar]
- 51.Li Q, Wang Y, Bryant SH. A novel method for mining highly imbalanced high-throughput screening data in PubChem. Bioinformatics. 2009 December 15;25(24):3310–16. doi: 10.1093/bioinformatics/btp589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu XH, Ma XH, Tan CY, Jiang YY, Go ML, Low BC, et al. Virtual screening of Abl inhibitors from large compound libraries by support vector machines. J Chem Inf Model. 2009 Sep;49(9):2101–10. doi: 10.1021/ci900135u. [DOI] [PubMed] [Google Scholar]
- 53.Liu XH, Song HY, Zhang JX, Han BC, Wei XN, Ma XH, et al. Identifying Novel Type ZBGs and Nonhydroxamate HDAC Inhibitors Through a SVM Based Virtual Screening Approach. Mol Inf FIELD. 2010;29(5):407–20. doi: 10.1002/minf.200900014. Full Journal Title:Molecular Informatics. [DOI] [PubMed] [Google Scholar]
- 54.Ma XH, Wang R, Yang SY, Li ZR, Xue Y, Wei YC, et al. Evaluation of virtual screening performance of support vector machines trained by sparsely distributed active compounds. J Chem Inf Model. 2008 Jun;48(6):1227–37. doi: 10.1021/ci800022e. [DOI] [PubMed] [Google Scholar]
- 55.Rao H, Li Z, Li X, Ma X, Ung C, Li H, et al. Identification of small molecule aggregators from large compound libraries by support vector machines. J Comput Chem. 2010;31(4):752–63. doi: 10.1002/jcc.21347. [DOI] [PubMed] [Google Scholar]
- 56.Liao Q, Wang J, Webster Y, Watson IA. GPU Accelerated Support Vector Machines for Mining High-Throughput Screening Data. Journal of Chemical Information and Modeling. 2009;49(12):2718. doi: 10.1021/ci900337f. [DOI] [PubMed] [Google Scholar]
- 57.Wang L, XX Q. Develope and validate SVM and KNN based predictive models for classification of the functional type of Human 5HT1A ligands. J Comput Info Modeling. 2010 submitted. [Google Scholar]
- 58.Okuno Y, Tamon A, Yabuuchi H, Niijima S, Minowa Y, Tonomura K, et al. GLIDA: GPCR--ligand database for chemical genomics drug discovery--database and tools update. Nucleic Acids Res. 2008 Jan;36(Database issue):D907–12. doi: 10.1093/nar/gkm948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Rohrer SG, Baumann K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J Chem Inf Model. 2009 Jan 22; doi: 10.1021/ci8002649. [DOI] [PubMed] [Google Scholar]
- 60.Rogers D, Brown RD, Hahn M. Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. Journal of Biomolecular Screening. 2005;10(7):682–86. doi: 10.1177/1087057105281365. [DOI] [PubMed] [Google Scholar]
- 61.Gramatica P. Principles of QSAR models validation: internal and external. QSAR & Combinatorial Science. 2007;26(5):694–701. [Google Scholar]
- 62.Tang Y, Zhang YQ, Chawla Nitesh V, Krasser S. SVMs modeling for highly imbalanced classification. IEEE transactions on systems, man, and cybernetics Part B, Cybernetics : a publication of the IEEE Systems, Man, and Cybernetics Society. 2009;39(1):281–8. doi: 10.1109/TSMCB.2008.2002909. [DOI] [PubMed] [Google Scholar]
- 63.PubCHEM-Fingerpints. PubChem fingerprint is downloaded. cited; Available from: ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt.
- 64.Groom K. NIH Chemical Genomics Center: Small-Molecule Screening for Investigating Fundamental Biological Questions. NIH Chemical Genomics Center - Promega Notes 99. 2008 wwwpromegacom.
- 65*.Guha R, Schurer SC. Utilizing high throughput screening data for predictive toxicology models: protocols and application to MLSCN assays. J Comput Aided Mol Des. 2008 Jun-Jul;22(6-7):367–84. doi: 10.1007/s10822-008-9192-9. [DOI] [PubMed] [Google Scholar]
- 66.Ekins S, Williams AJ. Precompetitive preclinical ADME/Tox data: set it free on the web to facilitate computational model building and assist drug development. Lab Chip. 2010 Jan 7;10(1):13–22. doi: 10.1039/b917760b. [DOI] [PubMed] [Google Scholar]
- 67.Afantitis A, Melagraki G, Sarimveis H, Koutentis PA, Markopoulos J, Igglessi-Markopoulou O. A novel QSAR model for predicting induction of apoptosis by 4-aryl-4H-chromenes. Bioorg Med Chem. 2006;14(19):6686–94. doi: 10.1016/j.bmc.2006.05.061. [DOI] [PubMed] [Google Scholar]
- 68.Kaiser KLE, Niculescu SP, Schultz TW. Probabilistic neural network modeling of the toxicity of chemicals to Tetrahymena pyriformis with molecular fragment descriptors. SAR QSAR Environ Res. 2002;13(1):57–67. doi: 10.1080/10629360290002217. [DOI] [PubMed] [Google Scholar]
- 69.Mazzatorta P, Vracko M, Jezierska A, Benfenati E. Modeling Toxicity by Using Supervised Kohonen Neural Networks. Journal of Chemical Information and Computer Sciences. 2003;43(2):485–92. doi: 10.1021/ci0256182. [DOI] [PubMed] [Google Scholar]
