

Abstract
In terms of the reduced potential energy barrier ζ = ΔuTS/kT, the rate coefficients for chemical reactions are usually expressed as proportional to e-ζ. The coupling between vibrational modes of the medium to the reaction coordinate leads to a proportionality of the regularized gamma function of Euler Q(a,ζ) = Γ(a,ζ)/Γ(a), with a being the number of modes coupled to the reaction coordinate. In this work, the experimental rate coefficients at various temperatures for several chemical reactions were fitted to the theoretical expression in terms of Q(a,ζ) to determine the extent of its validity and generality. The new expression affords lower deviations from the experimental points in 29 cases out of 38 and it accounts for the curvature in the logarithmic plots of rate coefficients versus inverse temperature. In the absence of tunneling, conventional theories predict the curvature of these plots to be identically zero.
1. Introduction
Curvatures in logarithmic plots of enzyme-catalyzed reactions were detected and discussed as early as 1949 by Kistiakowsky and Lumry [1] in a paper on the hydrolysis of urea by urease, and described as widespread by Maier and Tappel [2] in 1955. Among the many examples of curved Arrhenius plots known for enzyme-catalyzed processes are the cleaving of RNA by deoxyribozyme [3], the activation of spinach chloroplast fructose-1,6-biphosphatase [4], the catalytic activity of urocanase from Pseudomonas putida [5], adenylate cyclase from Saccharomyces cerevisiae [6], turnip peroxidase, bovine intestinal phosphatase [2], the kinetics of Shiff base formation between a cholesterol ozonolysis product and dimyristoyl phosphatidyl ethanolamine [7], the activity of cyanobacterial ADP-glucose pyrophosphorylase from Anabaena [8], and the hydrolysis of various amides catalyzed by α-chymotrypsin [9]. Among the explanations put forward to rationalize the observed curvature are the temperature-induced conformational change in the reactant [10], the assumption that the measured rate coefficient actually is a combination of elementary rate coefficients for a multiple-step reaction [6], or the notion that the derivative of ΔHTS with respect to temperature is negative, i. e. 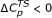 [11]. The curvature of lnkr versus 1/T is thus viewed as a result of a more complex microscopic mechanism in turn composed of individual elementary steps that do follow Transition State Theory (TST) behavior. The shortcomings of TST have been discussed by Pineda and Schwartz [12], who describe promoting vibrations coupled to the reaction coordinate. While their computational analysis spans a time scale of tens of picosecond [13], this work focuses on the low-frequency, long-range motions of the whole protein on a time scale of up to one second for α-chymotrypsin. The link between enzyme dynamics and hydrogen-transfer reactions was also discussed by Knapp and Klinman [14] in the context of the environmentally coupled hydrogen tunneling model. Their analysis rests on the relatively large deBroglie wavelength for hydrogen, and was tested on a thermophilic alcohol dehydrogenase and soybean lipoxygenase-1. Among the extended formulations of TST, the ensemble-averaged variational transition-state theory with multidimensional tunneling (EA-VTST/MT) [15] was successfully applied to many proton- or hydride-transfer reactions, as the interconversion of L-alanine and D-alanine catalyzed by alanine racemase [16], and the hydride transfer from nicotinamide adenine dinucleotide to flavin mononucleotide catalyzed by morphinone reductase [17]. Another way of interpreting the curvature of Arrhenius plots has been described by Masgrau and González-Lafont [18], who postulate the temperature dependence of the activation energy. Their analysis shows that curvature is present even in the absence of variational or tunneling effects, and the calculated Arrhenius plots for two gas-phase reactions are convex at low temperature and concave at high temperatures. Quite interestingly, the experimental Arrhenius plots taken into account in our work, which is not by any means exhaustive, are, without exception, convex at any temperature, with the curvature being even more negative at higher temperatures. This difference could be a consequence of the condensed-phase environment of the reactions investigated in this work to evaluate the curvatures of Arrhenius plots.
[11]. The curvature of lnkr versus 1/T is thus viewed as a result of a more complex microscopic mechanism in turn composed of individual elementary steps that do follow Transition State Theory (TST) behavior. The shortcomings of TST have been discussed by Pineda and Schwartz [12], who describe promoting vibrations coupled to the reaction coordinate. While their computational analysis spans a time scale of tens of picosecond [13], this work focuses on the low-frequency, long-range motions of the whole protein on a time scale of up to one second for α-chymotrypsin. The link between enzyme dynamics and hydrogen-transfer reactions was also discussed by Knapp and Klinman [14] in the context of the environmentally coupled hydrogen tunneling model. Their analysis rests on the relatively large deBroglie wavelength for hydrogen, and was tested on a thermophilic alcohol dehydrogenase and soybean lipoxygenase-1. Among the extended formulations of TST, the ensemble-averaged variational transition-state theory with multidimensional tunneling (EA-VTST/MT) [15] was successfully applied to many proton- or hydride-transfer reactions, as the interconversion of L-alanine and D-alanine catalyzed by alanine racemase [16], and the hydride transfer from nicotinamide adenine dinucleotide to flavin mononucleotide catalyzed by morphinone reductase [17]. Another way of interpreting the curvature of Arrhenius plots has been described by Masgrau and González-Lafont [18], who postulate the temperature dependence of the activation energy. Their analysis shows that curvature is present even in the absence of variational or tunneling effects, and the calculated Arrhenius plots for two gas-phase reactions are convex at low temperature and concave at high temperatures. Quite interestingly, the experimental Arrhenius plots taken into account in our work, which is not by any means exhaustive, are, without exception, convex at any temperature, with the curvature being even more negative at higher temperatures. This difference could be a consequence of the condensed-phase environment of the reactions investigated in this work to evaluate the curvatures of Arrhenius plots.
In this series of tests, we extend the analysis of the nature of the temperature dependence of rate constants to any reaction, not necessarily involving hydrogen transfer or enzyme-catalyzed. We consider the possibility that the plots deviating from Arrhenius behavior actually are relevant to elementary steps, and attempt to give a unitary rationalization of the above observations using a recently proposed relation between the rate coefficient and the potential energy barrier. It was shown for a model system that the coupling between vibrational modes is crucial to the energy transfer to the reaction coordinate [19]. Within this cooperative mechanism, the energy transfer from the low-frequency thermally excited vibrational modes to the reaction coordinate is the leading effect that promotes the barrier crossing from the Michaelis complex to product. Once the coupling between the medium and the reacting system is taken into account, the unimolecular rate coefficient for the process is given by [20]
| (1) |
with ζ = ΔuTS/kT being the reduced potential energy barrier. In (1) the quantities
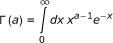 ,
, 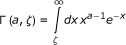 , and
, and 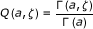
are the complete, incomplete, and regularized gamma functions of Euler, respectively. The vibrational partition functions 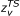 and zυ are relevant to the transition structure and the reagent, ν is the intrinsic response frequency of the reacting system once the threshold energy is acquired. The corresponding expression for bimolecular reactions in solution was subsequently given [21] as
and zυ are relevant to the transition structure and the reagent, ν is the intrinsic response frequency of the reacting system once the threshold energy is acquired. The corresponding expression for bimolecular reactions in solution was subsequently given [21] as
| (2) |
with kd the diffusion rate coefficient, and η a non-dimensional ratio of response frequencies. A bimolecular reaction is envisioned as two reactant species A and B with total energy E colliding with formation of a vibrationally excited species C*, which in turn can revert back to reactants or evolve to products P with the energy-dependent unimolecular rate constants k-1(E) and k2(E), respectively. The ratio of the response frequencies v-1/v2 is the parameter η. The systems following (1-2) exhibit a number a of active modes energetically coupled to the reaction coordinate [22]. In these cases, the non-exponential dependence of the rate constant of the potential energy leads to curved Arrhenius and Eyring plots. On the other hand, the unimolecular expression given by TST provides the exponential dependence
| (3) |
from the reduced barrier or, allowing for a more general system-dependent response frequency,
| (4) |
This work aims to establish whether the validity of (1) and (2) extends beyond the successful interpretations of the hydrolysis of amides by α-chymotrypsin [20] and the alkylation of 3-bromopyridine by iodomethane in acetonitrile [21]. To this effect, the rate coefficient at different temperatures for various unimolecular, bimolecular, and enzyme-catalyzed reactions were examined, restricting the analysis to sets of data with at least four experimental points. By no means does the data discussed in this work cover the extensive literature reporting Arrhenius and Eyring plots for a wide variety of reacting systems. Whenever possible, a statistical analysis on the standard deviations of the activation parameters induced by the error bars of the rate coefficients was carried out.
2. Method
Neglecting the weak temperature dependence of the partition functions, we may express (1, 2, and 4) as
| (5) |
and
| (6) |
with 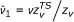 or
or 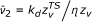 for a unimolecular or bimolecular process, respectively. This approximation is further justified by the consideration in the following treatment of the relative variation of the rate coefficient with respect to ζ through the first and second derivatives of ln k, given by
for a unimolecular or bimolecular process, respectively. This approximation is further justified by the consideration in the following treatment of the relative variation of the rate coefficient with respect to ζ through the first and second derivatives of ln k, given by
| (7) |
The second derivative of ln ka of is in turn
| (8) |
and its curvature
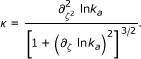 |
(9) |
Typically, a - 1 <ζ, |∂ζ ln ka| << 1, and the sign of κ is negative. For a = 1 (7) and (8, 9) reduce to the Arrhenius behavior where
| (10) |
and
| (11) |
Consequently, if (6) holds true, we should observe plots of ln k1 versus ζ with constant slope and zero curvature. On the other hand, (5) gives a nonzero curvature of ln ka, as shown in Figure 1 for the activation parameters of the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase. To obtain the parameters a,  , and ΔuTS in (5), unconstrained nonlinear regressions of experimental rate constants for various reactions were performed minimizing the deviation
, and ΔuTS in (5), unconstrained nonlinear regressions of experimental rate constants for various reactions were performed minimizing the deviation
| (12) |
Figure 1.

