

Abstract
Background
There is a clear need to develop biomarkers for Parkinson disease (PD) diagnosis, differential diagnosis of parkinsonian disorders, and monitoring disease progression. We and others have demonstrated that a decrease in DJ-1 and/or α-synuclein in the cerebrospinal fluid (CSF) is a potential index for PD diagnosis, but not for PD severity.
Methods
Using highly sensitive and quantitative Luminex assays, we measured total tau, phosphorylated tau, amyloid beta peptide 1-42 (Aβ1-42), Flt3 ligand and fractalkine levels in CSF in a large cohort of PD patients at different stages as well as healthy and diseased controls. The utility of these five markers was evaluated for disease diagnosis and severity/progression correlation alone, as well as in combination with DJ-1 and α-synuclein. The major results were further validated in an independent cohort of cross-sectional PD patients as well as in PD cases with CSF samples collected longitudinally.
Findings
The results demonstrated that combinations of these biomarkers could differentiate PD patients not only from normal controls but also from patients with Alzheimer disease and multiple system atrophy. Particularly, with CSF Flt3 ligand, PD could be clearly differentiated from multiple system atrophy, a disease that overlaps with PD clinically, with excellent sensitivity (99%) and specificity (95%). In addition, we identified CSF fractalkine/Aβ1-42 that positively correlated with PD severity in cross-sectional samples as well as with PD progression in longitudinal samples.
Interpretation
We have demonstrated that this panel of seven CSF proteins could aid in PD diagnosis, differential diagnosis, and correlation with disease severity and progression.
Introduction
Diagnosis of Parkinson disease (PD) currently relies almost entirely on clinical acumen. Moreover, differential diagnosis from other parkinsonian disorders, such as essential tremor, multiple system atrophy (MSA) and progressive supranuclear palsy (PSP), can be rather challenging due to overlapping symptoms, particularly during the early disease stages1, 2. While developing neuroimaging and olfaction tests may prove useful as biomarkers of PD3-6, there is still limited knowledge about their specificity among neurodegenerative diseases, and these studies have not proven to be reliable and/or cost effective at distinguishing patients with different forms of neurodegenerative parkinsonism7-9. Indeed, currently there is no established laboratory test or biomarker that can reliably and specifically identify PD, particularly at its early stages. Furthermore, there is no effective biomarker to robustly track PD progression or assess treatment response.
Cerebrospinal fluid (CSF), being much more accessible, less costly than imaging, and reflecting metabolic and pathological states of the central nervous system (CNS) more directly than any other body fluids, is an ideal source for PD biomarker discovery. So far the two markers that have been tested most extensively in CSF are DJ-1 and α-synuclein (α-syn)10-14, two proteins intimately involved in familial and sporadic PD pathogenesis15. In a recent study involving a large cohort of subjects, we confirmed that a decrease in CSF DJ-1 and/or α-syn yielded 90-92% sensitivity and 58-70% specificity when PD subjects aged ≥65 years were compared to healthy controls and patients with Alzheimer disease (AD). Additionally, the decrease in concentration of either biomarker did not correlate with PD severity or progression14. However, both results await independent validation, and it is unclear whether DJ-1 and α-syn can differentiate PD from other diseases that also give rise to parkinsonism.
In a prior investigation, we also demonstrated that a panel of CSF markers including amyloid beta peptide 1-42 (Aβ1-42) and total tau (t-tau), when used in combination, could robustly distinguish PD from controls and from AD16. In the current study, we quantified the levels of these markers, along with phosphorylated tau (181P) (p-tau), as well as Flt3 ligand and fractalkine - two inflammatory markers that appeared to relate to PD or other parkinsonian disorders specifically in a separate CSF cytokine profiling study (unpublished data) - in CSF samples from a larger cohort using well-established Luminex assays. In this study, we asked whether these markers could assist with differentiating PD from other parkinsonian conditions as well as correlating with PD severity and/or progression, approximated by the UPDRS (Unified Parkinson Disease Rating Scale) motor scores, with samples collected cross-sectionally and longitudinally.
Patients and Methods
1. Participants
The study was approved by the Institutional Review Boards of all participating institutions. All individuals provided informed consent, and underwent an evaluation that consisted of medical history, physical and neurological examinations, laboratory tests, and neuropsychological assessments. For discovery, 137 normal controls, 126 PD, 50 AD and 32 MSA patients (a total of 345 subjects) were included in this investigation (among them, 132 control, 117 PD and 50 AD subjects were included in a previous DJ-1 and α-syn study14). For validation, an independent cohort with 44 sporadic PD patients was included. Additionally, a total of 39 PD cases from the Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism (DATATOP) study17, 18 with CSF samples collected longitudinally were also included as another independent validation. More details on the inclusion and exclusion criteria for normal controls and patients with PD, MSA or AD can be found in the Supporting Methods. Demographic information is listed in Table 1 for all subjects/patients.
Table 1.
