

Abstract
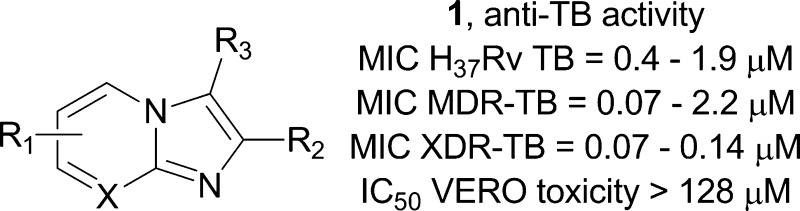
A set of nine 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamides and one 2,6-dimethylimidazo[1,2-a]pyrimidine-3-carboxamide were synthesized. The compounds were evaluated for their in vitro antituberculosis activity versus replicating, nonreplicating, multi- and extensive drug resistant Mtb strains. The MIC90 values of seven of these agents were ≤1 μM against the various tuberculosis strains tested. A representative compound of this class (1) was screened against seven nontubercular strains as well as other nonmycobacteria organisms and demonstrated remarkable microbe selectivity. A transcriptional profiling experiment of Mtb treated with compound 1 was performed to give a preliminary indication of the mode of action. Lastly, the in vivo ADME properties of compounds 1, 3, 4, and 6 were assessed. The 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamides are a druglike and synthetically accessible class of anti-TB agents that have excellent selective potency against multi- and extensive drug resistant TB and encouraging pharmacokinetics.
Keywords: Antituberculosis; imidazo[1,2-a]pyridine-3-carboxamides; MDR-TB; XDR-TB
Tuberculosis (TB) is a serious global health risk. More than one-third of the human population is infected, resulting in an estimated 1 700 000 deaths in 2006 (1.5 million in HIV-negative people and 0.2 million in HIV-positive people).1 Moreover, there were a staggering 14 400 000 cases estimated worldwide in 2006, with 83% of the total cases located in the African, South-East Asia, and Western Pacific regions.1Mycobacterium tuberculosis, the causative agent of TB, is an airborne pathogen that can be spread from one person to another by close contact. Because it can lie dormant in a latent state for many years, it is a silent killer among the poor, HIV-infected, immune-compromised, and the elderly. To make matters worse, multiple drug resistant TB [MDR-TB, strains that are resistant to first line drugs isoniazid (INH) and rifampin] and extensively drug resistant TB (XDR-TB, strains that are resistant to INH and rifampin, as well as any fluoroquinolone and at least one of three injectable second-line drugs, such as amikacin, kanamycin, or capreomycin) are on the rise.2 Most alarming is the emergence of extremely drug resistant TB “XXDR-TB” (the proposed designation for TB that is resistant to all first- and second-line TB drugs), which is now documented.3 In 2008, there were 12 898 cases of TB provisionally reported in the United States.4
A focus of our laboratories is to facilitate the decline of TB by the identification of therapeutically effective anti-TB agents to augment the long dosing regimen of first-line drugs.5 Herein, we call attention to the in vitro potency of the imidazo[1,2-a]pyridine-3-carboxamide scaffold. To our knowledge, the anti-TB activity of the 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide class is unprecedented. Imidazo[1,2-a]pyridine-3-nitroso derivatives were reported in 2004 to impart notable anti-TB activity (MIC = 3.1 μg/mL vs H37Rv-TB) concurrent with notable toxicity to VERO cells (IC50 = 3.6 μg/mL).6 Also in 2004, rationally designed imidazo[1,2-a]pyridine-3-hydrazones7 were reported but were all inactive against H37Rv-TB at 6.25 μg/mL. Most recently, in 2009, functionalized 3-amino-imidazo[1,2-a]pyridines were reported as in vitro Mtb glutamine synthetase inhibitors but without assessment of the in vitro activity versus H37Rv-TB.8 While reports on the syntheses of imidazo[1,2-a]pyridine-3-carboxamides date to 1965,9 the 2,7-dimethylimidazo[1,2-a]pyridine architecture is atypical within the cannon of medicinal chemistry literature and is unprecedented within the TB lexicon.