Plot of the curvature of ln ka versus ζ for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase, as given by (9).
with the simplex search method of Lagarias [23]. The form (12) of σ was chosen in order to have the same response for xi and 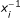 . A typical value of σ = 2.10-2 corresponds to a 15% average deviation. For comparison, constrained minimizations with a = 1 for all reactions (equivalent to Arrhenius plots) were also performed, and the results are given in the supporting information [Additional file 1].
. A typical value of σ = 2.10-2 corresponds to a 15% average deviation. For comparison, constrained minimizations with a = 1 for all reactions (equivalent to Arrhenius plots) were also performed, and the results are given in the supporting information [Additional file 1].
3. Results and Discussion
The set of reactions taken into consideration includes 13 unimolecular, 13 enzyme-catalyzed, and 12 bimolecular processes. Among the unimolecular reactions, rate coefficients for a series of thermolyses of N-benzyl-N-nitrosopivalamides 4-R-C6H4-CH2-N(N = O)-C( = O)-C(CH3)3 and N-benzyl-N-nitrosotosylamides C6H5-CH2-N(N = O)-SO2-C6H4-4-CH3 were reported by Darbeau [24]. These nitrosoamides undergo a rearrangement with elimination of N2 to give the corresponding benzyl esters. Other unimolecular processes taken into consideration are the hydrolysis of trans-dinitrobis(ethylenediamine) cobalt III nitrate in various acids [25], the rearrangement of bis-(4-chlorophenyl) thioncarbonate to bis-(4-chlorophenyl) thiolcarbonate [26], the solvolysis of methyldiphenylsulfonium ion in water and ethanol [27]. The temperature dependence of the rate coefficients for the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO (mole fraction of water 0.973) was investigated with particular care by Euranto [11], whose results stand alone both for the number of experimental points and their accuracy. The unimolecular rate constants for the enzyme-catalyzed processes considered in this work are the oxidation of xanthine to uric acid by xanthine oxidase [28], the hydrolysis of amides catalyzed by α-chymotrypsin [9], and the hydride transfer from NADPH to 7,8-dihydrofolate catalyzed by Escherichia coli dihydrofolate reductase in various solvents [29]. In the logarithmic plots for enzyme-catalyzed processes the deviations from linearity are more evident. The bimolecular processes include the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PMDETA) in acetonitrile [30], the hydration of CO2 by water and OH- [31], and the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate in 90% acetone [32]. The data obtained by the regression procedures are collected in Tables 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12, where it is shown that in 29 out of 38 cases, the minima of σ are located in regions where a > 1.
Table 1.
Activation parameters for the thermolyses of N-benzyl-N-nitrosoamides
| Amide | R | Solvent | a | 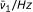 |
ΔuTS / kcal mol-1 | σ | 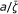 |
|---|---|---|---|---|---|---|---|
| pivalamide | MeO | DMSO | 388 (100.28) |
1.74 10-2 (1.90 10-2) |
245.62 (59.91) |
8.88 10-2 (1.77 10-2) |
0.96 (1.94 10-2) |
| " | Me | " | 213 (111.62) |
4.04 10-2 (1.58) |
143.96 (67.23) |
3.87 10-2 (2.43 10-2) |
0.90 (7.98 10-2) |
| " | H | " | 9 (4.12) |
2.82 105 (1.48 106) |
22.76 (3.23) |
4.02 10-2 (3.32 10-2) |
0.24 (6.79 10-2) |
| " | NO2 | " | 168 | 5.02 10-2 | 119.54 | 5.35 10-2 | 0.86 |
| tosylamide | H | " | 211 | 4.73 10-3 | 174.47 | 1.14 10-1 | 0.88 |
| " | H | toluene | 450 | 2.44 10-3 | 346.84 | 5.63 10-2 | 0.95 |
Standard deviations are given in parentheses.
Table 2.
Activation parameters for the hydrolysis of trans-dinitrobis(ethylenediamine) cobalt III ion
| Acid | a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| HBr 3.547 M | 120 (6.87) |
0.24 (6.87 10-2) |
94.42 (4.25) |
1.47 10-2 (5.13 10-3) |
0.81 (1.02 10-2) |
| H2SO4 2.575 M | 82 (3.62) |
0.66 (0.23) |
70.69 (2.20) |
7.08 10-2 (4.69 10-3) |
0.73 (9.65 10-3) |
| HNO3 6.148 M | 28 (2.92) |
2.70 102 (8.95 102) |
36.19 (1.77) |
5.83 10-2 (4.59 10-3) |
0.48 (2.58 10-2) |
Standard deviations are given in parentheses.
Table 3.
Activation parameters for the rearrangement of bis-(4-chlorophenyl) thioncarbonate to bis-(4-chlorophenyl) thiolcarbonate (without solvent)
| a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|
| 85 | 2.02 10-1 | 125.29 | 6.61 10-3 | 0.72 |
Table 4.
Activation parameters for the solvolysis of methyldiphenylsulfonium ion in H2O and EtOH
| Solvent | a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| H2O | 1 | 3.36 1010 (1.22 1010) |
26.51 (0.22) |
4.04 10-2 (1.06 10-2) |
2.57 10-2 (2.13 10-4) |
| EtOH | 77 (13.75) |
1.00 10-2 (3.48 10-2) |
72.51 (8.76) |
1.01 10-2 (4.98 10-3) |
0.69 (3.87 10-2) |
Standard deviations are given in parentheses.
Table 5.
Activation parameters for the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO
| a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|
| 35 (4.14 10-2) |
0.50 (1.71 10-3) |
29.19 (2.30 10-2) |
2.36 10-3 (2.94 10-5) |
0.70 (2.73 10-4) |
Table 6.
Activation parameters for the oxidation of xanthine by xanthine oxidase
| a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|
| 572 | 2.91 101 | 349.86 | 1.34 10-1 | 0.99 |
Table 7.
Activation parameters for hydrolyses catalyzed by α-chymotrypsin
| Substrate | a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| Suc-Phe-pNAa | 133 (36.81) |
1.62 (28.06) |