Demographics of study participants and CSF marker levels in diagnostic groups*
| Ctl | PD | MSA | AD | P values | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of cases** | 137 | 126 | 32 | 50 | PD | MSA | AD | PD | PD | MSA |
| (age≥50 only) | (105) | (117) | (28) | (50) | vs | vs | vs | vs | vs | vs |
| [age≥50 & HGB<200 ng/mL] | [92] | [86] | [20] | [38] | Ctl | Ctl | Ctl | MSA | AD | AD |
|
| ||||||||||
| Age (age≥50 only) | 58.9±18.4 (67.4±10.5) | 63.8±10.4 (65.4±9.1) | 60.3±9.0 (62.4±7.7) | 68.1±9.5 (68.1±9.5) | ||||||
|
| ||||||||||
| Gender (F/M) (age≥50 only) | 61/76 (53/52) | 33/93 (29/88) | 11/21 (11/17) | 19/31 (19/31) | ||||||
|
| ||||||||||
| t-tau (pg/mL) | 62.3±20.2 | 54.6±14.8 | 46.7±18.9 | 95.0±37.5 | 0.026 | 0.004 | 0.001 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| p-tau (pg/mL) | 29.4±17.6 | 21.4±7.6 | 22.4±10.7 | 57.3±25.8 | <0.001 | >0.1 | <0.001 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| Aβ1-42 (pg/mL) | 404.2±184.0 | 332.9±127.4 | 312.0±120.1 | 209.2±73.4 | 0.003 | 0.022 | <0.001 | >0.1 | <0.001 | 0.013 |
|
| ||||||||||
| Flt3 ligand (pg/mL) | 47.2±14.4 | 51.7±13.4 | 21.5±9.5 | 47.7±13.4 | >0.1 | <0.001 | >0.1 | <0.001 | >0.1 | <0.001 |
|
| ||||||||||
| Fractalkine (pg/mL) | 257.5±105.7 | 281.9±114.4 | 277.9±153.3 | 281.1±108.7 | >0.1 | >0.1 | >0.1 | >0.1 | >0.1 | >0.1 |
|
| ||||||||||
| DJ-1 (ng/mL; HGB<200 ng/mL) | 41.0±14.4 | 28.2±7.5 | 23.8±7.5 | 41.1±15.5 | <0.001 | <0.001 | >0.1 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| α-syn (ng/mL; HGB<200 ng/mL) | 0.49±0.17 | 0.38±0.10 | 0.30±0.09 | 0.55±0.15 | <0.001 | <0.001 | >0.1 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| p-tau/t-tau | 0.46±0.14 | 0.40±0.10 | 0.49±0.12 | 0.61±0.16 | 0.002 | >0.1 | <0.001 | 0.008 | <0.001 | <0.001 |
|
| ||||||||||
| p-tau/Aβ1-42 | 0.091±0.078 | 0.074±0.051 | 0.074±0.024 | 0.291±0.127 | >0.1 | >0.1 | <0.001 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| t-tau/Aβ1-42 | 0.18±0.10 | 0.18±0.09 | 0.16±0.06 | 0.48±0.17 | >0.1 | >0.1 | <0.001 | >0.1 | <0.001 | <0.001 |
|
| ||||||||||
| Fractalkine/Aβ1-42 | 0.76±0.51 | 0.97±0.59 | 0.96±0.59 | 1.51±0.80 | >0.1 | >0.1 | <0.001 | >0.1 | <0.001 | 0.001 |
Data shown are mean ± S.D. Only subjects with age ≥ 50 were included for the analyses. Comparisons were made using one-way ANOVA followed by Tukey’s HSD post-hoc tests. Conservatively, values with P<0.001 should be regarded as significant to minimize errors related to multiple comparisons. Only the ratios discussed in the text are listed here. The receiver operating characteristic values of all markers including other calculated ratios/combinations can be found in Supporting Table 2.
More than 90% of the subjects/patients were included in our previous study14 where DJ-1 and α-syn results were reported.
α-syn, α-synuclein; Aβ1-42, amyloid beta peptide 1-42; AD, Alzheimer disease; ANOVA = analysis of variance; CSF, cerebrospinal fluid; Ctl, healthy control; HGB, hemoglobin; HSD, honestly significant difference; MSA, multiple system atrophy; PD, Parkinson disease; p-tau, phosphorylated tau; t-tau, total tau.
2. CSF Samples and Hemoglobin Test
All CSF samples were obtained by lumbar puncture as described previously14. A more detailed discussion of lumbar puncture procedure, CSF processing, patient acceptability and other related issues can be found in the Supporting Methods. The hemoglobin (HGB) levels in CSF samples were chosen as an index of the degree of red blood cell contamination of CSF and were measured as described14.
3. Luminex Assays
CSF Aβ1-42, t-tau and p-tau levels were measured using the INNO-BIA AlzBio3 kit obtained from Innogenetics (Gent, Belgium) following the manufacturer’s instructions except that the CSF samples were diluted 1:4 in Diluent before performing the assay. CSF Flt3 ligand and fractalkine levels were measured using a human cytokine/chemokine kit (MPXHCYTO-60K; Millipore, Billerica, MA, USA) according to the manufacturer’s overnight protocol with minor modifications. The total initial volume in each well was 100 μl instead of 75 μl. CSF samples (10 μl/well for fractalkine and 50μl/well for Flt3 ligand) were diluted with 0.1% bovine serum albumin (BSA)/phosphate buffered saline (PBS) (pH7.4) before assay. CSF DJ-1 and α-syn levels were measured as previously described14. Most of the DJ-1 and α-syn data were published recently14, but we have now expanded these analyses (see the Supporting Methods for details).
All CSF samples were analyzed using a LiquiChip Luminex 200™ Workstation (Qiagen, Valencia, CA, USA). Details on batch-to-batch variation control can be found in the Supporting Methods.
4. Statistical Analysis
All analyses were performed with PASW Statistics (previously SPSS) 18.0 (SPSS Inc, Chicago, IL, USA). To assess differences between groups, one-way analysis of variance (ANOVA) followed by the post-hoc Tukey honestly significant difference (HSD) test was used, and correlations were evaluated using linear regression analysis (Pearson’s correlation), as well as Spearman’s nonparametric correlation to control for potential contributions secondary to outliers. The analyses were also done using an analysis of covariance (ANCOVA) model to check our results after controlling for possible confounding variables - i.e., age, gender, or HGB concentration. Forward stepwise logistic regression analysis was used to screen for the best predictors in the pairwise group comparisons. Models for biomarker combinations were determined as linear combinations of biomarkers using the coefficients from logistic regression models.
A Receiver Operating Characteristic (ROC) curve was used to calculate the relationship between sensitivity and specificity for the disease group versus healthy or disease controls, and hence evaluate the diagnostic performance of the analytes, either individually or in combinations. The “optimum” cutoff value from the ROC curve is determined by anchoring the sensitivity to be 90-95%, with the exception in a few cases that the sensitivity is below 90% but the sum of sensitivity and specificity is maximal. Values with P<0.05 were regarded as significant, while values with P<0.001 were used for all of our major findings (Table 1 and Table 2) to minimize errors related to multiple comparisons.
Table 2.
