

Abstract
This is an in-depth review of the history of quinacrine as well as its pharmacokinetic properties and established record of safety as an FDA-approved drug. The potential uses of quinacrine as an anti-cancer agent are discussed with particular attention to its actions on nuclear proteins, the arachidonic acid pathway, and multi-drug resistance, as well as its actions on signaling proteins in the cytoplasm. In particular, quinacrine's role on the NF-κB, p53, and AKT pathways are summarized.
Nomenclature and chemical grouping
Quinacrine (IUPAC name 4-N-(6-chloro-2-methoxyacridin-9-yl)-1-N,1-N-diethylpentane-1,4-diamine) is a heterocyclic three-ring compound (Figure 1A), and an acridine (Figure 1B) derivative (9-aminoacridine). It is readily available as quinacrine dihydrochloride, the dihydrochloride salt of quinacrine, for clinical use. The interest in quinacrine stems from its long history of therapeutic uses, as will be discussed in the following sections, and in particular its potential antineoplastic activities.
Figure 1.
Structures of acridine and the acridine family member quinacrine. (A) Chemical structure of the acridine family member quinacrine. (B) Chemical structure of acridine.
Quinacrine formulations and isomers are known by numerous designations some of which are: acrichine, Atabrine®, atebrine, atebrin, mepacrine, quinacrine dihydrochloride, quinacrine dihydrochloride dihydrate, quinacrine dihyrochloride (R)-isomer, quinacrine dihyrochloride (S)-isomer, quinacrine dimesylate, quinacrine hydrochloride, quinacrine monoacetate, quinacrine monohydrochloride, quinacrine monomesylate, quinacrine (R)-isomer, quinacrine (S)-isomer, and 6-chloro-9-[[4-(diethylamino)-1 methylbutyl]amino]-2-methoxyacridine. The most commonly used designations for quinacrine are mepacrine, quinacrine hydrochloride, quinacrine dihydrochloride, and the registered name Atabrine®. Quinacrine is one of several known aminoacridines which include, for example, acridine orange, acriflavine, aminacrin, amsacrine, ethacridine, nitracrine, proflavine and tacrine and which have a range of biological and therapeutic applications. Table 1 summarizes some of the key biological and therapeutic applications of these compounds.
Table 1.
Selected aminoacridines and their typical applications
| Acridine orange | A cationic cytochemical stain specific for cell nuclei, especially DNA. It is used as a supravital stain and in fluorescence cytochemistry. It may cause mutations in microorganisms. |
|---|---|
| Acriflavine | 3,6-Diamino-10-methylacridinium chloride mixture. with 3,6-acridinediamine. Fluorescent dye used as a local antiseptic and also as a biological stain. It intercalates into nucleic acids thereby inhibiting bacterial and viral replication. |
| Aminacrine | A highly fluorescent anti-infective dye used clinically as a topical antiseptic and experimentally as a mutagen, due to its interaction with DNA. It is also used as an intracellular pH indicator. |
| Amsacrine | Aminoacridine derivative that is a potent intercalating antineoplastic agent. It is effective in the treatment of acute leukemias and malignant lymphomas, but has poor activity in the treatment of solid tumors. It is frequently used in combination with other antineoplastic agents in chemotherapy protocols. It produces consistent but acceptable myelosuppression and cardiotoxic effects. |
| Ethacridine | A topically applied anti-infective agent. |
| Nitracrine | Acridine antineoplastic agent used in mammary and ovarian tumors. It inhibits RNA synthesis. |
| Proflavine | 3,6-Diaminoacridine. Topical antiseptic used mainly in wound dressings. |
| Tacrine | A cholinesterase inhibitor that crosses the blood-brain barrier. Tacrine has been used to counter the effects of muscle relaxants, as a respiratory stimulant, and in the treatment of Alzheimer's disease and other central nervous system disorders. |
| Quinacrine | An acridine derivative formerly widely used as an antimalarial but superseded by chloroquine in recent years. It has also been used as an anthelmintic and in the treatment of giardiasis and malignant effusions as well as a form of contraception/sterilization. It is used in cell biological experiments as an inhibitor of phospholipase A2. |
Modified from National Library of Medicine - Medical Subject Headings; 2009 MeSH; MeSH Descriptor Data. http://www.nlm.nih.gov/cgi/mesh/2009/MB_cgi?mode=&term=Aminoacridines&field=entry#TreeD03.494.046.250; retrieved July 2010.
History of quinacrine
Originally developed as pigments and dyes, the pharmalogical properties of acridine compounds were first investigated by Ehrlich and Benda in 1912, as antiprotozoal agents that act upon trypanosome parasites and developed further by Carl Browning as antibacterial agents [1-3]. The use of acridines as antibacterial agents fell out of favor in the 1940's after the discovery and wide spread availability of penicillin to combat bacterial infections. However, from the 1940's to the present day acridines have found wide use as antimalarial agents with Atabrine® (quinacrine) being one of the acridine derivatives successfully used to combat the disease. Atabrine® was discovered as part of an intensive antimicrobial research program broadly based on biologically active dyes carried out in 1930's in the German laboratories of I.G. Farbenindustrie. The program covered the preparation and trial of over 12,000 compounds leading to the identification of pamaquine and quinacrine as potential therapeutic agents [4].