Since 2007, we have had a collaborative agreement with Dow AgroScience to screen their compound inventory for inhibitors of Mtb. This effort, coupled with a program to elaborate novel heterocylic anti-TB agents from fragment-based studies of mycobacterial siderophores,10 led to the identification of an ethyl 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxylate. This compound had weak activity against H37Rv TB (MIC ∼ 65 μM, average) but was nonetheless an attractive heterocyclic scaffold to optimize as we did previously with related heterocyclic classes.11
The simplest synthesis of the imidazo[1,2-a]pyridine-3-carboxylate ring system12 is the straightforward reaction of 2-amino-4-picoline with ethyl 2-chloroacetoacetate to give the desired heterocyclic scaffold in 78% yield (Scheme 1). Saponification with lithium hydroxide followed by acidic work up gave the free acid, which was then easily converted to various amide analogues through classical EDC-mediated couplings in good yields (70% for 1).
Scheme 1. Synthesis of Imidazo[1,2-a]pyridines.

Reagents: (a) Ethyl 2-chloroacetoacetate, DME, reflux, 48 h. (b) (1) LiOH, EtOH; (2) HCl, 56 h. (c) EDC, DMAP, R1, ACN, 16 h.
Our initial structure−activity relationship (SAR) strategy evaluated a representative panel of nine imidazo[1,2-a]pyridine-3-carboxamide analogues. The chosen imidazopyridine analogues included the classical Topliss13 set of benzyl, 4-methoxyphenyl, 4-methylphenyl, 4-chlorophenyl, and 3,4-dichlorophenyl amides. This set was augmented by ortho- and meta-methoxyphenyl analogues to probe possible steric effects. Next, because of potential metabolic issues associated with a benzylic methylene group, the corresponding aniline was prepared as well as a fluorine replacement for the chlorine. Finally, we explored the influence on potency by changing the imidazo[1,2-a]pyridine to an imidazo[1,2-a]pyrimidine core (as in 10).
Table 1 summarizes the in vitro anti-TB activity of these 10 analogues in three different media (GAS,14 GAST,15 and 7H1214), their potency against nonreplicating “latent” TB (LORA16), and an assessment of their toxicity by the VERO17 assay. All compounds were potent (MIC < 10 μM in the GAS assay media) with the exception of the aniline derived analogue (9, MIC > 128 μM), suggesting that in the imidazo[1,2-a]pyridine series the benzylic position is important for activity. Additionally, by running the TB assay in three different media (GAS, GAST, and 7H12), we eliminated concern that the activity of these compounds might be carbon source dependent, a flaw discovered in the pyrimidine-imidazoles reported by Pethe and colleagues at Novartis18 as the GAS and GAST assays use glycerol-alanine salts as the carbon source, while the 7H12 media use palmitic acid.
Table 1. In Vitro Evaluation of Compounds 1−10 against H37Rv-TB in Various Assays and Media (MIC90 in μM), Stability to Rat Liver (RLM) and Human Liver (HLM) Microsomes and VERO Cellular Toxicity (IC50 in μM).