Area under curve values, sensitivities, specificities and cut-off points of the best discriminating parameters for each differential diagnostic testing
| Differential diagnosis | Parameter | AUC | Cut-off | Sensitivity%* | Specificity%* |
|---|---|---|---|---|---|
| PD vs. MSA | Flt3L | 0.98 | 29.90 | 99 (93.7-100.0) | 95 (75.1-99.9) |
| PD vs. Control | DJ-1 & Flt3La | 0.84 | 26.11 | 94 (86.8-98.1) | 60 (48.8-70.5) |
| DJ-1 | 0.78 | 40 | 94 (86.8-98.1) | 50 (39.0-61.0) | |
| α-syn | 0.71 | 0.5 | 92 (83.9-96.7) | 38 (28.1-49.5) | |
| PD vs. AD | p-tau/Aβ1-42 | 0.94 | 0.135 | 93 (85.4-97.4) | 90 (75.2-97.1) |
| t-tau/Aβ1-42 | 0.94 | 0.297 | 92 (83.9-96.7) | 84 (68.7-94.0) | |
| α-syn & Aβ1-42b | 0.95 | 169.1 | 93 (85.4-97.4) | 84 (68.7-94.0) |
Cut-off refers to the selected value of the individual biomarker or the combination where the two groups can be separated at the indicated sensitivity and specificity. All the differential diagnostic testing listed was done with subjects younger than 50 and/or samples with high blood contamination (hemoglobin ≥ 200 ng/mL) excluded. P<0.001 for all the tests. Only the markers whose sensitivity was confirmed by an independent PD cohort are listed. A complete list of ROC analysis results can be found in Supporting Table 2. For the combinations of biomarkers, values provided were derived from models based on logistic regression analysis. The models were then subjected to ROC to determine AUC, cutoff, sensitivity and specificity. Model equations are as the following:
2 [DJ-1] – [Flt3L];
895 [α-syn] - [Aβ1-42].
Confidence intervals (at 95% confidence level) are shown for sensitivity and specificity. The formulas used were obtained from http://statpages.org/confint.html.
α-syn, α-synuclein; Aβ1-42, amyloid beta peptide 1-42; AD, Alzheimer disease; AUC, area under the ROC curve; Flt3L, Flt3 ligand; MSA, multiple system atrophy; p-tau, phosphorylated tau; PD, Parkinson disease; t-tau, total tau. ROC, receiver operating characteristic.
Results
1. Effects of Age, Gender and Blood Contamination
Consistent with previous studies19, 20, CSF levels of t-tau, along with p-tau, tended to increase with age for all groups studied, with statistical significance achieved in MSA and PD patients for t-tau (P<0.05) and in controls and PD patients for p-tau (P<0.001) (see Supporting Fig 1A, 1B). Levels of Flt3 ligand and fractalkine also increased with age in control (fractalkine: P<0.05; Flt3 ligand: P<0.0001), PD (Flt3 ligand: P<0.0001), and AD (Flt3 ligand: P<0.05) groups (see Supporting Fig 1D, 1E). Similar to other observations19, 21, 22, concentrations of Aβ1-42 were stable with respect to age (see Supporting Fig 1C). No association of the levels of any markers with gender was found (data not shown).
Because contamination of blood in CSF could have a significant impact on the levels of some proteins, including DJ-1 and α-syn14, HGB levels were evaluated in all CSF samples to control for this variable. No association was observed between levels of CSF HGB and t-tau, p-tau or Flt3 ligand; however, Aβ1-42 and fractalkine levels started to increase appreciably at high HGB concentrations (Supporting Fig 2). The reported plasma/serum levels of soluble fractalkine in healthy controls range from ~100 to >1000 pg/mL23-25, but it appears that the effects of blood contamination on CSF fractalkine values were diminished when Aβ1-42 levels were also included in the calculation (see fractalkine/Aβ1-42 discussion in “PD Severity/Progression Correlations”).
2. Group Differences of CSF Aβ1-42, t-tau/p-tau, Flt3 ligand and Fractalkine Levels
As discussed above, tau, Flt3 ligand and fractalkine levels in human CSF were dependent on age. Additionally, the early onset of PD could be secondary to genetic mutations and our AD patients were all older than 50; therefore, the subjects younger than 50 were excluded from this analysis to match the mean and the range of age in all groups.
As expected, significant lower levels of Aβ1-42, accompanied by significant higher levels of t-tau and p-tau, were seen in AD samples in comparison to controls (see Table 1). Concentrations of Aβ1-42 in PD and MSA groups were comparable, but significantly higher than those in AD (P<0.001, PD vs. AD; P=0.01, MSA vs. AD; ANOVA) while much lower than those found in control subjects (P=0.003, PD vs. control; P=0.02, MSA vs. control) (see Table 1). In contrast to those in AD, levels of t-tau were lower in PD and MSA groups than those in controls (P=0.026, PD vs. control; P=0.004, MSA vs. control). The t-tau levels in both PD and MSA patients were significantly lower than those in AD patients (P<0.001). Alterations in p-tau mirrored those of t-tau, but significantly lower p-tau levels were found in the PD group when compared with controls (P<0.001) (see Table 1).
CSF fractalkine levels tended to be higher in PD, MSA and AD groups versus age-matched controls, but no statistical significance was observed. Samples from MSA patients had significantly lower Flt3 ligand concentrations than the other groups (P<0.001, MSA vs. control, MSA vs. PD, or MSA vs. AD) (see Table 1).
The group differences were also evaluated in an ANCOVA model and the conclusions persisted after adjustment for demographic factors (age, gender), CSF HGB and CSF total protein level (data not shown).
3. Diagnostic Utility of Individual or Combinations of Biomarkers
3.1 PD versus CONTROL
In one of our recent investigations, both α-syn and DJ-1 concentrations were lower on average in PD patients as compared to controls and AD patients, with a high diagnostic sensitivity and modest specificity for patients with PD versus controls14. Thus, we first investigated whether adding Aβ1-42, t-tau, p-tau, Flt3 ligand and fractalkine would enhance the diagnostic accuracy of DJ-1 and/or α-syn. Logistic regression analysis suggested that among all seven markers, DJ-1 was still the best predictor when samples with high blood contamination (HGB ≥ 200 ng/mL) were excluded (Fig. 1A and see Table 2; the performance of all individual markers and several combinations can be found in Supporting Table 2; see Table 1 for the numbers of samples with HGB<200 ng/mL). Whenever DJ-1 (and α-syn for that matter) was considered, no significant improvement in diagnostic sensitivity or specificity could be achieved by adding any of the other five markers, whether all patients or only those with age ≥65 were included in calculations (see Supporting Table 2). The only exception was that DJ-1 plus Flt3 ligand slightly enhanced the ROC values (sensitivity 94% and specificity 60%) (see Fig. 1A and Table 2).