Quinacrine was re-discovered in American laboratories as "American Atabrine" during the Second World War when an alternative to quinine was needed for the treatment of malaria [4]. The outcomes from the use of quinacrine in the armed forces demonstrated it to be superior to quinine and made it the official medicine for the treatment of malaria [5] until 1945 when it was substituted by chloroquine [6]. Before the substitution, millions of military personal took Atabrine® for prophylaxis. This allowed physicians in the US armed forces to conduct extensive follow-up studies and provide health professionals with detailed information on the side effects and toxicity of quinacrine, making it among the best studied drugs ever introduced. Three million soldiers took the drug for up to four years in the controlled setting of the military service where arguably compliance and follow up rates are much better than in a typical study in the civilian population [7-9].
Throughout the years, the use of quinacrine has continued attaining FDA-approval for the treatment of diseases such as malaria, giardiasis [10-13] and tapeworm infection [14-16]. Its therapeutic effectiveness has also been demonstrated in controlled studies in combating refractory lupus erythematosus [17-22], rheumatoid arthritis [21,23], and as an adjuvant cancer therapy [24,25]. In addition, quinacrine has been used as an intrapleural sclerosing agent to prevent recurrence of pleural effusion or pneumothorax in patients at high risk of recurrence, resulting in painless pleurodesis and remission of fluid and/or air collections [26-30]. Quinacrine has also been used for regional cancer therapy of pericardial and abdominal effusions with an ~25-50% responses rate [31]. Due to its effectiveness as a sclerosing agent, quinacrine has also been utilized for contraceptive purposes. It produces an asymptomatic fibrosis and occlusion of the fallopian canal [32-35]. It should be noted that for some of these conditions quinacrine has been superseded by other agents, however the use of quinacrine has to date not become contraindicated due to safety concerns. Quinacrine is currently being clinically tested in the treatment of Creutzfeldt-Jakob disease through the National Institute of Aging (NIA) [ClinicalTrials.gov Identifier: NCT00183092] and through the Medical Research Council, in the PRION-1 trial, [ClinicalTrials.gov Identifier: NCT00104663]. In addition, a trial has recently been completed in the treatment of androgen-independent prostate cancer through the University of Chicago and Cleveland Biolabs [ClinicalTrials.gov Identifier: NCT00417274].
Pharmacokinetics of quinacrine
The typical route of quinacrine administration is orally with water after a meal [36]. The drug can also be administered intralesionally/paralesionally [21,25,37,38], intramuscularly, rectally, intravenously [21], transcervically [34], and interstitially [26-30,39].
It is rapidly absorbed from the gastrointestinal tract following oral administration [40] with plasma levels increasing 2-4 hours after administration and reaching a peak in 8-12 hours [7,21]. Plasma concentration increases rapidly during the first week and equilibrates (94%) by the fourth week. Quinacrine is also rapidly absorbed and distributed after intrapleural, intralesion/paralesion, and intrauterine administration [41,42]. The plasma levels of quinacrine remain low in comparison to tissue concentrations. Peak plasma concentrations of up to 140 ng/ml (0.32 μM) for quinacrine have been documented on a standard malaria regimen [8]. It is distributed throughout the body and its liberation from different tissue compartments is slow. The highest concentrations are found in the liver, spleen, lungs and adrenal glands, with liver concentrations reaching 20,000 times that of plasma. The lowest concentrations of the drug are found in the brain, heart and skeletal muscle [6,8]. Quinacrine is also heavily deposited in the skin, fingernails and hair [21]. Spinal fluid concentrations are 1-5% of plasma levels. 80-90% of the drug is bound to plasma proteins when given at therapeutic doses and the half life of the drug is five to fourteen days depending on the dosing regimen [41,43]. Although small amounts are excreted in bile, sweat, and saliva [21,40], the major route of quinacrine elimination is via the renal system which may be enhanced by acidification and reduced by alkalinization [6,7].
Reported quinacrine toxicity
Quinacrine has the advantage of a long history of clinical use in the treatment of malaria, so that human tolerances are well known. In addition quinacrine has displayed tissue specificity making its toxicity tolerable in different therapeutic situations [21,44-46]. The following sections give an overview of the toxicity of quinacrine as it is applicable in the clinical setting.
General toxicity
Mostly minor or reversible adverse reactions include transient symptoms of mild headache, dizziness, or gastrointestinal symptoms (diarrhea, anorexia, nausea, abdominal cramps) which decrease with a reduction in dosage [21]. These symptoms occur in half of the patient population receiving 100 mg of quinacrine daily while almost all patients treated with higher doses experience some sort of adverse reaction. Some infrequent serious side effects of quinacrine have been reported and will be covered in the following sections.
Gastroenterological and hepatic toxicity
Persistent abdominal cramping or diarrhea has been reported for patients receiving the drug. These symptoms are readily dealt with by co-administration of bismuth-containing suspensions or antispasmodic agents. Long-term high-dose malarial suppressive therapy was occasionally associated with reversible hepatitis presumably due to quinacrine's tendency to concentrate in the liver. Transient lupus associated quinacrine hepatitis and peritonitis have also been reported, although these symptoms are attributed to doses three times that of the recommended dose [47,48].
Ophthalmologic and central nervous system toxicity
Quinacrine has very low risk of retinal toxicity [49,50]. At doses over 500 mg the drug has the potential to induce in rare cases a hypersensitivity reaction resulting in corneal edema, which is reversible [51,52]. Cortical stimulatory effects of quinacrine were documented in a study of a group of healthy volunteers given doses of quinacrine ranging between 200 to 1,200 mg daily for ten days [53]. At higher doses symptoms may include restlessness, vertigo, insomnia, nightmares, hyperirritability, psychosis and convulsions. Although toxic psychosis following quinacrine administration has been reported [54-56], large scale studies reveal this to be a rare and quickly reversible event. However it must be noted that a study of over 7,500 US soldiers given quinacrine (100 mg/day) in World War II revealed a 0.4% incidence of toxic psychosis [57]. Further investigations revealed twenty eight (0.1%) CNS-toxic cases among 30,000 treated for malaria [58].