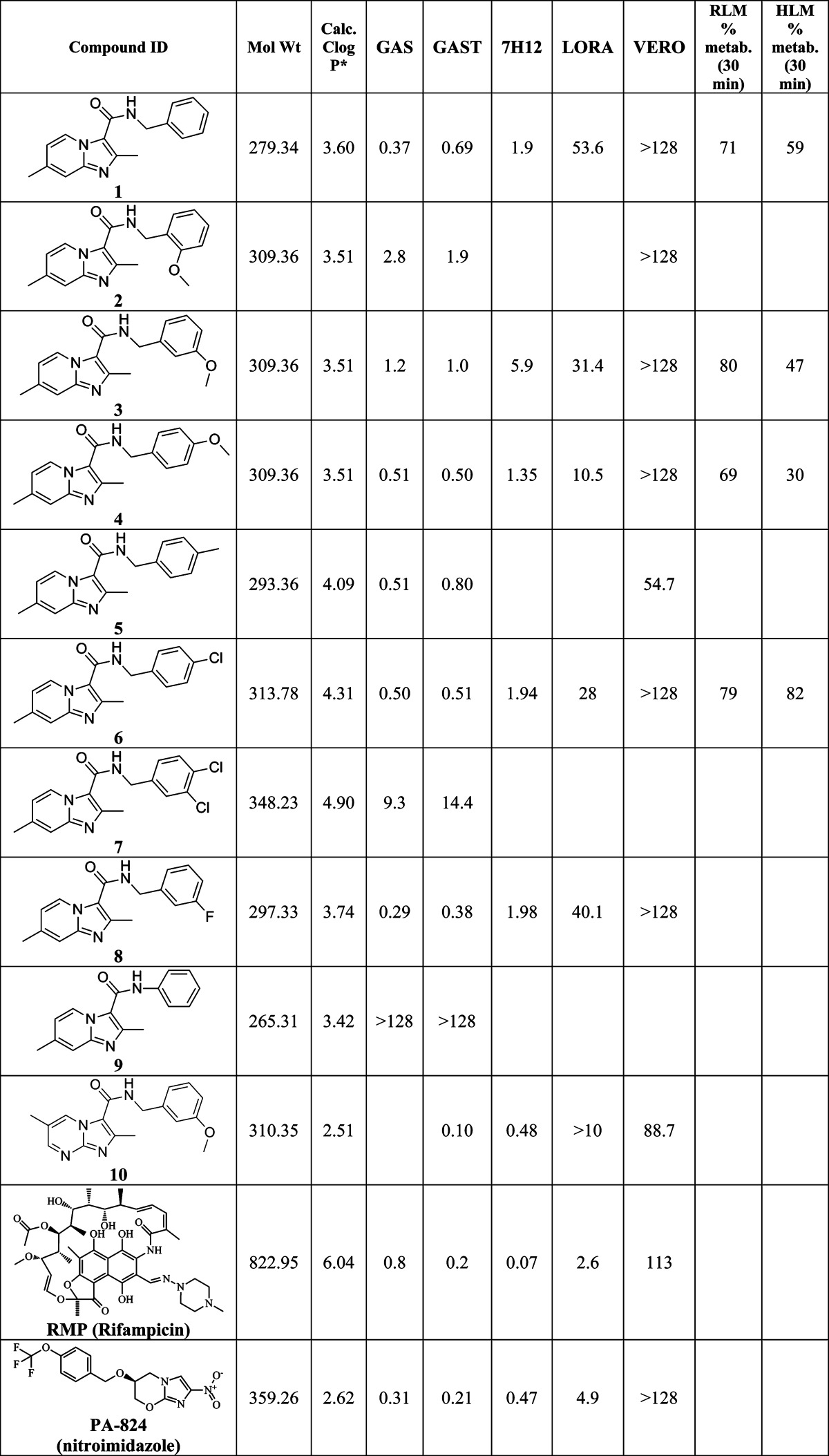 |
Calculated ClogP by ChemDraw version 12.0. GAS, glycerol-alanine-salts media; GAST, iron deficient glycerol-alanine-salts with Tween 80 media; 7H12, 7H9 broth base media with BSA, casein hydrolysate, catalase, palmitic acid; LORA, low oxygen recover assay; VERO, African green monkey kidney cell line; RLM, rat liver microsomes; and HLM, human liver microsomes. Values reported are the average of three individual measurements.