94.58 (21.59) |
5.61 10-2 (1.78 10-2) |
0.85 (5.21 10-2) |
| Suc-Ala-Phe-pNA | 221 (21.75) |
0.31 (6.65 10-2) |
143.24 (12.60) |
3.24 10-2 (1.26 10-2) |
0.93 (1.05 10-2) |
| Suc-Ala-Ala-Pro-Phe-pNA | 157 (36.31) |
8.22 102 (2.94 103) |
103.56 (20.94) |
6.16 10-2 (1.90 10-2) |
0.91 (3.43 10-2) |
Standard deviations are given in parentheses.
a pNA = p-nitroanilide
Table 8.
Activation parameters for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in various solvents
| Solvent | a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| H2O | 1 | 2.73 108 (1.26 108) |
8.29 (0.23) |
1.65 10-2 (1.14 10-2) |
7.02 10-2 (1.92 10-3) |
| MeOH 17% | 24 (18.08) |
1.46 104 (4.78 104) |
20.95 (9.92) |
8.20 10-3 (1.03 10-2) |
0.67 (0.11) |
| MeOH 33% | 1 | 1.46 107 (8.52 106) |
6.72 (0.27) |
1.35 10-2 (1.41 10-2) |
8.66 10-2 (3.60 10-3) |
| MeOH 50% | 1 | 3.04 106 (3.89 106) |
6.37 (0.42) |
3.67 10-2 (1.74 10-2) |
8.98 10-2 (6.18 10-3) |
| glycerol 17% | 1 | 3.45 109 (8.72 108) |
9.74 (0.14) |
7.22 10-2 (7.56 10-3) |
5.98 10-2 (8.51 10-4) |
| glycerol 33% | 96 (18.15) |
1.06 103 (1.13 103) |
62.22 (10.18) |
2.39 10-2 (7.37 10-3) |
0.90 (3.09 10-2) |
| glycerol 50% | 169 (20.18) |
3.87 102 (5.56 101) |
104.67 (11.37) |
3.37 10-2 (8.57 10-3) |
0.94 (1.09 10-2) |
| sucrose 17% | 3 (1.00) |
4.65 106 (9.44 106) |
8.80 (0.48) |
1.96 10-2 (1.76 10-2) |
0.20 (5.53 10-2) |
| sucrose 30% | 108 (44.96) |
4.38 102 (1.10 103) |
64.78 (24.83) |
5.79 10-2 (9.23 10-3) |
0.97 (6.75 10-2) |
Standard deviations are given in parentheses.
Table 9.
Calculated (Q(a,ζH)/Q(a,ζD)) and experimental kinetic deuterium isotope effects at 20 C for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in various solvents
| Solvent | a | KIEa | KIEexp |
|---|---|---|---|
| H2O | 1 | 7.15 | 2.70 |
| MeOH 17% | 24 | 2.25 | 2.73 |
| MeOH 33% | 1 | 7.15 | 2.60 |
| MeOH 50% | 1 | 7.15 | 2.70 |
| glycerol 17% | 1 | 7.15 | 2.65 |
| glycerol 33% | 96 | 1.37 | 2.65 |
| glycerol 50% | 169 | 1.23 | 2.66 |
| sucrose 17% | 3 | 5.68 | 3.13 |
| sucrose 30% | 108 | 1.22 | 3.22 |
The corresponding TST value is 7.15.
Table 10.
Activation parameters for the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PMDETAa) in acetonitrile
| Halide | a | 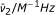 |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| BrANb | 10 | 9.69 102 | 11.90 | 2.99 10-2 | 0.45 |
| MBriB | 24 | 2.95 101 | 18.03 | 3.29 10-2 | 0.72 |
| EtBriB | 11 | 1.70 102 | 11.57 | 6.94 10-2 | 0.51 |
| MBrP | 3 | 5.39 103 | 9.00 | 4.61 10-2 | 0.19 |
| ClPN | 26 | 6.11 | 21.98 | 5.30 10-2 | 0.68 |
| PEBr | 1 | 3.00 105 | 8.71 | 6.67 10-2 | 6.65 10-2 |
| ClAN | 11 | 5.01 101 | 13.97 | 4.84 10-2 | 0.46 |
| AllBr | 1 | 8.00 105 | 9.84 | 9.18 10-2 | 5.88 10-2 |
| BrPN | 1 | 2.11 106 | 6.71 | 3.44 10-2 | 7.26 10-2 |
a PMDETA = N,N,N',N',N''-pentamethyldiethylenetriamine.
b the alkyl halides include three α-bromoesters, four α-halonitriles, benzyl bromide, and allyl bromide. For the exact structures see [30].
Table 11.
Activation parameters for the hydration of carbon dioxide in water
| Reaction | a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|---|
| CO2 + H2O | 30 (16.35) |
1.50 102 (2.61 103) |
35.95 (9.70) |
5.76 10-2 (2.38 10-2) |
0.49 (0.10) |
| CO2 + OH- | 1 | 1.95 1015 (4.49 1014) |
15.33 (0.13) |
1.02 10-1 (1.27 10-2) |
0.038 (3.28 10-4) |
Standard deviations are given in parentheses.
Table 12.
Activation parameters for the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate in 90% acetone
| a | |
ΔuTS / kcal mol-1 | σ | |
|---|---|---|---|---|
| 6 (5.15 10-2) |
1.06 106 (3.50 104) |
29.95 (8.16 10-2) |
3.15 10-2 (4.12 10-3) |
0.14 (8.36 10-4) |
Standard deviations are given in parentheses.
Concerning the physical interpretation of the above analysis, we envision a reacting system as a system composed of s oscillators with the reduced vibrational energy n
| (13) |
computed according to the Debye model [19]. In (13) we have xj = xc(j+1), xc = hωc/2πkT, ωc = 2πc/dc, with dc the diameter of the cluster, c the speed of sound in the medium, and s =⌊ωD/ωc⌋ with ωD the Debye frequency. Thus, a reaction occurs if the energy ζ is localized in one coordinate, according to TST, or in a coordinates if (5) is valid. We may have a qualitative picture of the relative probability of the two events if we consider each unit kT independent of others. In reality the localization of energy into a group of oscillators is a function of how much energy is already in that group, and the more energy is localized, the more intense will be the flow out. In this approximation of independent events, the probability to have energy ζ localized in a group of a oscillators out of a total of s is given by a binomial distribution with the probability of each event p = a/sv
| (14) |
The quantity sv = 3ρV - 6 is the number of vibrational degrees of freedom in a region of given composition, nuclei number density ρ, and volume V. Assuming, for the sake of exemplification, the following functional forms a =αsv/(sv + β), ζ = ζ0 + γa, we may estimate C(n,ζ). Since the calculations involve the evaluation of Euler's gamma function for large arguments, the result is approximate and the distribution is normalized only in the region of small values of dc. Treating both n and ζ as continuous variables, we may evaluate the cumulative distribution function
| (15) |
Plots of the total energy and F(n) are given as a function of the cell size dc in Figure 2 for ζ0 matching the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in 17% methanol.
Figure 2.