Hematologic toxicity
The most serious potential toxicity of quinacrine is aplastic anemia. The incidence of aplastic anemia in World War II soldiers increased after the drugs introduction, but still remained quite low (0.003%) [59]. Reported cases of aplastic anemia have been associated with patients receiving more than the recommended daily dose and long treatment periods without having blood counts checked [60-65]. In considering this toxicity it is important to note that the potential lethality of aplastic anemia is readily preventable due to the early signs of skin rash. Moreover, hypoplastic anemia can be identified with frequent routine blood tests [60]. In the more modern clinical setting 300 mg/day of quinacrine has been administered and found to be reasonably tolerated with no reported incidence of hematologic toxicity [66].
Dermatologic toxicity
In a study of 120,000 Australian soldiers serving during the Second World War only 1.6 percent developed rashes from quinacrine treatment. Eighty percent were eczematous and twenty percent were lichenoid or exfoliative [67]. Lichen planus was observed in 1 of 2,000 soldiers given 100 mg/day and in 1 of 500 given 200 mg/day. The dermatitis quickly resolved upon cessation of drug administration. Quinacrine can produce a yellow stain in the skin as well as areas of discoloration appearing like "black and blue marks" or bruises presumably due to melanin binding [67-69]. Slate-colored pigmentation of the palate and subungual areas were described in soldiers treated with quinacrine hydrochloride by Lippard and Kauer [69]. Hyperpigmentation of the oral mucosa, typically restricted to the hard palate has since been reported by many others [70]. These marks consist of membrane bound intracellular granules of quinacrine that contain large amounts of iron and some sulphur [67,71-76]. At the doses currently used, approximately half of the patients receiving the drug develop increased pigmentation and in half of these patients, an asymptomatic yellow stain is evident, which is reversible upon reduction to an average daily dose of <50 mg of the drug [21].
Carcinogenicity/Tumorigenicity
There have been no studies conducted to investigate the tumorigenicity of orally administered quinacrine in humans. The data that exist document the use of quinacrine in female sterilization. Retrospective analysis revealed that patients administered with intrauterine quinacrine had a slight but not statistically significant increase in the incidence of cancer compared to a control population. However, the studies concluded that there was no evidence for an excess risk of cancer development [34,77-79].
Conflicting tumorigenicity data have been reported in short term (up to 30 days) animal studies. Studies in female mice and rats have shown that quinacrine (at doses of 30 mg/kg and 22.5 mg/kg, respectively) enhanced growth of implanted tumor cells and decreased survival [80,81]. However, other studies in male mice have shown that quinacrine at doses between 20-25 mg/kg suppress the growth of transplanted tumors and increase the rate of survival [82-84]. These inconsistencies may be explained by a recent study that reveals that dosing schemes in mice that are not equivalent to that used in humans lead to tumor formation [10]. While tumor formation in mice receiving dosages that are equivalent to those currently used in the clinic are equivalent to that of control animals [10].
LD50 established in animal studies
The LD50 of quinacrine hydrochloride for rats is 900 mg/kg by oral administration [85,86]. The LD50 for the i.p. route for rats has not been estimated, but the experiments of Keeler and Richardson [87] suggest that it is approximately 250 mg/kg.
Mechanisms of quinacrine as an anti-cancer agent
Most of the efforts in anti-cancer drug discovery have so far been focused on identifying drugs which target a single protein. Currently there is an increasing recognition for the need of rationally designed drugs that act on several different proteins and pathways [88-90]. Hence "polypharmacology", the term used for drugs that bind to and modulate multiple targets, thereby eliciting several clinical effects, is an exciting and developing area of cancer research [91]. The anti-cancer mechanism of quinacrine is complex, with many potential cellular targets. This "shotgun" nature of the drug is what may make it attractive in the treatment of some cancers. The following sections describe the different anti-cancer mechanisms elicited and signaling pathways modulated by quinacrine.
Quinacrine intercalates into DNA
DNA is generally considered to be one of the biological targets for acridine anticancer compounds. There are three general modes of binding that characterize the compound interactions with double-stranded DNA: intercalation, groove binding and covalent binding [92-94]. Synthetic or natural acridine drugs display varying chemical and biological properties but they share the common property of DNA intercalation. This is due to the presence of an acridine "backbone" that confers a planar structure to the molecules, allowing them to intercalate into DNA by stacking between base pairs. The intercalation of several acridines has been demonstrated whereby the flat polyaromatic chromophore inserts between the base pairs of double-helical DNA. This process is driven by stacking and charge-transfer interactions between the aromatic systems of the acridine compounds and the DNA bases, resulting in unwinding of the helix [95,96]. The acridine derivative quinacrine is no exception, it also binds to DNA by intercalation [95,97-109]. It should be noted that intercalation is not the only type of interaction quinacrine has with DNA, another involves the diaminobutyl side chain which interacts with the minor groove of the DNA and is involved in the stabilization of the double helix against thermal strand separation [110,111].