Figure 1. Receiver operating characteristic (ROC) analysis of cerebrospinal fluid biomarkers.
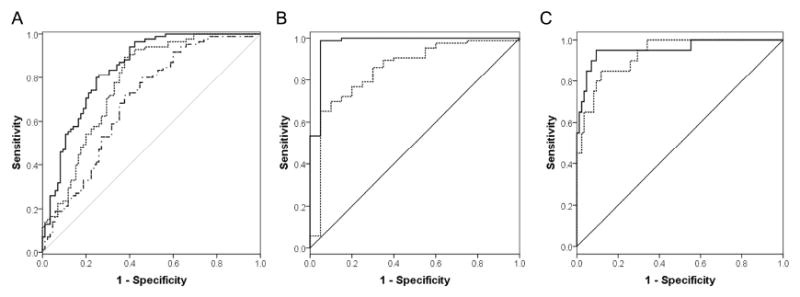
(A) For Parkinson disease (PD) patients versus controls, the combination of DJ-1 and Flt3 ligand (solid) and the combination of phosphorylated tau (p-tau), total tau (t-tau) and amyloid beta peptide 1-42 (Aβ1-42) (dot-dash) were the best discriminating parameters along with DJ-1 (dot) and α-synuclein (not shown) alone (see also Table 2). (B) For PD versus multiple system atrophy (MSA), Flt3 ligand (solid) and the combination of α-synuclein and p-tau% (p-tau/t-tau) (dot) were the best discriminating parameters. (C) For MSA patients versus controls, Flt3 ligand (solid) along with the combination of DJ-1 and p-tau% (dot) were the best discriminating parameters.
3.2 PD versus MSA
As mentioned, it is often difficult to differentiate PD clinically from other parkinsonian disorders, particularly at the early disease stages, and there is no established laboratory test or biomarker that can assist in differential diagnosis in a regular hospital setting. Here, with CSF Flt3 ligand, MSA patients were differentiated from PD patients with excellent sensitivity (99%) and specificity (95%). A high sensitivity (90%) and a reasonable specificity (71%) could also be achieved using a combination of α-syn and p-tau% (a ratio of p-tau and t-tau) when samples with high blood contamination were excluded (see Fig. 1B, Table 2, and Supporting Table 2).
3.3 MSA versus CONTROL
As to MSA patients versus controls, high sensitivity and specificity could be achieved with Flt3 ligand alone (sensitivity 95%, specificity 90%) or a combination of DJ-1 and p-tau% (sensitivity 95%, specificity 70%) when samples with high blood contamination were excluded (see Fig. 1C and Supporting Table 2).
3.4 AD versus CONTROL and PD/MSA
As a confirmation of previous data26, the CSF p-tau/Aβ1-42 or t-tau/Aβ1-42 ratio could discriminate AD patients from controls with high sensitivity and specificity (see Supporting Table 2). We identified that, with the same ratios, high sensitivity (92-95%) and specificity (84-90%) could be achieved for AD versus PD or MSA patients with or without excluding samples with high blood contamination (see Table 2, Supporting Table 2 and data not shown). Similarly, a combination of α-syn and Aβ1-42 yielded high sensitivity and specificity for differentiating the two movement disorders from AD (see Table 2, Supporting Table 2).
4. Validation of the Diagnostic Markers
To validate the candidate diagnostic markers, we also measured the biomarker levels in an independent cohort of PD patients (35 cases with age ≥50 and CSF HGB <200 ng/ml). The models and cutoff values obtained from the discovery investigation were applied to calculate the diagnostic values. Notably, part of the validation set of PD patients was collected at the same site where the MSA cohort was collected, with the same exclusion criteria applied. The markers whose sensitivities have been confirmed are listed in Table 2.
For PD versus MSA, with CSF Flt3 ligand alone, 32 of 35 clinically diagnosed sporadic PD patients were correctly classified (sensitivity = 91.4%, specificity = 95.0%). The sensitivity observed in the discovery set was largely confirmed. Note that the specificity was the same because the same MSA cohort was used.
For PD versus controls, the classification with DJ-1 alone resulted in 97.0% sensitivity and 50.0% specificity. However, combining DJ-1 with Flt3 ligand didn’t improve the classification in this validation set (91.4% sensitivity and 60.0% specificity). On the other hand, the classification became slightly better with α-syn alone (100% sensitivity and 38.4% specificity).
The sensitivity for PD versus AD was also largely confirmed with the independent PD cohort. 88.6% sensitivity was obtained with p-tau/Aβ1-42, 91.4% sensitivity with t-tau/Aβ1-42, and 100% sensitivity with the combination of α-syn and Aβ1-42.
5. PD Severity/Progression Correlations
Once we were able to distinguish the major disease groups from one another, we turned our examinations to the relationship of these proteins with PD severity and progression (indexed by UPDRS scores). Both linear regression and nonparametric correlation analyses revealed that the CSF fractalkine/Aβ1-42 ratio increased significantly with increasing UPDRS scores (r=0.252, P<0.01, Pearson’s correlation; r=0.159, P<0.05, Spearman’s nonparametric correlation) (Fig. 2A). Similarly, the fractalkine/Aβ1-42 ratio also correlated with Hoehn and Yahr (H&Y) stages, which are another way to approximate PD severity (data not shown). It should be noted that this correlation was independent of CSF HGB levels. Specifically, statistical significance could still be achieved (P<0.05) when samples with HGB ≥500 ng/mL (where fractalkine and Aβ1-42 started to increase substantially as a function of HGB, as shown in Supporting Fig 1C, 1E) were excluded. This result was expected because the fractalkine/Aβ1-42 ratio was not correlated with HGB levels in these subjects (r=0.148, P>0.05). Finally, no apparent correlation was found between CSF fractalkine/Aβ1-42 and disease duration of PD patients (data not shown). This is not entirely surprising because although H&Y stages and UPDRS scores, on average, correlate with disease duration, disease duration may not reflect the actual disease severity or disease progression as exemplified by the cohort of DATATOP study (discussed below).
Figure 2. Correlation of CSF fractalkine/Aβ1-42 with Parkinson disease severity and rate of progression.