Plots of the total vibrational energy in a cluster versus the cluster size dc, the energy per vibrational degree of freedom, and the cumulative binomial distribution functions for one active mode (green) and a active modes (blue). The reduced reaction barrier is taken from the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in 17% methanol. (a) Total energy in units of kT for a cluster solute-solvent of size 10 ≤ dc ≤ 80 Å according to the Debye model. (b) Ratio of the vibrational energy given by (13) in a water cluster over its number of vibrational degrees of freedom. (c-d) Cumulative binomial distribution functions as a function of dc for p = 1/sv (green) and p = a/sv (blue).* The reduced reaction barrier is taken from the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in 17% methanol with α = 200 and β = 105. * Note that in (d) the blue curve lies above the green curve only for dc approximately above 20 Å, indicating the presence of a threshold size for the cooperative effect to take over.
If a classical oscillators among the set of s are coupled to the reaction coordinate, we may estimate that the average energy akT is available to overcome the reaction barrier. In systems with optimal catalysis, the energy akT matches the potential energy barrier, giving the condition a/ζ ≈ 1. In all cases with a/ζ < 1, the coupling is not optimal and the rates are correspondingly slower. In all the Tables we report the average value of ζ in the experimental temperature interval [T1, T2] calculated as
| (16) |
The high potential energy barriers obtained with the minimization of σ are a consequence of the augmented size of the reacting system with respect to the substrate molecule. Although the barrier of 143.96 kcal mol-1 obtained with (5) (second entry in Table 1) might be considered unusual, its high value is due to the participation of the solvent to the reaction coordinate. The gas-phase barriers obtained computationally may thus be regarded as lower limits to a much higher barrier in the condensed phase, augmented by the increase in potential energy due to change in coordinates of the medium to reach the transition structure of a large substrate-solvent cluster.
An efficient catalytic system displays a prompt frequency response (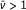 Hz) and a value of
Hz) and a value of 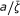 approaching unity. If both the frequency and are lower than unity the reaction rate is less than ideal.
approaching unity. If both the frequency and are lower than unity the reaction rate is less than ideal.
In order to estimate the error bars on the parameters obtained by nonlinear regression to (5) and (6), we reoptimized all the parameters in 10000 runs after allowing the rate constants to vary according to a normal distribution with the experimentally determined variance (the averaged variances for the rate coefficients of the reactions under consideration are: 16.9% for the thermolyses N-alkyl-N-nitrosopivalamides, 5% for the hydrolysis of dinitrobis(ethylenediamine) cobalt III ion, 4.0% for the solvolysis of methyldiphenylsulfonium ion, 0.04% for the hydrolysis of methyl trifluoroacetate, 8.7% for the hydrolyses catalyzed by α-chymotrypsin, 4.7% for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase, 7.1% for the hydration of CO2, and 1.9% for the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate). The standard deviations for each activation parameter were determined and are given in Tables 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12 in parentheses. In all cases where the standard deviations are large, the parameters must be regarded as mere estimates. The Arrhenius-type plots give much narrower error bars with respect to the plots obtained with (5). For example, the standard deviation on reaction barriers with a = 1 lie all below 2 kcal mol-1, while, with the available experimental precision, some values of a exhibit a 50% error bar. Thus the accuracy required in order to obtain small error bars on the activation parameters with (5) is demanding. This degree of accuracy is matched by the excellent set of rate coefficients for the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO, affording the paramenters in Table 5, which exhibit standard deviations in stark contrast with their counterparts in the remaining Tables [11].
The sets of data for the thermolyses of N-benzyl-N-nitrosoamides (Figures 3-4, Table 1) include only four experimentally measured rate coefficients for each reaction, and the values of σ are relatively high (from 3.87·10-2 to 1.14·10-1). With the exception of the unsubstituted pivalamide (which we consider an anomaly), the response frequencies for pivalamides are about 10-2 Hz, while for tosylamides they fall below this value. The response frequencies are higher for the hydrolysis of trans-dinitrobis(ethylenediamine) cobalt III nitrate (Figure 5, Table 2), but the coupling is less effective, as exemplified by the lower values of (0.48 ÷ 0.81). The values of the parameters for the rearrangement of bis-(4-chlorophenyl) thioncarbonate to bis-(4-chlorophenyl) thiolcarbonate (Figure 6, Table 3) fall between the two previous cases, with a response frequency of the order of 10-1 Hz and 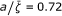 . The solvolysis of methyldiphenylsulfonium ion in water is the first case where the minimum of σ is given by a regression with a = 1, i.e. an Arrhenius plot (all the linear plots are shown in green in the Figures). The corresponding hydrolysis in ethanol exhibits a curved plot (Figure 7, Table 4) with low values of both and . Although the average curvature can in principle only be assigned with confidence in those cases where the experimental determinations exhibit sufficient accuracy, as in the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO (Figure 8), we might tentativley interpret the alternation between zero and non zero curvature as a result of the high sensitivity of the coupling between the substrate and the medium to the values of the parameters governing the dynamics of the cluster (see the effect of the parameter γ in Figure 2). This sensitivity was demonstrated for a model system of two oscillators [19] driven by an external force according to the Debye theory of solids. The oxidation of xanthine to uric acid by xanthine oxidase affords