Parameters such as fluorescence quantum yield (i.e. absorption/emission spectra), binding constant, and flexibility in the quinacrine/polynucleotides complexes have been found to strongly depend on the DNA sequence [102,112,113]. In general, a clear difference has been found between the fluorescence quenching of quinacrine when comparing adenine (A)-thymine (T) rich polynucleotides to guanine (G)-cytosine (C) containing ones. Fluorescence emission is enhanced in AT polymers, and a marked quenching observed in GC polymers [98,100,101,103,106-108,114]. Fluorescence-assayed preferential binding studies of quinacrine to DNA reveal that the neighbor base sequences influence the binding of quinacrine. In particular the sites where a GC base pair is involved were found to display high affinities [115]. The high affinity of quinacrine for DNA via intercalation can be hampered by denaturation or depurination [114,116].
Acridine interaction with nuclear enzymes - potential mechanisms for anti-tumor effects
It has been demonstrated that DNA intercalation is necessary but not sufficient for the antitumor activities of acridines [109,117,118]. Although the chemotherapeutic potency of acridines is partly determined by the strength of DNA binding [117,119-121], the antitumor properties of acridines are not solely due to their DNA binding, but also stem from specific interactions with certain enzymes. Hence the toxicities of acridines are not largely due to an unspecific toxicity associated with DNA damage or binding.
Telomerases as acridine targets
The two major classes of enzymes which have been considered as targets for these intercalating anticancer drugs are telomerases [122-126] and topoisomerases [127-131]. Topoisomerases have been well described as the target of many DNA-binding anti-cancer drugs while telomerases have more recently been the center of attention.
Telomerases are not active in normal somatic cells after birth. However, perhaps as many as 80-90% of cancer cells have reactivated telomerase [132]. Turning on this enzyme complex prevents or reverses telomere degradation and contributes to the growth of a malignant clone [133]. The inhibition of telomerase in cancer cells leads to growth arrest and ultimately cell death [132-134]. Acridines have been shown to help form or stabilize four-stranded intramolecular quadruplex structures (G-quadruplexes or G-quartets) from the guanine-rich DNA sequences of telomeres, which inhibit telomerase activity [123,126]. The formation of G-quadruplexes in telomeric DNA and the subsequent inhibition of telomerase make these conformations important as anti-cancer targets, and the drugs that help to form or stabilize them candidates for chemotherapeutic agents [135,136].
Topoisomerases as acridine targets
Tumor cells are thought to over-express topoisomerase enzymes to enhance cellular proliferation. As the degree of topoisomerase poisoning and inhibition is a function of the amount of the enzyme present, this mechanism provides a potentially selective mode for killing of tumor cells [130,137]. By inhibiting the re-ligation activity of topoisomerase enzymes, acridines convert topoisomerases into DNA damaging agents leading to cellular toxicity and death [117,127-131].
Quinacrine and telomerases
The mechanism of action of quinacrine on telomerase activity is not well described. Dominick et al. found that purified E. aediculatus, T. thermophila, and human telomerase was inhibited by quinacrine [138]. The banding patterns of the telomerase products generated in the presence of quinacrine were, however, not consistent with typical quadruplex stabilizing compounds which tend to cause enrichment of products associated with four repeats of the telomeric sequence [138,139]. Hence the exact process of quinacrine-induced inhibition of telomerase remains unclear. It should be noted that a 50 μM concentration of quinacrine was used to achieve telomerase inhibition, a dose well above the concentration needed to see the cytotoxic effects of quinacrine.
Quinacrine and topoisomerase
Quinacrine is suggested to be a topoisomerase inhibitor as it displays intercalative activity and structural similarity to other acridines. Furthermore, quinacrine, like topoisomerase poisons, inhibits DNA repair [140-142]. It also inhibits excision repair processes in E. coli [143,144] and in human fibroblasts exposed to ultraviolet light [142,145-147]. In addition, quinacrine also sensitizes cultured HA1 cells (a sub-line of Chinese hamster ovary cells) to killing by X-rays and it prevents the repair of single strand breaks [148]. It has further been shown to sensitize cells when applied at the time of irradiation or shortly beforehand and to prevent the enzymatic rejoining of single strand breaks [148]. Hence it has been postulated that the observed radiosensitization is attributable to its capacity to inhibit such repair processes, in which topoisomerases are implicated [141,142,147,149]. It has also been hypothesized [142,146,147] that densely packed chromatin structures must be transiently loosened by topoisomerase to render the DNA damage sites accessible to excision repair enzymes, particularly for access of repair endonuleases that excise the damaged site [150-152].
It must be noted that the majority of reports which suggest that quinacrine is a topoisomerase inhibitor do not provide direct experimental evidence supporting the topoisomerase inhibitory activity of quinacrine. In one set of studies the effect of quinacrine on topoisomerase is assumed from experiments where quinacrine inhibits UV-induced DNA repair with a Kd of 38.1 μM for reparative DNA synthesis [142] and 781 μM for inhibition of DNA incision [146]. Hence in many studies the effect of quinacrine on topoisomerase is assumed to be due to the effect on DNA repair and when quinacrine is shown to inhibit this nuclear enzyme, the concentration required to induce this effect is quite high.