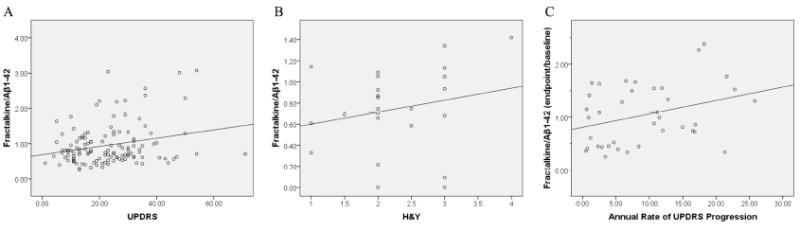
(A) CSF fractalkine and Aβ1-42 levels in cross-sectional CSF samples were measured by Luminex and the fractalkine/Aβ1-42 ratio correlated with disease severity as measured by UPDRS motor scores (r=0.252, P<0.01) using linear regression analysis with Pearson’s correlation. (B) A similar trend was observed between the fractalkine/Aβ1-42 ratio and Hoehn and Yahr (H&Y) stages in a small independent PD cohort (UPDRS not available); however, the correlation was not statistically significant (r=0.216, P>0.05). (C) In an independent longitudinally collected sample set, the fold change of fractalkine/Aβ1-42 (endpoint/baseline) was also well correlated with the annual rate of UPDRS (motor) progression (r=0.325, P<0.05, Pearson’s correlation).
To confirm and extend this result, CSF biomarker levels were measured in two independent cohorts of PD subjects. First, a small independent cross-sectional PD cohort was analyzed using the fractalkine/Aβ1-42 ratio against PD severity, as determined by H&Y stages. The trend of alterations (Fig. 2B) was clearly similar to that shown in Fig. 2A; however, the correlation was not statistically significant (r=0.216; P>0.05). One explanation is the lower statistical power in a smaller cohort. Alternatively, it might be that H&Y stages rather than UPDRS scores (not available in this cohort) were used. H&Y stages are categorical and thus less powerful in this type of statistical analysis.
The second independent cohort was a set of longitudinally collected PD samples from the DATATOP study17, 18. 39 subjects randomized to placebo were included in our study. As shown in Fig. 2C, the fold change of fractalkine/Aβ1-42 (endpoint values / baseline values) significantly correlated with the annual rate of UPDRS progression (r=0.325, P<0.05, Pearson’s correlation; r=0.299, P<0.05, Spearman’s correlation). Notably, in this longitudinal cohort, the alternation of fractalkine alone (i.e., without a need to factor in the changes in Aβ1-42) was well correlated with the clinical progression of PD patients assessed by UPDRS motor scores (r=0.389, P<0.05). These results are consistent with our observation in cross-sectional samples, i.e., in general, a greater increase in CSF fractalkine, along with decreased Aβ1-42 levels, correlated with a higher UPDRS score. The difference between the cross-sectional and longitudinal samples is likely due to the fact that repeated measures in the same subject can reduce variability.
Discussion
One question often encountered in a biomarker study is whether an alteration in a given biomarker is secondary to drug effects. In our previous study, preliminary examination suggested that pharmacotherapy (particularly dopamine-specific drugs) had no apparent effect on CSF DJ-1 or α-syn levels14. Similar analyses for CSF Aβ1-42, t-tau, p-tau, Flt3 ligand and fractalkine were performed in the current study. Notably, in our cohort, 16 of the 126 Parkinson cases were de novo and so not treated with any anti-parkinsonism drugs when the CSF samples were obtained. Another three patients were treated with non-dopamine drugs only14. The CSF protein levels in the de novo patients were not significantly different from patients treated with all anti-parkinsonism drugs or dopamine-specific drugs (L-dopa and dopamine agonists) only. Additionally, the results were not changed when these de novo patients were eliminated from the analysis (data not shown). Although not a focus of our study, these data suggest that pharmacotherapy with anti-parkinsonian medications, particularly those target dopaminergic neurotransmission, have no apparent effect on CSF Aβ1-42, t-tau, p-tau, Flt3 ligand or fractalkine levels. Another potentially confounding issue in a typical biomarker investigation is that patients (PD in this case) may experience comorbidity directly or indirectly related to the disease or its treatment. The presence of potential comorbid conditions that might influence CSF protein levels will need to be investigated in future studies after the major classes of comorbid conditions/diseases are systematically studied.
With these caveats in mind, several major advances were achieved in the current study: 1) CSF Aβ1-42, t-tau and p-tau behaved differently in PD and MSA than in AD; 2) identification of Flt3 ligand alone could differentiate PD from MSA with high sensitivity and specificity; and 3) identification of the fractalkine/Aβ1-42 ratio as a marker that positively correlated with PD severity (UPDRS score) in cross-sectional samples as well as with PD progression in a cohort where CSF samples were collected longitudinally. It should be stressed that our two best PD diagnosis markers, DJ-1 and α-syn, did not correlate with PD severity in our recent study14.
Reduced concentrations of Aβ1-42 in CSF, combined with increased t-tau and p-tau levels, have been generally accepted as diagnostic criteria for AD 26, 27. Here, we observed lower Aβ1-42 levels, though to a less extent than in AD, in both PD and MSA patients (both considered synucleinopathies). This is in line with the suggestion that α-syn and Aβ might interact with each other, thereby potentially contributing to an overlapping pathocascade of AD and PD in some cases28. The decrease in Aβ1-42 levels might also relate to a decrease in overall production of total Aβ as a result of synaptic dysfunction in PD and MSA. Remarkably, however, in contrast to AD patients, we found that tau levels decreased in both PD and MSA, arguing for a clear difference between synucleinopathies and tauopathies (AD is largely considered a disease with tauopathy). The mechanisms underlying a lowered tau in PD and MSA are currently unclear. In addition to a lower tau level in PD patients, we have recently demonstrated that the abnormal CSF p-tau/Aβ1-42 changes, typically seen in AD patients, only occurred in a minority of PD patients (<25%) and were not different from age-matched controls29. This suggests that the potential contribution of typical mechanisms of AD to PD progression appears to occur in a minority of patients with PD, particularly in those who develop cognitive impairment.