- 70.Nidhi, Glick M, Davies JW, Jenkins JL. Prediction of Biological Targets for Compounds Using Multiple-Category Bayesian Models Trained on Chemogenomics Databases. J Chem Inf Model. 2006;46(3):1124–33. doi: 10.1021/ci060003g. [DOI] [PubMed] [Google Scholar]
- 71.Crettaz P, Benigni R. Prediction of the Rodent Carcinogenicity of 60 Pesticides by the DEREKfW Expert System. J Chem Inf Model. 2005;45(6):1864–73. doi: 10.1021/ci050150z. [DOI] [PubMed] [Google Scholar]
- 72.Veith GD. On the nature, evolution and future of quantitative structure-activity relationships (QSAR) in toxicology. SAR QSAR Environ Res. 2004;15(5-6):323–30. doi: 10.1080/10629360412331297380. [DOI] [PubMed] [Google Scholar]
- 73.Poroikov V, Filimonov D, Lagunin A, Gloriozova T, Zakharov A. PASS: identification of probable targets and mechanisms of toxicity. SAR QSAR Environ Res. 2007;18(1-2):101–10. doi: 10.1080/10629360601054032. [DOI] [PubMed] [Google Scholar]
- 74.Edelstein M, Buchwald F, Richter L, Kramer S. Integrating background knowledge from internet databases into predictive toxicology models. SAR QSAR Environ Res. 2010;21(1 & 2):21–35. doi: 10.1080/10629360903560579. [DOI] [PubMed] [Google Scholar]
- 75.Zhu H, Rusyn I, Richard A, Tropsha A. Use of Cell Viability Assay Data Improves the Prediction Accuracy of Conventional Quantitative Structure-Activity Relationship Models of Animal Carcinogenicity. Environmental Health Perspectives. 2008;116(4):506. doi: 10.1289/ehp.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu H, Sedykh A, Wright FA, Rusyn I, Tropsha A. Abstract of Papers, 238th ACS National Meeting, Washington, DC, United States, August 16-20, 2009. 2009. Using quantitative high-throughput screening (qHTS) results as biological descriptors to assist quantitative structure activity relationship (QSAR) modeling of rat acute toxicity. COMP-339. [Google Scholar]
- 77.Williams-DeVane CR, Wolf MA, Richard AM. DSSTox chemical-index files for exposure-related experiments in ArrayExpress and Gene Expression Omnibus: enabling toxico-chemogenomics data linkages. Bioinformatics. 2009 Mar 1;25(5):692–4. doi: 10.1093/bioinformatics/btp042. [DOI] [PubMed] [Google Scholar]
- 78.Barupal DK, Wohlgemuth G, Fiehn O. Abstracts of Papers, 239th ACS National Meeting, San Francisco, CA, United States, March 21-25, 2010. 2010. Functional and structural network modeling of metabolomics datasets. CINF-11. [Google Scholar]
- 79.DrugBank. DrugBank is a database containing nearly 4800 drug entries including >1,350 FDA-approved small molecule drugs, 123 FDA-approved biotech (protein/peptide) drugs, 71 nutraceuticals and >3,243 experimental drugs. DrugBank is supported by David Wishart, Departments of Computing Science & Biological Sciences, University of Alberta. cited; Available from: http://www.drugbank.ca/
- 80.DSSTox. Environmental Protection Agency's (EPA) Distributed Structure-Searchable Toxicity (DSSTox) Database. cited; Available from: http://www.epa.gov/ncct/dsstox/. (AM Richard) ftp://ftp.epa.gov/dsstoxftp/
- 81.Judson RRA, Dix D, Houck K, Elloumi F, Martin M, Cathey T, Transue TR, Spencer R, Wolf M. ACToR--Aggregated Computational Toxicology Resource. Toxicol Appl Pharmacol. 2008 Nov 15;233(1) doi: 10.1016/j.taap.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 82.Symyx. Symyx' Metabolite and Toxicity database. cited; Available from: http://www.symyx.com/products/databases/bioactivity/index.jsp.
- 83.Aureus-AurSCOPE-ADME/DDI. Aureus AurSCOPE ® ADME/DDI databse provides the drug-drug interaction and metabolic properties of drugs. 2010 cited; Available from: http://www.aureus-pharma.com/Pages/Products/Aurscope_DDI.php.
- 84.PharmaPendium. PharmaPendium online resource from Elsevier enables to search all FDA/CDER/FOI archived FDA drug approval review data as well as an EMEA EPAR approval document database. 2010 cited; Available from: www.pharmapendium.com.
- 85.Chen B, Wild D, Guha R. PubChem as a Source of Polypharmacology. Journal of Chemical Information and Modeling. 2009;49(9):2044. doi: 10.1021/ci9001876. [DOI] [PubMed] [Google Scholar]
- 86.Schuler GD, Epstein JA, Ohkawa H, Kans JA. Entrez: molecular biology database and retrieval system. Methods in Enzymology. 1996;266:141–62. doi: 10.1016/s0076-6879(96)66012-1. Computer Methods for Macromolecular Sequence Analysis. [DOI] [PubMed] [Google Scholar]
- 87.Zhou Y, Zhou B, Chen K, Yan SF, King FJ, Jiang S, et al. Large-Scale Annotation of Small-Molecule Libraries Using Public Databases. Journal of Chemical Information and Modeling. 2007;47(4):1386. doi: 10.1021/ci700092v. [DOI] [PubMed] [Google Scholar]