Although relatively few research reports exist that study the direct role of quinacrine in topoisomerase inhibition, a recent report revealed a lack of detectable topoisomerase interaction for quinacrine at doses up to 11 μM [109]. In another study, an indirect measure of topoisomerase activity, the P4 DNA unknotting assay, revealed that a concentration of 50 μM quinacrine was required to inhibit topoisomerase II P4 unknotting activity. However, in the same study the lowest IC50 for growth inhibition was attained in a cell line where drug resistance should have been encountered if the mechanism of action was due to topoisomerase inhibition [153]. Also in the same investigation, no DNA breakage and no DNA-protein binding was observed at lower doses of quinacrine which were observed to have an inhibitory effect on in vitro growth [153]. The notion that relatively high amounts of quinacrine are needed to interfere topoisomerase was shown by dose-dependent inhibition of topoisomerase enzyme activity, with 30-40% inhibition at 20 μM and 80-90% inhibition at 100 μM [141]. In addition, the high (>700 μM) concentration of quinacrine needed to induce DNA incision observed by Thielmann et al. [146] hints that enzymes involved in DNA repolymerization and not topoisomerase may be involved. Taken together these finding indeed support the role of other nuclear enzymes in the anti-tumor effect observed by quinacrine. From the body of evidence in the literature it is valid to assume that the stifled DNA repair observed with quinacrine is mediated by the inhibition of other enzymes, for instance repair-specific UV endonucleases, DNA helicases [154], or DNA polymerases [147], but not topoisomerases. One can also assume that at lower doses the effect of quinacrine may not be attributed to its interaction with the DNA and inhibition of nuclear enzymes as detailed further in later section of this review.
Quinacrine effects on DNA and RNA polymerases
The literature describing the mechanism of quinacrine's anti-tumor effect suggests that two candidate families of nuclear enzymes, DNA polymerase and to a less extent RNA polymerase, may be involved in the mechanism of quinacrine's radiosensitizing ability. Effective nucleotide excision repair requires DNA gaps be filled by reparative DNA synthesis. In principle, all DNA polymerases found in the nucleus may play a role in this gap-filling. The effects of quinacrine on DNA and RNA polymerase reactions in vitro shed light on how quinacrine may inhibit enzymatic polymerization reactions in vivo and induce anti-tumor effects.
Early experiments have hinted at a mechanism of quinacrine preventing the action of DNA and RNA polymerase [155,156]. van Dyke et al. [155] demonstrated that quinacrine inhibits the incorporation of tritiated adenosine triphosphate primarily into RNA and DNA of the erythrocyte-free malaria parasite. In Tetrahymena, 32 μM quinacrine inhibits the synthesis of DNA (almost completely), RNA (70%), and protein (50%), and almost completely blocks the incorporation of labeled acetate into lipid components [156]. Evidence of quinacrine inhibition of DNA and RNA polymerase has also been obtained in E. coli K12, with RNA polymerase inhibition being less sensitive than DNA polymerase to quinacrine inhibition [110,111]. A Kd in excess of 10 μM is reported by O'Brien et al. [110] and Hahn et al. [111]. More recent work revealed that when normal rat liver and Novikoff hepatoma DNA polymerases α, δ, and ε were treated with a dose range of 0.1 μM to 200 μM quinacrine, the drug preferentially inhibited the DNA polymerases from the malignant cells [157]. The IC50 values of quinacrine inhibition were 15.2 μM, 22.6 μM, and 11.4 μM for DNA polymerase α, δ, and ε, respectively, that were isolated from hepatoma, compared to that of 92.5, 200, and 146 μM for DNA polymerase α, δ, and ε isolated from normal rat liver [157]. The observed differences in DNA polymerase inhibition most likely reflects differences in the weakening effect on DNA-protein interactions [157], which in turn suggest a specific change in the DNA-binding domains of the individual polymerase enzymes. This hypothesis has been supported by the discovery of sequence changes of these DNA-binding domains for human and yeast DNA polymerases [158,159].
It should be noted that inhibition of DNA polymerases in other experiments is achieved at much higher concentrations of quinacrine. Inhibition of Hepatitis B virus DNA polymerase by quinacrine was only achieved at over 700 μM [160]. This agrees with the results of Thielmann et al. [146] where approximately the same concentration of quinacrine was needed to induce DNA incision in human fibroblasts. It should also be noted that using a different system to analyze the inhibitory effect of quinacrine on Hepatitis B virus DNA polymerase Hess et al. [161] found quinacrine only to be effective in the 20 to 50 mM range. Hence the cytotoxicity and anti-tumor effect of quinacrine achieved at lower dose well below those needed to generally inhibit polymerase activity must be attributed to other cellular mechanisms.
Interaction with and inhibition of proteins involved in multidrug resistance
Multidrug resistance (MDR) is a major obstacle to the effective treatment of cancer, as MDR proteins aid in the active transport of a broad range of anticancer drugs out of the cancer cells. This export is ATP-dependent, allowing efflux against concentration gradients. An important set of proteins involved in this export is the ATP-binding cassette transporter family, which includes P-glycoprotein (P-gp). P-gp is encoded by the MDR1 gene and its overexpression is one of the major underlying mechanisms of MDR. The upregulation of P-gp in cancer cells has made it an attractive therapeutic target for combating MDR. One hypothesis supposes that P-gp allows cells to achieve MDR by actively pumping the chemotherapeutic agent out of the cells, thereby reducing the toxic effect [162,163]. The interaction of acridine-based chemotherapeutics with P-gp thus inhibits not only their own efflux but may also the efflux of co-administered chemotherapeutics, as well as increasing uptake into cells [164-168]. The interaction of acridine derivatives with proteins involved in MDR is not related to their DNA intercalation capabilities and appears to be an exciting new strategy for chemotherapy [162,167,169].
Quinacrine is implicated in the reversal of the MDR phenotype from several studies. It has been shown to reverse drug resistance to vincristine in a MDR sub-clone of K562 cells (a human chronic myelogenous leukemia cell line) starting at 5 μM [170]. Furthermore, it has been demonstrated to induce cytotoxicity, but the exact mechanism of cell death was not investigated [170]. The effect of quinacrine in reversing the MDR phenotype in leukemia cell lines in vitro was also supported by other investigators who used approximately 6 μM of quinacrine to increase cellular uptake of vincristine. They observed a cytotoxic effect with approximately 1aμM of quinacrine treatment, reducing cell growth by 82% when used alone, and almost completely inhibiting growth when combined with vincristine [171]. These same investigators then went on to conduct in vivo experiments showing the reversal of vincristine resistance with addition of 50 or 80 mg/kg/day of quinacrine [171].