SAR analysis based on the whole cell assay readout indicated that the 3,4-dichloro analogue (7) had diminished activity (MICs of 9−14 μM) when compared to the 4-chloro (6, 0.5 μM) and 3-fluoro analogues (8, ∼0.3 μM). There appeared to be a slight preference for para-substitution in terms of potency (MIC = 2.8 μM for ortho- vs 1.2 μM for meta- vs 0.5 μM for para-methoxy analogue in the GAS assay media). Comparison of the imidazo[1,2-a]pyrimidine analogue (10) to the corresponding imidazo[1,2-a]pyridine analogue (3) indicated that the additional nitrogen in the heterocyclic core was well tolerated (submicomolar potency) although VERO toxicity (IC50 = 89 μM) was noted. Compounds 1 and 10 were rescreened in the presence of 4% BSA (bovine serum albumin) and 10% FBS (fetal equine serum), and their MICs were found to shift less than 2-fold by ATP and MABA readouts (see the Supporting Information), indicating that protein binding is not a problem.
Encouraged that six of the 10 analogues tested had submicromolar MIC values against the H37Rv Mtb strain, we next screened compounds 1 and 10 against a panel of single drug resistant strains (Table 2) against controls rifampicin (RIF) and isoniazid (INH) and then three of the more promising compounds (1, 3, and 8) against a panel of MDR and XDR clinical strains (Table 2). The difference in potencies of these compounds against the clinical strains may be due to the difference in growth media where growth inhibition for clinical strains was tested in media containing glucose as well as glycerol as the carbon source, as well as the fact that many clinical strains exhibit poor growth in vitro since they are not adapted to laboratory conditions and media, which may affect their apparent susceptibility to certain inhibitors.
Table 2. Potency of Imidazo[1,2-a]pyridines (1, 3, and 8) and Imidazo[1,2-a]pyrimidine (10) against Single Drug Resistant Strains, MDR-TB and XDR-TB Strains (MIC90 in μM)a.
| control/compound ID |
||||||
|---|---|---|---|---|---|---|
| strains resistant to drugs | RMP | INH | 1 | 3 | 8 | 10 |
| RMP | >1 | 0.23 | 0.28 | 1.49 | ||
| INH | 0.01 | >8 | 0.33 | 5.84 | ||
| kanamycin | 0.02 | 0.43 | 1.07 | 1.02 | ||
| streptomycin | 0.02 | 0.23 | 1.02 | 5.84 | ||
| MDR-HRESP | 2.24 | 1.01 | 0.26 | |||
| MDR-HREZSP | 1.12 | 0.06 | 0.06 | |||
| MDR-HCPTh | 1.12 | 0.13 | 0.26 | |||
| MDR-HREKP | 0.28 | 0.13 | 0.26 | |||
| MDR-HRERbb | 0.14 | ≤0.03 | 0.13 | |||
| MDR-HRERbb | 0.14 | 0.03 | 0.34 | |||
| MDR-HRERbb | 0.28 | 0.06 | 0.26 | |||
| MDR-HREZSKPTh | 0.07 | ≤0.03 | 0.06 | |||
| MDR-HREZRbTh | 0.14 | 0.06 | 0.06 | |||
| XDR-HRESPOCTh | 0.07 | 0.03 | 0.07 | |||
| XDR-HREPKOTh | 0.07 | 0.02 | 0.03 | |||
| XDR-HRESPO | 0.14 | 0.02 | 0.03 | |||
Abbreviations: H = isoniazid, R = rifampicin, E = ethambutol, Z = pyrazinamide, S = streptomycin, C = cycloserine, Th = ethionamide, K = kanamycin, P = p-aminosalicylic acid, Rb = rifabutin, Th = thioacetazone, and O = ofloxacin.
Different clinical strains.20 Values reported are the average of three individual measurements.