the plot that deviates the most from linearity (Figure 9, Table 6), even though the actual calculated average curvature is not high, due to the large span of ζ values. In this case we obtain a relatively large frequency and a value of near unity. Also the hydrolysis of amides by α-chymotrypsin (Figure 10, Table 7) may be interpreted along the same lines, large values of both the response frequencies and the parameter . The reactions catalyzed by Escherichia coli dihydrofolate reductase exhibits overall higher values for and lower (for an enzyme) values of (Figures 11-12, Table 8) with respect to the previous enzymes. Also, four reactions out of nine have zero curvature, albeit two of them with high values of σ. The experimental values of the deuterium kinetic isotope effects for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in Table 9 may be an indication that the cooperative mechanism is in effect for all solvents. In fact all KIE values are in the range 2.60 ÷ 3.22, well below the estimated value of 7.15 given by classical transition state theory for a difference of 800 cm-1 between the stretching frequencies C-H and C-D. The first bimolecular reaction of our analysis is the activation step of the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PDMETA) (PMDETA = N,N,N',N',N''-pentamethyldiethylenetriamine) in acetonitrile [30] (Figures 13-14, Table 10). The measured rate constants are relevant to the cleavage of the C-X bond (X = Cl, Br) of various halides. With respect to the unimolecular and enzyme-catalyzed reactions discussed previously, this process exhibits low values of a and correspondingly low values of the activation barriers. Values of σ are relatively high (never below 3·10-2, corresponding to an average deviation of 19%) and the values of are lower than optimal. Also, three processes have plots with zero curvature. Both the hydration of CO2 by water (Figure 15, Table 11) and the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate in 90% acetone (Figure 16, Table 12) are cases of nearly linear plots with low values of a. The numerical analysis however reveals the non-zero curvature. In these cases, the experimental error may decide between linear and non linear plots and we are not in a position to draw firm conclusions. Concerning the cases that exhibit a mixed behavior of zero and negative curvatures within the same class of reactions, we limit ourselves to the observation that the value of the deviation σ, averaged over the processes with zero curvature (6.04 10-2) is about twice the value averaged over the reactions of the same class with negative curvature (3.58 10-2).
. The solvolysis of methyldiphenylsulfonium ion in water is the first case where the minimum of σ is given by a regression with a = 1, i.e. an Arrhenius plot (all the linear plots are shown in green in the Figures). The corresponding hydrolysis in ethanol exhibits a curved plot (Figure 7, Table 4) with low values of both and . Although the average curvature can in principle only be assigned with confidence in those cases where the experimental determinations exhibit sufficient accuracy, as in the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO (Figure 8), we might tentativley interpret the alternation between zero and non zero curvature as a result of the high sensitivity of the coupling between the substrate and the medium to the values of the parameters governing the dynamics of the cluster (see the effect of the parameter γ in Figure 2). This sensitivity was demonstrated for a model system of two oscillators [19] driven by an external force according to the Debye theory of solids. The oxidation of xanthine to uric acid by xanthine oxidase affords the plot that deviates the most from linearity (Figure 9, Table 6), even though the actual calculated average curvature is not high, due to the large span of ζ values. In this case we obtain a relatively large frequency and a value of near unity. Also the hydrolysis of amides by α-chymotrypsin (Figure 10, Table 7) may be interpreted along the same lines, large values of both the response frequencies and the parameter . The reactions catalyzed by Escherichia coli dihydrofolate reductase exhibits overall higher values for and lower (for an enzyme) values of (Figures 11-12, Table 8) with respect to the previous enzymes. Also, four reactions out of nine have zero curvature, albeit two of them with high values of σ. The experimental values of the deuterium kinetic isotope effects for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in Table 9 may be an indication that the cooperative mechanism is in effect for all solvents. In fact all KIE values are in the range 2.60 ÷ 3.22, well below the estimated value of 7.15 given by classical transition state theory for a difference of 800 cm-1 between the stretching frequencies C-H and C-D. The first bimolecular reaction of our analysis is the activation step of the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PDMETA) (PMDETA = N,N,N',N',N''-pentamethyldiethylenetriamine) in acetonitrile [30] (Figures 13-14, Table 10). The measured rate constants are relevant to the cleavage of the C-X bond (X = Cl, Br) of various halides. With respect to the unimolecular and enzyme-catalyzed reactions discussed previously, this process exhibits low values of a and correspondingly low values of the activation barriers. Values of σ are relatively high (never below 3·10-2, corresponding to an average deviation of 19%) and the values of are lower than optimal. Also, three processes have plots with zero curvature. Both the hydration of CO2 by water (Figure 15, Table 11) and the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate in 90% acetone (Figure 16, Table 12) are cases of nearly linear plots with low values of a. The numerical analysis however reveals the non-zero curvature. In these cases, the experimental error may decide between linear and non linear plots and we are not in a position to draw firm conclusions. Concerning the cases that exhibit a mixed behavior of zero and negative curvatures within the same class of reactions, we limit ourselves to the observation that the value of the deviation σ, averaged over the processes with zero curvature (6.04 10-2) is about twice the value averaged over the reactions of the same class with negative curvature (3.58 10-2).
Figure 3.