The only direct test of the role of quinacrine as an inhibitor of P-gp has been conducted by using a multidrug resistant human T-cell leukemic cell line which expresses P-gp as a doublet that can be photaffinitty labeled by the analog of vinblastine, N(p-azido-[3-125I]salicyl)-N'-β- amlnoethylvindesine ([125I]NASV) [172]. [125I]NASV specifically binds to P-gp and the inhibition of its binding was used as a read out for the affinity of quinacrine for P-gp. In this study the binding affinity of quinacrine to P-gp was correlated to its ability to increase vinblastine sensitivity. It is, however, noteworthy that, although in an earlier investigation published by the same authors quinacrine increased the toxicity of vinblastine (12-fold) and vincristine (15-fold) at 5 μM and had an IC50 of 14 μM when administered alone [173], in this subsequent study a 50 μM dose only partially reduced [125I]NASV labeled P-gp [172].
The study of quinacrine's role in MDR has not been limited to leukemia but it has also been analyzed in MDR cells from the ovary and prostate cancer. Quinacrine was reported to affect MDR Chinese hamster ovary (CHO) cells at 6 μM in studies measuring the uptake of labeled palmitoyl carnitine and palmitoyl lysophosphatidyicholine. They were more rapidly taken up by the MDR cells and this uptake was reversed after quinacrine treatment back to the rates observed with the parental cell line, hence implicating quinacrine in reversing the MDR [174]. It also enhanced the activity of paclitaxel in hormone-refractory prostate cancer cells both in vitro and in vivo [175]. Quinacrine itself displayed IC50 values of 3.1 μM for PC-3, of 4.7 μM for PC-3M (a MDR sub-clone of the cell line) and of 3.5 μM for DU145 cells. Combination therapy of quinacrine and paclitaxel were determined to be synergistic in both, in vitro and in vivo (mouse xenografts) experiments. The exact mechanism of this synergistic effect was not studied however, the authors attributed to quinacrine's effect on phospholipase A2 (PLA2)[175].
Disruption of the arachidonic acid pathway
Manipulations of the arachidonic acid pathway (Figure 2) have received considerable attention in the chemoprevention of cancer [176-180]. Agents which inhibit this pathway have been demonstrated to hold promise in the chemoprevention of prostate, gastrointestinal, lung as well as esophageal cancer [177,181-184]. Although cyclooxygenase has been the focus of many anti-neoplastic agents targeting the arachidonic acid pathway [185], other components of the pathway could potentially also be promising targets. One such putative target, PLA2, hydrolyzes the sn-2-acyl bond of membrane phospholipids to produce arachidonic acid (Figure 2), which has been implicated in a variety of signal transduction events, including those regulating malignant cell proliferation [186,187]. Histological studies suggest that membrane phospholipase A2 expression levels are associated with tumor aggressiveness in gastric [188] and breast cancers [189].
Figure 2.

The arachidonic acid pathway and its role in tumor promotion. (A) Potential inhibitory role of quinacrine in the arachidonic acid pathway. (B) Potential role of arachidonic acid pathway in tumor promotion. Adapted from http://en.wikipedia.org/wiki/File:Eicosanoid_synthesis.png; retrieved January 2010 and Ulrich [278].
Disruption of the arachidonic acid pathway by quinacrine via inhibition of PLA2, leads to a wide array of effects. The inhibition of PLA2 [190-193] occurs via quinacrine's binding to membrane phospholipids (primarily phosphatidylethanolamine), and subsequent intercalation into the membrane [194-198] and inhibition of PLA2 membrane binding and activity [190-193,199]. The decrease in arachidonic acid due to PLA2 inhibition [199] in turn results in the inhibition of leukotrienes (LOX activity) and prostanoids (COX activity), as well as eicosanoids (MOX/CYP450 activity) [193,194,200-210]. This is of interest since recent reports implicate an enhanced activity of arachidonic acid pathway proteins in preventing apoptosis and promoting tumor progression in head and neck cancer [211-222].
In platelets, the conversion of arachidonic acid to thromboxane is suppressed by quinacrine [21,206,210,223]. Thromboxane is a major factor in blocking the release of arachidonic acid from cellular phospholipases. In addition, thromboxane is involved in angiogenesis and the development of tumor metastasis [224-226]. Quinacrine also decreases prostaglandin E2 (PGE2) production in a dose-dependent manner. Prostaglandin, PGE2 and the COX2-PGE2 pathway/arachidonic acid pathway play an important role in the induction of the pro-inflammatory response and ultimately tumorigenesis [227-229]. PGE2 levels have been implicated in angiogenesis, tumor growth and invasion, apoptosis resistance and suppression of anti-tumor immunity via suppression of T and NK cells, and amplifying Treg [227,230-232]. The upregulation and pro-oncogenic actions of PGE2 have been demonstrated in head and neck cancer [222,233-237].