The excellent activity found when these imidazo[1,2-a]pyridine agents were tested against the drug resistant strains compared favorably to the published MIC values of the nitroimidazole clinical candidate PA-82419 (MICs against MDR-TB from 0.03 to 0.25 μg/mL or 0.08 to 0.7 μM, comparatively). Furthermore, the improved, and indeed outstanding, potency of these agents against the various drug resistant strains suggests that they inhibit a novel target. Selectivity screening against various nontubercular mycobacteria revealed that compounds 1 and 10 are also inhibitors of Mycobacterium avium, Mycobacterium bovis BCG, and Mycobacterium kansasii but not inhibitors of Mycobacterium smegmatis, Mycobacterium abscessus, Mycobacterium chelonae, and Mycobacterium marinum (Table 3). This unusual selectivity prompted us to further screen compounds 1, 3, 4, 6, 8, and 10 against a panel of representative nonmycobacterial organisms. Compounds 1, 3, 4, 6, 8, and 10 were all found to be inactive against the Gram-positive strain of Staphylococcus aureus (MIC > 128 μM), the Gram-negative strain of Escherichia coli (MIC > 128 μM), and the fungus Candida albicans (MIC > 128 μM), further suggesting a mycobacterium specific target of these agents.
Table 3. Nontubercular Mycobacteria Activity and Selectivity of Imidazo[1,2-a]pyridine (1) and Imidazo[1,2-a]pyrimidine (10) (MIC90 in μM)a.
| compound ID | TB-H37Rv | M. abscessus | M. chelonae | M. marinum | M. avium | M. kansasii | M. bovis BCG | M. smegmatis |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.07 | >50 | >50 | >50 | 1.32 | 1.32 | 0.33 | >50 |
| 10 | 5.94 | >50 | >50 | >50 | 12.00 | 12.00 | 2.78 | >50 |
| RMP | 0.05 | 162.3 | 150.00 | <0.78 | <0.78 | <0.78 | <0.78 | 162.3 |
Values reported are the average of three individual measurements.
The in vivo pharmacokinetics (PK) of compounds 1, 3, 4, and 6 were evaluated in Sprague−Dawley rats by oral (po) and intravenous (iv) routes of administration at 10 and 1 mg/kg dosing levels, respectively (Table 4). Compound 4 had moderate in vitro rat microsomal stability (69% metabolized, t1/2 = 19 min) and also displayed promising PK by having the lowest in vivo clearance (28 mL/min/kg, t1/2 = 0.28 hours by iv). The aqueous solubility for compounds 1, 3, 4, and 6 was measured at 181, 149, 148, and 25 μM, respectively, in phosphate-buffered saline (PBS) at pH 7.4. Additional in vivo ADME properties including terminal half-life (t1/2ss), the area under the curve (AUC), the volume of distribution (Vd) and volume of distribution at steady state (Vdss) for compounds 1, 3, 4, and 6 can be found in the Supporting Information. Encouraged by the potency, PK, and favorable oral bioavailability of these imdazo[1,2-a]pyridine agents, we intend to evaluate various analogues in vivo by the murine gamma knockout (GKO) infection model, and the results will be reported in due course.
Table 4. In Vivo PK Evaluation of Imidazo[1,2-a]pyridinesa.
| compound ID | po Cmax (ng/mL) | po Tmax (h) | iv t1/2 (h) | iv clearance (mL/min/kg) | % F |
|---|---|---|---|---|---|
| 1 | 3012 | 0.25 | 0.35 | 91 | 76 |
| 3 | 3140 | 0.25 | 0.33 | 43 | 43 |
| 4 | 5741 | 0.25 | 0.28 | 28 | 50 |
| 6 | 1995 | 0.31 | 0.4 | 51 | 49 |
Values reported are the average of three individual measurements.