Logarithmic plots of rate coefficients for the thermolyses of N-benzyl-N-nitrosopivalamides 4-R-C6H4-CH2-N(N = O)-C( = O)-C(CH3)3 in DMSO. The experimental points are taken from ref. [24].
Figure 4.

Logarithmic plots of rate coefficients for the thermolyses of N-benzyl-N-nitrosotosylamides C6H5-CH2-N(N = O)-SO2-C6H4-4-CH3 in DMSO (a) and toluene (b). The experimental points are taken from ref. [24].
Figure 5.

Logarithmic plots of rate coefficients for the hydrolysis of dinitrobis(ethylenediamine) cobalt III ion in HBr 3.547 M (a), H2SO4 2.575 M (b), and HNO3 6.148 M (c). The experimental points are taken from ref. [25].
Figure 6.

Logarithmic plots of rate coefficients for the rearrangement of bis-(4-chlorophenyl) thioncarbonate to bis-(4-chlorophenyl) thiolcarbonate. The experimental points are taken from ref. [26].
Figure 7.

Logarithmic plots of rate coefficients for the solvolysis of methyldiphenylsulfonium ion in H2O (a) and EtOH (b). The experimental points are taken from ref. [27].
Figure 8.

Logarithmic plot of rate coefficients for the neutral hydrolysis of methyl trifluoroacetate in H2O/DMSO. The experimental points are taken from ref. [11].
Figure 9.

Logarithmic plot of rate coefficients for the oxidation of xanthine by xanthine oxidase. The experimental points are taken from ref. [28].
Figure 10.

Logarithmic plots of rate coefficients for the hydrolysis of Suc-Phe-pNA (a), Suc-Ala-Phe-pNA (b), and Suc-Ala-Ala-Pro-Phe-pNA (c) catalyzed by α-chymotrypsin. The experimental points are taken from ref. [9].
Figure 11.

Logarithmic plots of rate coefficients for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in water and methanol at various concentrations. The experimental points are taken from ref. [29].
Figure 12.

Logarithmic plots of rate coefficients for the hydride transfer catalyzed by Escherichia coli dihydrofolate reductase in glycerol and sucrose at various concentrations. The experimental points are taken from ref. [29].
Figure 13.

Logarithmic plots of rate coefficients for the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PMDETA) in acetonitrile (see ref. 30 for the actual structures). The experimental points are taken from ref. [30].
Figure 14.

Logarithmic plots of rate coefficients for the atom transfer radical polymerization of alkyl halides catalyzed by Cu(I)Br(PMDETA) in acetonitrile. The experimental points are taken from ref. [30].
Figure 15.

Logarithmic plots of rate coefficients for the hydration of carbon dioxide in water by H2O (a) and OH- (b). The experimental points are taken from ref. [31].
Figure 16.