Quinacrine as an inducer of p53 and inhibitor of the NF-κB and AKT pathways
The deleterious roles of p53 inactivation [238,239] and nuclear factor-kB (NF-κB) hyperactivation [240,241] have been well established in human cancers. They lead to inhibition of cell death and promotion of oncogenesis. Cross talk between these two pathways has been identified and studied (Figure 3). It has been reported that p53 and NF-κB repress each other's activity by competing for transcriptional proteins such as p300 and CREB-binding protein (CBP) [242]. One signaling protein known to influence this competition is IKKα [243,244]. In particular, IKKα has been implicated in phosphorylating and directing CBP to participate in either the p53 or NF-κB pathway [243,244]. Another well studied signaling protein, AKT, can both activate IKKs as well as phosphorylate and enhance the transcriptional activity of p65 (NF-κB complex protein) [245-247]. In addition it has been demonstrated that AKT-mediated phosphorylation of MDM2 inhibits p53 stabilization [248]. Hence, it is likely that inhibitors of AKT activation could be utilized as anti-cancer agents for the inactivation of p53 and the inhibition NF-κB signaling.
Figure 3.
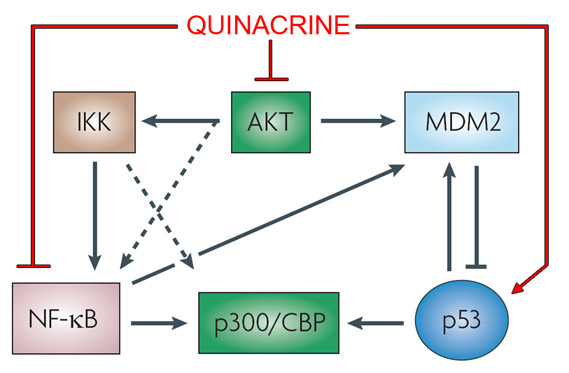
Cross talk between NF-κB and p53. There are many lines of crosstalk between the p53 and NF-κB pathways. A few of these are highlighted, such as AKT and the transcriptional co-activator proteins CREB-binding protein (CBP) and the related p300 protein. AKT can activate both IκB kinases (IKKs) and phosphorylate p65. AKT-mediated MDM2 phosphorylation can also inhibit p53 stabilization. Due to competition for the limited pool of CBP/p300, this protein also plays a crucial role in determining which pathway dominates in terms of cellular outcome. In addition, NF-κB has been shown to directly upregulate the levels of MDM2 mRNA and hence the protein. One promising aspect of quinacrine is its simultaneous ability to inhibit AKT, to induce the p53 pathway, and to inhibit the NF-κB pathway. Adapted from Dey et al. [279].
Besides the development of AKT inhibitors, there has been no concerted effort to rationally design drugs that can simultaneously activate p53 and inhibit NF-κB. The opposing nature of these pathways suggests that a drug which activates p53 and simultaneously inhibits NF-κB would have significant clinical potential due to the fact that it is concomitantly modulating two critical cancer targets. In addition, a drug capable of affecting both of these pathways would also be a useful tool to study the interactions between the opposing p53 and NF-κB pathways. The literature [249] and further unpublished work from members of the Tumor Biology Group of NIDCD at NIH point to quinacrine as being such a drug (VanWaes et al., unpublished data).
Quinacrine and p53
Several recent investigations support the notion that quinacrine-induced cell death is caused by a mechanism that is independent of DNA damage [250-252]. Quinacrine has been demonstrated to stabilize p53 in a manner that differs from that of DNA-damage induced p53 stabilization [250,251]. Wang et al. have reported the ability of quinacrine and other acridine derivatives to activate wild-type p53 transcription in ovarian cancer, non-small cell-lung carcinoma and colon adenocarcinoma cell lines, independent of DNA damage and MDM2 [251]. This DNA damage- and MDM2-independent effect of quinacrine in activating wild-type p53 in a diverse set of cell lines was supported by Gurova et al. when they found that quinacrine activates p53 in renal cell carcinomas, non-small cell lung carcinoma, colon and breast carcinomas, prostate adenocarcinomas, and fibrosarcomas [250]. Friedman et al. extended these findings to reveal that quinacrine activates p53 in several different head and neck squamous cell carcinoma cell lines with wild-type p53 [249]. The cell death induced after quinacrine treatment was not only p53 dependent [250], but also involved Bcl-2-associated X protein (BAX) [251], thereby indicating an important role of the mitochondrial apoptosis pathway. This suggests that other signaling proteins may also be involved in the cell death induction by quinacrine. The mechanism of p53 activation by quinacrine and its ability to modulate other signaling proteins may minimize the toxic side effects seen with treatments using DNA-binding platinum agents, making it potentially a desirable anticancer agent.
Quinacrine and NF-κB
The unique mechanism of p53 upregulation which differs from the genotoxic upregulation of p53 was not investigated by Wang et al. [251], but Gurova et al. [250], found the induction of p53 to be linked to the inhibition of NF-κB. These results were later extended using a skin inflammation mouse model where the contact hypersensitivity (CHS) response to chemical allergen sensitization was evaluated [253]. In this mouse model the authors identified NF-κB to be critical in the development of the contact hypersensitivity response and demonstrated that quinacrine reduced CHS by inhibiting NF-κB activation and as well as cytokines (TNFα, IL-1β, and CCL21) that are dependent on NF-κB activation [253].
NF-κB is also a key regulator of cytokine-induced expression of endothelial cellular adhesion molecules (CAMs) [254,255]. The inhibitory effect of quinacrine on NF-κB in this context was supported in experiments where NiCl2- and CoCl2-induced cellular activation of ICAM-1 was inhibited by quinacrine [256]. The effect was attributed to PLA2 because the enzyme causes the generation of platelet activation factor and eicosanoids [257], which are thought to play a role in the activation of NF-κB [256].