Finally, curious as to the mechanism of action of these agents, we performed transcriptional profiling experiments of M. tuberculosis treated with compound 1, and comparison to the existing database of drug-induced transcriptional profiles indicated that this compound inhibited an aspect of energy generation in the cell (see the Supporting Information). Thus, compound 1 resulted in up-regulation of the cytochrome bd oxidase, which is the high oxygen-affinity respiratory enzyme21 observed to be up-regulated during oxygen restriction as well as inhibition of respiration by agents such as cyanide, sodium azide, the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), and the nitric oxide-releasing pro-drug PA-824.22 In addition, this compound up-regulated the phosphoenolpyruvate carboxykinase, which plays an important role in modulating carbon flow during cellular energy restriction23 and has previously been observed to be up-regulated by stresses such as hypoxia, sodium azide, valinomycin, nigericin, carbonyl cyanide rn-chlorophenylhydrazone, cyanide, PA-824, and the ATP inhibitor dicyclohexylcarboxydiimide, that limit energy generation through respiration.22
All of the data suggest that we have discovered a class of compounds with promising attributes of synthetic accessibility, no redox active moieties,19 impressive potency, and selectivity toward replicating MDR and XDR Mtb strains. This class has good in vivo ADME properties that potentially can be improved through further analogue generation. Additionally, compound 1 appears to act by a novel mechanism of action based on transcriptional profiles to known anti-TB agents. With new anti-TB agents desperately needed, we offer the imidazo[1,2-a]pyridine class as a potential therapeutic for further development.
Acknowledgments
We thank Prof. Jennifer DuBois and Dr. Jed Fisher for profound scientific discussions. The excellent technical assistance of Baojie Wan and Yuehong Wang with anti-TB assays at UIC is greatly appreciated. Finally, we thank Gail Cassell and the Lilly Tuberculosis Drug Discovery Initiative for their continued support of this project.
Supporting Information Available
Full experimental details for compounds synthesized, descriptions of assays, PK data, and transcriptional profiling as well as copies of relevant NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
G.C.M. participated in the design, performed the syntheses, drafted the manuscript, and facilitated all interactions. L.D.M. participated in the design and coordinated interactions through Dow AgroSciences. P.A.H. facilitated microsome and PK assessment. H.B. performed MDR and XDR anti-TB assays and the transcriptional profiling. S.C. and S.G.F. provided anti-TB and selectivity assays. M.J.M. drafted the manuscript and participated in the design and direction of the project.
Funding was provided by NIH AI054193, Dow AgroSciences, and NSF CHE-0741793. This research was supported in part by the Intramural Research Program of the NIH, NIAID, and by Grant 2R01AI054193 from the National Institutes of Health (NIH) and in part by intermediates provided from Dow AgroSciences. We thank the University of Notre Dame, especially the Mass Spectrometry and Proteomics Facility (Bill Boggess, Michelle Joyce, and Nonka Sevova), which is supported by Grant CHE-0741793 from the NSF.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Global tuberculosis control: Surveillance, planning, financing: WHO report 2008. WHO/HTM/TB/2008.
- Sacchettini J. C.; Rubin E. J.; Freundlich J. S. Drugs versus bugs: In pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2008, 6, 41–52. [DOI] [PubMed] [Google Scholar]
- Migliori G. B.; De Iaco G.; Besozzi G.; Centis R.; Cirillo D. M. First tuberculosis cases in Italy resistant to all tested drugs. Eurosurveillance 2007, 12, 3194. [DOI] [PubMed] [Google Scholar]