Logarithmic plots of rate coefficients for the solvolysis of 4-methylbenzhydryl 4-nitrobenzoate in 90% acetone. The experimental points are taken from ref. [32].
Although the high values of the standard deviations for the activation parameters obtained with (5) demand caution in their interpretation, we may qualitatively take the average curvature to measure the amount of deviation from TST caused by the cooperative mechanism, and outline four types of reactions: (a) systems with a limited coupling between the medium and the substrate that follow Arrhenius behavior. In this group the conventional mechanism of exclusive energy transfer to the reaction coordinate is the fastest, with a resulting linear logarithmic plot (Figure 2c); (b) systems with moderate to good coupling (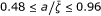 ) and low response frequencies (10-3 ÷ 10-2 Hz). As the ability of the medium to effectively couple modes to the reaction coordinate grows, the coupled mechanism prevails for sufficiently large clusters (Figure 2d); (c) systems with moderate coupling (
) and low response frequencies (10-3 ÷ 10-2 Hz). As the ability of the medium to effectively couple modes to the reaction coordinate grows, the coupled mechanism prevails for sufficiently large clusters (Figure 2d); (c) systems with moderate coupling (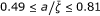 ) and high response frequencies (10-1 ÷ 10-2 Hz); (d) lastly, we may have the predominance of the cooperative mechanism with high response frequencies (10-1 ÷ 103 Hz), and optimal coupling with
) and high response frequencies (10-1 ÷ 10-2 Hz); (d) lastly, we may have the predominance of the cooperative mechanism with high response frequencies (10-1 ÷ 103 Hz), and optimal coupling with 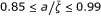 in enzyme-catalyzed reactions.
in enzyme-catalyzed reactions.
4. Conclusions
1. When the vibrational coupling between the substrate and the medium is taken into account, the rate coefficients of unimolecular and bimolecular reactions exhibit a dependence of the reduced potential energy barrier ζ = ΔuTS/kT proportional to the regularized gamma function of Euler Q(a,ζ).
2. The deviations obtained from the experimental rate constants as a function of temperature using equation (5) are in many cases lower with respect to the standard TST equation.
3. The corresponding activation energies exhibit considerably higher values with respect to the values obtained with TST, a fact ascribed to the much larger size of the system under consideration, which adds the contribution of the reaction medium to the intrinsic gas-phase barrier. Response frequencies are correspondingly lower.
4. The standard deviations on the activation parameters obtained using equation (5) are of the same order of magnitude as the parameter themselves, a fact that puts more stringent requirements on the accuracy of the experimentally determined rate coefficients.
5. The reactions subjected to the above analysis may be divided into four groups according to the extent and the features of the substrate-medium coupling: (a) no coupling, linear logarithmic plots of kr versus ζ; (b) coupling with low response frequencies; (c) coupling with high response frequencies; (d) optimal coupling (a/ζ ≈ 1) with very high response frequencies (enzyme catalysis).
6. The proposed expression for the rate constants of reactions in the condensed phase does not yet allow direct calculations of rate coefficients of chemical reactions, in that the physical parameters in the expression (the reduced potential energy barrier ζ, the intrinsic response frequency ν of the reacting system, and the number a of active modes energetically coupled to the reaction coordinate) remain computationally elusive. However, its statistical nature is able to account for the negative curvatures of experimental Arrhenius plots through the intrinsically non-exponential dependence of the rate constants from the reduced potential energy barrier.
5. Competing interests
The author declares that he has no competing interests.
Supplementary Material
Arrhenius plots. The supporting information reports the Arrhenius activation parameters for all the reactions discussed in the text. The parameters were obtained with the same regression procedure outlined in the paper with the constraint a = 1. The corresponding standard deviations are also reported when available.
References
- Kistiakowsky GB, Lumry R. Anomalous temperature effects in the hydrolysis of urea by urease. J Am Chem Soc. l949;71(6):2006–2013. doi: 10.1021/ja01174a029. [DOI] [Google Scholar]
- Maier VP, Tappel AL, Volma DH. Reversible inactivation of enzymes at low temperatures. Studies of temperature dependence of phosphatase- and peroxidase-catalyzed reactions. J Am Chem Soc. 1955;77(5):1278–1280. doi: 10.1021/ja01610a054. [DOI] [Google Scholar]
- Bonaccio M, Credali A, Peracchi A. Kinetic and thermodynamic characterization of the RNA-cleaving 8-17 deoxyribozyme. Nucleic Acid Research. 2004;32(3):916–925. doi: 10.1093/nar/gkh250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley E, Wolosiuk RA. The effect of organic solvents on the activation and the activity of spinach chloroplast fructose-1,6-biphosphatase. J Biol Chem. 1985;260(7):3978–3983. [PubMed] [Google Scholar]
- Hug DH, Hunter JK. Effect of temperature on urocanase from a psychrophile, Pseudomonas putida. Biochemistry. 1974;13(7):1427–1431. doi: 10.1021/bi00704a017. [DOI] [PubMed] [Google Scholar]
- Londesborough J, Varimo K. The temperature-dependence of adenylate cyclase from baker's yeast. Biochem J. 1979;181(3):539–543. doi: 10.1042/bj1810539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach D, Wachtel E, Miller IR. Kinetics of Schiff base formation between the cholesterol ozonolysis product 3β-hydroxy-5-oxo-5,6-secocholestan-6-al and phosphatidylethanolamine. Chemistry and Physics of Lipids. 2009;157(1):51–55. doi: 10.1016/j.chemphyslip.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Gómez-Casati DF, Preiss J, Iglesias AA. Studies on the effect of temperature on the activity and stability of cyanobacterial ADP-glucose pyrophosphorylase. Arch Biochem Biophys. 2000;384(2):319–326. doi: 10.1006/abbi.2000.2101. [DOI] [PubMed] [Google Scholar]