Of note, the data for CSF Aβ peptides and tau proteins in PD have been inconsistent. While several previous studies also reported lower levels of CSF Aβ1-42 in PD patients than in controls22, 30-33, others reported normal Aβ1-42 levels in PD34-37. Similarly, while we, as well as others, observed lower levels of tau in PD than in controls16, 38, 39, there are also studies reporting negative results31-34, 36, 40-42. There are no clear explanations for these discrepancies currently; but it cannot be overemphasized that many of the previous studies had limited numbers of subjects (often fewer than 20-30). In the current investigation, we included a large cohort of subjects, providing more power to address a few major variables that might have confounded some of the earlier investigations, including age; aggregation of Aβ is an age-dependent process, and tau tended to increase during aging as demonstrated in the current investigation (see Supporting Fig 1). Other issues to consider include differences in detection technologies and reagents (e.g., antibodies) employed in different investigations. Regarding the detection technology, we measured the CSF levels of the biomarkers by the bead-based Luminex technology, which typically shows much higher sensitivity, throughput and efficiency when compared to enzyme-linked immunosorbent assay (ELISA) or Western blotting. Finally, it should be stressed that, as a quality assurance, our data on AD versus control are completely consistent with almost all prior studies, with values determined within the reported ranges26, 27.
The second major advance of the current investigation relates to the observation that CSF Flt3 ligand differentiated PD from MSA with high sensitivity and specificity (see Fig. 1B and Table 2). Flt3 ligand was identified in a separate pilot CSF cytokine profiling study (with pooled samples) where the levels of 42 cytokines/chemokines were measured in CSF using Luminex technology. Among the 33 proteins detectable in CSF, Flt3 ligand was found to be significantly lower in MSA patients (unpublished data). Flt3 ligand, a cytokine known to act as a neurotrophic and anti-apoptotic factor in the CNS, can induce the proliferation, differentiation and survival of neurons and glia43-45. Thus, a significant decrease of its CSF levels in MSA patients could reflect a decrease of the protein in the CNS that might be responsible for the degeneration of oligodendroglial myelin in MSA.
On the other hand, a combination of α-syn and p-tau% (p-tau/t-tau) could also differentiate PD from MSA reasonably. CSF α-syn levels decreased in both PD and MSA, with the reduction in MSA being more significant (see Table 1). Though detailed mechanisms underlying this phenomenon remain to be determined, we speculate this observation might suggest that there is more widespread or faster neurodegeneration in MSA than in PD, because low CSF α-syn levels might reflect a reduction of ‘free’ α-syn circulating in the CSF7, which is likely due to α-syn aggregation or mis-metabolism. A difference in p-tau% between the two diseases is a quite novel observation, and warrants further investigation not only for diagnostic utility but also for its involvement in disease processes.
The correlation of CSF fractalkine/Aβ1-42 with PD severity/progression is another major discovery of this investigation. Two critical issues related to this correlation should be discussed. First, we selected the UPDRS motor score as an index for PD severity/progression in this study, as it has been utilized effectively to reflect clinical progression of motor impairments in patients with PD; however, the correlation between UPDRS and CSF biomarkers might not need to be particularly strong. This is because the clinical evaluation (UPDRS) reflects progression of disease in the nigrostriatal dopaminergic system, while the CSF biomarkers we measured are indices of pathology in the whole brain; a significant amount of pathology in PD affects other brain areas as PD advances1, 2. It is also unknown whether a strong linear relationship exists between worsening in the UPDRS and the progressive degeneration of the nigrostriatal system, a facet of disease that might be more accurately reflected by some biochemical markers. Furthermore, treatment of PD with drugs that target dopaminergic neurotransmission usually improves signs and symptoms (i.e., UPDRS score), but might not modulate CSF protein biomarkers as demonstrated in our previous14 and current investigations. This important issue will be addressed, at least in part, by correlating CSF biomarkers with structural and functional neuroimaging data in future investigations that are currently underway.
The other question about the correlation is what it means biologically. Fractalkine is a chemokine constitutively expressed in neurons throughout the CNS46. It has been suggested to be responsible for sustaining normal microglial activity through interaction with its receptor CX3CR1, which is also highly expressed in the CNS, primarily by microglia46, 47. Inflammatory changes and glial involvement have been suggested in PD pathogenesis and disease progression15, 48. The fact that fractalkine levels increased with PD severity (see Fig. 2A) and in relation to progression in the fast-progressing patients (see Fig. 2C), suggests that it might reflect pro-inflammatory signaling and microglial activation secondary to neuronal damage. This is in line with reports demonstrating that genetic ablation of microglial Cx3cr1, which is critical in neuron-microglia communication, prevented neuron loss in a mouse model of AD49 and reduced ischemic damage and inflammation50. However, the effects of fractalkine could be more than just a mediator that damages neurons, because activated microglia may function as a “double-edged sword”48, 51. Indeed, fractalkine was also reported to be important for neuroprotection, because Cx3cl (fractalkine) or Cx3cr1 deficient mice demonstrated enhanced neurotoxicity52. If this protective role of fractalkine were true in humans, the increased CSF levels of fractalkine in advanced and faster-progressing PD patients might be interpreted as a response to injury. Either way, these results suggest that more studies are needed to clarify the involvement of fractalkine in PD pathogenesis and progression.
It should be stressed that aside from the few caveats discussed earlier, there were additional limitations associated with the current investigation. First, we had a relatively small number of MSA subjects, so the related conclusions concerning this group need to be validated using a larger set of samples. Similarly, we also had limited numbers of longitudinal samples to validate the PD severity/progression findings, and the conclusions also need to be validated independently. Another limitation is that the sensitivity/specificity of the markers can be negatively impacted by imperfect clinical classifications of diseases or disease stages. This study included multiple markers in multiple groups. Although several measures were employed to minimize the chance of a Type I error, the data should still be interpreted with caution. Finally, some of the comparisons in this study are relevant to clinical decision-making (e.g., distinguishing PD from MSA), but others, such as distinguishing PD from AD, are important in biomarker validation but less so clinically. Future studies should include evaluating these biomarkers in patients with questionable or very early PD, essential tremor, and overlap disorders such as PSP.