- Maher D.; Blanc L.; Raviglione M. WHO policies for tuberculosis control. Lancet 2004, 363, 1911–1911. [DOI] [PubMed] [Google Scholar]
- Pratt R.; Robison V.; Navin T.; Bloss E. Centers for Disease Control and Prevention. Trends in Tuberculosis—United States. MMWR 2009, 58, 249–253.19300406 [Google Scholar]
- Anaflous A.; Benchat N.; Mimouni S.; Abouricha S.; Ben-Hadda T.; El-Bali A.; Hacht B. Armed Imidazo [1,2-a] Pyrimidines (Pyridines): Evaluation of Antibacterial Activity. Lett. Drug Des. Discovery 2004, 1, 35–44. [Google Scholar]
- Kasimogullari B. O.; Cesur Z. Fused Heterocycles: Synthesis of Some New Imidazo[1,2-a]-pyridine Derivatives. Molecules 2004, 9, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell L. R.; Nilsson M. T.; Gising J.; Lagerlund O.; Muthas D.; Nordqvist A.; Karlen A.; Larhed M. Functionalized 3-amino-imidazo[1,2-a]pyridines: A novel class of Mycobacterium tuberculosis glutamine synthetase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 4790–4793. [DOI] [PubMed] [Google Scholar]
- Lombardino J. G. Preparation and New Reactions of Imidazo[1,2-a]pyridines. J. Org. Chem. 1965, 30, 2403–2407. [Google Scholar]
- Moraski G. C.; Chang M.; Villegas-Estrada A.; Franzblau S.; Möllmann U.; Miller M. J. Structure-Activity Relationship of New Antituberculosis Agents Derived from Oxazoline and Oxazole Benzyl Esters. Eur. J. Med. Chem. 2010, 45, 1703–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraski G. C.; Franzblau S. G.; Miller M. J. Utilization of the Suzuki Coupling to Enhance the Antituberculosis Activity of Aryl Oxazoles. Heterocycles 2009, 80, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritzky A. R.; Xu Y. -J.; Tu H. Regiospecific Synthesis of 3-Substituted Imidazo[1,2-a]pyridines, Imidazo[1,2-a]pyrimidines, and Imidazo[1,2-c]pyrimidine. J. Org. Chem. 2003, 68, 4935–4937. [DOI] [PubMed] [Google Scholar]
- Topliss J. G. Utilization of operational schemes for analog synthesis in drug design. J. Med. Chem. 1972, 15, 1006–1011. [DOI] [PubMed] [Google Scholar]
- Collins L.; Franzblau S. G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Voss J. J.; Rutter K.; Schroeder B. G.; Su H.; Zhu Y.; Barry C. E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S. H.; Warit S.; Wan B.; Hwang C. H.; Pauli G. F.; Franzblau S. G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzari K.; Zhou Z.; Pan D.; Liu H.; Hongmanee P.; Franzblau S. G. In Vitro and In Vivo Activities of Macrolide Derivatives against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Sequeira P. C.; Agarwalla S.; Rhee K.; Kuhen K.; Phong W. Y.; Patel V.; Beer D.; Walker J. R.; Duraiswamy J.; Jiricek J.; Keller T. H.; Chatterjee A.; Tan M. P.; Ujjini M.; Roa S. P. S.; Camacho L.; Bifani P.; Mak P. A.; Ma I.; Barnes S. W. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nature Commun. 2010, 57, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N; Kreiswirth B. N.; Barry C. E.; Baker W. R. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [DOI] [PubMed] [Google Scholar]
- Jeon C. Y.; Hwang S. H.; Min J. H.; Prevots D. R.; Goldfeder L. C.; Lee H.; Eum S. Y.; Jeon D. S.; Kang H. S.; Kim J. H.; Kim B. J.; Kim D. Y.; Holland S. M.; Park S. K.; Cho S. N.; Barry C. E. 3rd; Via L. E. Extensively drug-resistant tuberculosis in South Korea: Risk factors and treatment outcomes among patients at a tertiary referral hospital. Clin. Infect. Dis. 2008, 46, 42–49. [DOI] [PubMed] [Google Scholar]
- Kana B. D.; Weinstein E. A.; Avarbock D.; Dawes S. S.; Rubin H.; Mizrahi V. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J. Bacteriol. 2001, 24, 7076–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshoff H. I.; Myers T. G.; Copp B. R.; McNeil M. R.; Wilson M. A.; Barry C. E. 3rd The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 38, 40174–40184. [DOI] [PubMed] [Google Scholar]
- Marrero J.; Rhee K. Y.; Schnappinger D.; Pethe K.; Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.