- Stein RL, Case A. Mechanistic origins of the substrate selectivity of serine proteases. Biochemistry. 2003;42(11):3335–3348. doi: 10.1021/bi020668l. [DOI] [PubMed] [Google Scholar]
- Truhlar DG, Kohen A. Convex Arrhenius plots and their interpretation. Proc Natl Acad Sci. 2001;98(3):848–851. doi: 10.1073/pnas.98.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva LT, Euranto EK, Cleve NJ. On the accuracy and significance in determination of the temperature dependence of activation energy in neutral ester hydrolysis and solvolytic substitution reactions. Acta Chem Scand. 1983;B37(2):85–92. [Google Scholar]
- Pineda JRET, Schwartz SD. Protein dynamics and catalysis: the problems of transition state theory and the subtlety of dynamic control. Phil Trans R Soc B. 2006;361(1472):1433–1438. doi: 10.1098/rstb.2006.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D, Basner J, Núñez S, Schwartz SD. Computational and theoretical methods to explore the relation between enzyme dynamics and catalysis. Chem Rev. 2006;106(8):3170–3187. doi: 10.1021/cr0503052. [DOI] [PubMed] [Google Scholar]
- Knapp MJ, Klinman JP. Environmentally coupled hydrogen tunneling. Linking catalysis to dynamics. Eur J Biochem. 2002;269(13):3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- Pu J, Ma S, Gao J, Truhlar DG. Small temperature dependence of the kinetic isotope effect for the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase. J Phys Chem B. 2005;109(18):8551–8556. doi: 10.1021/jp051184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major DT, Gao J. A combined quantum mechanical and molecular mechanical study of the reaction mechanism and α-amino acidity in alanine racemase. J Am Chem Soc. 2006;128(50):16345–16357. doi: 10.1021/ja066334r. [DOI] [PubMed] [Google Scholar]
- Pang J, Hay S, Scrutton NS, Sutcliffe MJ. Deep tunneling dominates the biologically important hydride transfer reaction from NADH to FMN in morphinone reductase. J Am Chem Soc. 2008;130(22):7092–7097. doi: 10.1021/ja800471f. [DOI] [PubMed] [Google Scholar]
- Masgrau L, González-Lafont A, Lluch JM. The curvature of the Arrhenius plots predicted by conventional canonical transition-state theory in the absence of tunneling. Theor Chem Acc. 2003;110(5):352–357. [Google Scholar]
- Canepa C. A stationary-wave model of enzyme catalysis. J Comput Chem. 2010;31(2):343–350. doi: 10.1002/jcc.21264. [DOI] [PubMed] [Google Scholar]
- Canepa C. Rates of catalyzed processes in enzymes and other cooperative media. J Phys Chem B. 2003;107(18):4437–4443. doi: 10.1021/jp0276698. [DOI] [Google Scholar]
- Canepa C. Rate-determining cooperative effects of bimolecular reactions in solution. J Phys Chem A. 2006;110(49):13290–13294. doi: 10.1021/jp063120n. [DOI] [PubMed] [Google Scholar]
- Canepa C, Bach RD. A rate expression for enzymatic reactions. The dynamical relation between coupled oscillators at the active site and the rate enhancement of enzyme-catalysed processes. Phys Chem Chem Phys. 2001;3(18):4072–4081. [Google Scholar]
- Lagarias JC, Reeds JA, Wright MH, Wright PE. Convergence properties of the Nelder-Meade simplex method in low dimensions. SIAM Journal of Optimization. 1998;9(1):112–147. doi: 10.1137/S1052623496303470. [DOI] [Google Scholar]
- Darbeau RW, Perez EV, Brantley M. A study of thermal activation of N-nitrosoamides. Letters in Organic Chemistry. 2006;3(9):689–698. doi: 10.2174/157017806778700051. [DOI] [Google Scholar]
- Lambert DG, Mason JG. The kinetics of hydrolysis of the dinitrobis (ethylenediamine) cobalt (III) ion in various concentrated acid solutions. J Am Chem Soc. 1966;88(8):1633–1636. doi: 10.1021/ja00960a009. [DOI] [Google Scholar]
- Powers DH, Tarbell DS. The Schönberg rearrangement. Kinetics of the rearrangement of bis-(4-chlorophenyl) thioncarbonate to the thiolcarbonate. J Am Chem Soc. 1956;78(1):70–71. doi: 10.1021/ja01582a021. [DOI] [Google Scholar]
- Kevill DN, Anderson SW. Correlation of the rates of solvolysis of the methyldiphenylsulfonium ion. J Org Chem. 1986;51(25):5029–5032. doi: 10.1021/jo00375a059. [DOI] [Google Scholar]
- Mondal MS, Mitra S. Kinetics and thermodynamics of the molecular mechanism of the reductive half-reaction of xanthine oxidase. Biochemistry. 1994;33(34):10305–10312. doi: 10.1021/bi00200a010. [DOI] [PubMed] [Google Scholar]
- Loveridge EJ, Tey L-H, Allemann RK. Solvent effects on catalysis by Escherichia coli dihydrofolate reductase. J Am Chem Soc. 2010;132(3):1137–1143. doi: 10.1021/ja909353c. [DOI] [PubMed] [Google Scholar]
- Seeliger F, Matyjaszewski K. Temperature effect on activation rate constants in ATRP: new mechanistic insights into the activation process. Macromolecules. 2009;42(16):6050–6055. doi: 10.1021/ma9010507. [DOI] [Google Scholar]
- Wang X, Conway W, Burns R, McCann N, Maeder M. Comprehensive study of the hydration and dehydration reactions of carbon dioxide in aqueous solution. J Phys Chem A. 2010;114(4):1734–1740. doi: 10.1021/jp909019u. [DOI] [PubMed] [Google Scholar]
- Goering HL, Hopf H. Stereochemistry of ion-pair return associated with solvolysis of para-substituted benzhydryl p-nitrobenzoates. J Am Chem Soc. 1971;93(5):1224–1230. doi: 10.1021/ja00734a033. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Arrhenius plots. The supporting information reports the Arrhenius activation parameters for all the reactions discussed in the text. The parameters were obtained with the same regression procedure outlined in the paper with the constraint a = 1. The corresponding standard deviations are also reported when available.