In summary, in this primarily discovery study, we demonstrated that by measuring seven CSF biomarkers (DJ-1, α-syn, Flt3 ligand, fractalkine, Aβ1-42, t-tau and p-tau), PD patients can be differentiated from not only normal controls but also patients with AD and MSA. It is also possible to correlate markers in this panel with PD severity, and with disease progression in CSF samples collected longitudinally. However, these results need to be validated in a different, and hopefully even larger, cohort of patients, preferably from an independent group.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health [ES004696 (J.Z.) from NIEHS, NS057567 (J.Z.), NS060252 (J.Z.), NS062684 (T.J.M., C.P.Z., J.B.L. and J.Z.) from NINDS, AG025327 (J.Z.), AG033398 (J.Z.), AG005136 (E.R.P and T.J.M.), AG008017 (K.A.C. and J.F.Q.) from NIA and UL1RR025014 (K.C.C) from NCRR]; Dana Foundation (J.F.Q.); Parkinson’s Disease Foundation (C.P.Z.); Michael J. Fox Foundation (C.P.Z. and J.Z.); Friends of Alzheimer’s Research (E.R.P.); Alzheimer’s Association of Western and Central Washington (E.R.P); Department of Veterans Affairs (C.P.Z. and E.R.P.).
We thank the Parkinson Study Group DATATOP investigators for their efforts in collecting CSF samples, Ms. Peggy Auinger and Dr. Michael McDermott (University of Rochester) for their significant contribution to the current study on the issues related to DATATOP samples. We also deeply appreciate those who have donated their CSF for our studies.
References
- 1.Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18(Suppl 6):S2–12. doi: 10.1002/mds.10557. [DOI] [PubMed] [Google Scholar]
- 2.Litvan I, Halliday G, Hallett M, et al. The etiopathogenesis of Parkinson disease and suggestions for future research. Part I. J Neuropathol Exp Neurol. 2007;66:251–257. doi: 10.1097/nen.0b013e3180415e42. [DOI] [PubMed] [Google Scholar]
- 3.Paviour DC, Price SL, Jahanshahi M, et al. Regional brain volumes distinguish PSP, MSA-P, and PD: MRI-based clinico-radiological correlations. Mov Disord. 2006;21:989–996. doi: 10.1002/mds.20877. [DOI] [PubMed] [Google Scholar]
- 4.Vaillancourt DE, Spraker MB, Prodoehl J, et al. High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology. 2009;72:1378–1384. doi: 10.1212/01.wnl.0000340982.01727.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang CC, Poston KL, Eckert T, et al. Differential diagnosis of parkinsonism: a metabolic imaging study using pattern analysis. Lancet Neurol. 2010;9:149–158. doi: 10.1016/S1474-4422(10)70002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross GW, Petrovitch H, Abbott RD, et al. Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol. 2008;63:167–173. doi: 10.1002/ana.21291. [DOI] [PubMed] [Google Scholar]
- 7.Eller M, Williams DR. Biological fluid biomarkers in neurodegenerative parkinsonism. Nat Rev Neurol. 2009;5:561–570. doi: 10.1038/nrneurol.2009.135. [DOI] [PubMed] [Google Scholar]
- 8.O’Keeffe GC, Michell AW, Barker RA. Biomarkers in Huntington’s and Parkinson’s Disease. Ann N Y Acad Sci. 2009;1180:97–110. doi: 10.1111/j.1749-6632.2009.04943.x. [DOI] [PubMed] [Google Scholar]
- 9.McKinnon JH, Demaerschalk BM, Caviness JN, et al. Sniffing out Parkinson disease: can olfactory testing differentiate parkinsonian disorders? Neurologist. 2007;13:382–385. doi: 10.1097/NRL.0b013e31815a351a. [DOI] [PubMed] [Google Scholar]
- 10.Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213:315–325. doi: 10.1016/j.expneurol.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Tokuda T, Salem SA, Allsop D, et al. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun. 2006;349:162–166. doi: 10.1016/j.bbrc.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 12.Ohrfelt A, Grognet P, Andreasen N, et al. Cerebrospinal fluid alpha-synuclein in neurodegenerative disorders-a marker of synapse loss? Neurosci Lett. 2009;450:332–335. doi: 10.1016/j.neulet.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Waragai M, Wei J, Fujita M, et al. Increased level of DJ-1 in the cerebrospinal fluids of sporadic Parkinson’s disease. Biochem Biophys Res Commun. 2006;345:967–972. doi: 10.1016/j.bbrc.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Hong Z, Shi M, Chung KA, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133:713–726. doi: 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Spec No 2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Sokal I, Peskind ER, et al. CSF multianalyte profile distinguishes Alzheimer and Parkinson diseases. Am J Clin Pathol. 2008;129:526–529. doi: 10.1309/W01Y0B808EMEH12L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parkinson_Study_Group. DATATOP: a multicenter controlled clinical trial in early Parkinson’s disease. Arch Neurol. 1989;46:1052–1060. doi: 10.1001/archneur.1989.00520460028009. [DOI] [PubMed] [Google Scholar]
- 18.Parkinson_Study_Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 19.Sjogren M, Vanderstichele H, Agren H, et al. Tau and Abeta42 in cerebrospinal fluid from healthy adults 21-93 years of age: establishment of reference values. Clin Chem. 2001;47:1776–1781. [PubMed] [Google Scholar]
- 20.Compta Y, Marti MJ, Ibarretxe-Bilbao N, et al. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov Disord. 2009;24:2203–2210. doi: 10.1002/mds.22594. [DOI] [PubMed] [Google Scholar]
- 21.Bibl M, Mollenhauer B, Lewczuk P, et al. Validation of amyloid-beta peptides in CSF diagnosis of neurodegenerative dementias. Mol Psychiatry. 2007;12:671–680. doi: 10.1038/sj.mp.4001967. [DOI] [PubMed] [Google Scholar]
- 22.Mollenhauer B, Bibl M, Esselmann H, et al. Tauopathies and synucleinopathies: do cerebrospinal fluid beta-amyloid peptides reflect disease-specific pathogenesis? J Neural Transm. 2007;114:919–927. doi: 10.1007/s00702-007-0629-4. [DOI] [PubMed] [Google Scholar]
- 23.Yasuda M, Ito T, Oono T, et al. Fractalkine and TGF-beta1 levels reflect the severity of chronic pancreatitis in humans. World J Gastroenterol. 2008;14:6488–6495. doi: 10.3748/wjg.14.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fevang B, Yndestad A, Damas JK, et al. Chemokines and common variable immunodeficiency; possible contribution of the fractalkine system (CX3CL1/CX3CR1) to chronic inflammation. Clin Immunol. 2009;130:151–161. doi: 10.1016/j.clim.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Kim TS, Lim HK, Lee JY, et al. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett. 2008;436:196–200. doi: 10.1016/j.neulet.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Hampel H, Burger K, Teipel SJ, et al. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimers Dement. 2008;4:38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 28.Mandal PK, Pettegrew JW, Masliah E, et al. Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem Res. 2006;31:1153–1162. doi: 10.1007/s11064-006-9140-9. [DOI] [PubMed] [Google Scholar]
- 29.Montine TJ, Shi M, Quinn JF, et al. CSF Abeta42 and tau in Parkinson’s disease with cognitive impairment. Mov Disord. 2010;25:2682–2685. doi: 10.1002/mds.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mollenhauer B, Trenkwalder C, von Ahsen N, et al. Beta-amlyoid 1-42 and tau-protein in cerebrospinal fluid of patients with Parkinson’s disease dementia. Dement Geriatr Cogn Disord. 2006;22:200–208. doi: 10.1159/000094871. [DOI] [PubMed] [Google Scholar]
- 31.Bibl M, Esselmann H, Mollenhauer B, et al. Blood-based neurochemical diagnosis of vascular dementia: a pilot study. J Neurochem. 2007;103:467–474. doi: 10.1111/j.1471-4159.2007.04763.x. [DOI] [PubMed] [Google Scholar]
- 32.Parnetti L, Tiraboschi P, Lanari A, et al. Cerebrospinal fluid biomarkers in Parkinson’s disease with dementia and dementia with Lewy bodies. Biol Psychiatry. 2008;64:850–855. doi: 10.1016/j.biopsych.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 33.Alves G, Bronnick K, Aarsland D, et al. CSF amyloid-{beta} and tau proteins, and cognitive performance, in early and untreated Parkinson’s Disease: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry. 2007;254:IV/2–IV/7. doi: 10.1136/jnnp.2009.199950. [DOI] [PubMed] [Google Scholar]
- 34.Sjogren M, Minthon L, Davidsson P, et al. CSF levels of tau, beta-amyloid(1-42) and GAP-43 in frontotemporal dementia, other types of dementia and normal aging. J Neural Transm. 2000;107:563–579. doi: 10.1007/s007020070079. [DOI] [PubMed] [Google Scholar]
- 35.Holmberg B, Johnels B, Blennow K, Rosengren L. Cerebrospinal fluid Abeta42 is reduced in multiple system atrophy but normal in Parkinson’s disease and progressive supranuclear palsy. Mov Disord. 2003;18:186–190. doi: 10.1002/mds.10321. [DOI] [PubMed] [Google Scholar]
- 36.Kanemaru K, Kameda N, Yamanouchi H. Decreased CSF amyloid beta42 and normal tau levels in dementia with Lewy bodies. Neurology. 2000;54:1875–1876. doi: 10.1212/wnl.54.9.1875. [DOI] [PubMed] [Google Scholar]
- 37.Verbeek MM, Abdo WF, De Jong D, et al. Cerebrospinal fluid Abeta42 levels in multiple system atrophy. Mov Disord. 2004;19:238–240. doi: 10.1002/mds.10687. author reply 240-231. [DOI] [PubMed] [Google Scholar]
- 38.Abdo WF, Bloem BR, Van Geel WJ, et al. CSF neurofilament light chain and tau differentiate multiple system atrophy from Parkinson’s disease. Neurobiol Aging. 2007;28:742–747. doi: 10.1016/j.neurobiolaging.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Molina JA, Benito-Leon J, Jimenez-Jimenez FJ, et al. Tau protein concentrations in cerebrospinal fluid of non-demented Parkinson’s disease patients. Neurosci Lett. 1997;238:139–141. doi: 10.1016/s0304-3940(97)00858-6. [DOI] [PubMed] [Google Scholar]
- 40.Sjogren M, Davidsson P, Tullberg M, et al. Both total and phosphorylated tau are increased in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2001;70:624–630. doi: 10.1136/jnnp.70.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jansen Steur E, Vermes I, de Vos RA. Cerebrospinal-fluid tau protein and aspartate aminotransferase in Parkinson’s disease. Lancet. 1998;351:1105–1106. doi: 10.1016/s0140-6736(05)79387-9. [DOI] [PubMed] [Google Scholar]
- 42.Kahle PJ, Jakowec M, Teipel SJ, et al. Combined assessment of tau and neuronal thread protein in Alzheimer’s disease CSF. Neurology. 2000;54:1498–1504. doi: 10.1212/wnl.54.7.1498. [DOI] [PubMed] [Google Scholar]
- 43.Drexler HG, Quentmeier H. FLT3: receptor and ligand. Growth Factors. 2004;22:71–73. doi: 10.1080/08977190410001700989. [DOI] [PubMed] [Google Scholar]
- 44.Chua SJ, Bielecki R, Wong CJ, et al. Neural progenitors, neurons and oligodendrocytes from human umbilical cord blood cells in a serum-free, feeder-free cell culture. Biochem Biophys Res Commun. 2009;379:217–221. doi: 10.1016/j.bbrc.2008.12.045. [DOI] [PubMed] [Google Scholar]
- 45.Brazel CY, Ducceschi MH, Pytowski B, Levison SW. The FLT3 tyrosine kinase receptor inhibits neural stem/progenitor cell proliferation and collaborates with NGF to promote neuronal survival. Mol Cell Neurosci. 2001;18:381–393. doi: 10.1006/mcne.2001.1033. [DOI] [PubMed] [Google Scholar]
- 46.Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 47.Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Fuhrmann M, Bittner T, Jung CK, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denes A, Ferenczi S, Halasz J, et al. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- 51.Schlachetzki JC, Hull M. Microglial activation in Alzheimer’s disease. Curr Alzheimer Res. 2009;6:554–563. doi: 10.2174/156720509790147179. [DOI] [PubMed] [Google Scholar]
- 52.Cardona AE, Pioro EP, Sasse ME, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.