NF-κB has been recently shown to also depend on arachidonic acid metabolites [258] and upstream inhibition by quinacrine has been proposed to inhibit the activation of NF-κB due to its inhibitory effects on PLA2 [259]. In a study evaluating the effect of lysophosphatidic acid (LPA) on endothelial cell activation, quinacrine blocked LPA-stimulated activation of NF-κB as well as the increase in expression of genes known to be dependent on the activation of the NF-κB transcription factor: E-selectin, ICAM-1, IL-8, and MCP-1 [260].
Another interesting line of investigation further revealed that reactive oxygen intermediates (ROI) are implicated in UVB-induced expression of TNFα in keratinocytes and that COX products and, more importantly, LOX products, also known as eicosanoids, which are themselves products of an oxidative metabolism, are the main ROI implicated in this induction [261]. The investigators hypothesize that eicosanoids likely exert their function through activation of NF-κB [261]. They also attributed the reduction of TNFα mRNA after quinacrine administration to the inhibitory activity of quinacrine on PLA2, based on reports showing that UVB can induce PLA2 in keratinocytes [262-264]. Another study investigating the source of oxygen radicals which activate Kupffer cell NF-κB after co-culture with AH70 cells attributed the attenuation of oxidative NF-κB activation to the PLA2 inhibitory activity of quinacrine [265].
However, because many studies do not directly implicate PLA2 inhibition by quinacrine as the mechanism of NF-κB inhibition and in light of more recent investigations challenging the notion that quinacrine acts primarily as an inhibitor of PLA2 [200,253,266], quinacrine's effect is presumably, at least partially, due to NF-κB inactivation via a mechanism other than PLA2 inhibition. Pupe et al. [261] present another intriguing mechanism for NF-κB inactivation as their experiments revealed quinacrine to inhibit UVB-induced IκBα degradation. However, this type of inhibition may be tumor-specific since another mechanism of NF-κB inhibition, nuclear translocation and sequestration of an inactive complex, has been well documented.
Unlike other NF-κB inhibitors, quinacrine does not inhibit the NF-κB pathway via cytoplasmic sequestration of p65. Instead, published experiments indicate a mechanism involving confinement of the p65 complex in the nucleus in an inactive state [250]. The increased presence of NF-κB in the nucleus after quinacrine exposure was supported by experiments revealing increased DNA binding of NF-κB after quinacrine treatment alone or in combination with TNFα [267]. These experiments also revealed that quinacrine plus TNFα induced greater NF-κB DNA binding than TNFα treatment alone. The lack of DNA binding inhibition of NF-κB after quinacrine treatment was also confirmed in a report by Fabbri et al. [268].
Down-regulation of p65 Ser536 phosphorylation by IKKα has been suggested as the primary mechanism of action for quinacrine [250]. The physiological importance of this inhibition was recently confirmed in the skin inflammation mouse model described in a previous section [253]. Further support of this mechanism was attained by Na et al. when hydrogen peroxide-induced phosphorylation of the p65 subunit of NF-κB was partially inhibited by quinacrine [269]. The effects of quinacrine on NF-κB are in line with its uses in the treatment of inflammatory diseases as a single agent or in combination with other medications [21]. As discussed further below, first hints of the mechanism by which quinacrine may inhibit the NF-κB pathway and promote the p53 pathway have come from studies with 9-aminoacridine (9AA), which implicated AKT and mTOR as targets for quinacrine [270].
Quinacrine and AKT
As shown in Figure 3, AKT is involved in the NF-κB and p53 signaling pathways [245,271,272]. AKT phosphorylates the p65 subunit of NF-κB at Ser536, ultimately stimulating NF-κB transcriptional activity [272,273]. In addition, AKT phosphorylates MDM2 on Ser166 [271]. AKT phosphorylation of MDM2 induces translocation of MDM2 into the nucleus and targets p53 for destruction [271]. Phosphorylated MDM2 also transports p53 from the nucleus to the cytoplasm where it is involved in the induction of p53 degradation through the proteasome. Therefore, AKT is a critical signaling protein involved in the suppression of p53 activity. This hypothesis has been supported by experiments demonstrating a correlation between AKT kinase activity and inhibition of p53 [272].
Guo et al. demonstrated that 9AA inhibits AKT activity and its phosphorylation at Ser473 [270]. They went on to show that this inhibition was not a direct effect of reduced PI3K activity and implicated mTOR in this inhibition. Hence, it seems that acridines like quinacrine may be involved in stopping a positive feedback loop between AKT and mTOR [270]. The inhibition of AKT activity by 9AA has also been confirmed by other investigators in a model of human T-cell leukemia virus-transformed cells [274]. Furthermore, in a study of the role of arachidonic acid metabolism and epidermal growth factor (EGF) receptor in neurotensin-induced prostate cancer cell growth, quinacrine's activity as an inhibitor of AKT was reaffirmed. These experiments revealed quinacrine to inhibit neurotensin-, and to a lesser degree EGF-stimulated phosphorylation of AKT [275].
The multiple actions of quinacrine and its established history of safety make it an attractive anti-neoplastic chemotherapeutic agent
Since its discovery as a potent antimalarial compound, quinacrine has been effective not only in the treatment, but also as a prophylaxis for malaria as well as a medication for a wide range of other disorders. Due to its anti-inflammatory activity in patients with autoimmune disorders quinacrine has been used to treat lupus erythematosus, rheumatoid arthritis, bronchial asthma and other inflammatory diseases. The safety of and bioavailability of quinacrine has been demonstrated as patients with these diseases used quinacrine for months at a time to control their symptoms. The pharmacokinetics and safety of quinacrine has been extensively studied as it was administered as a protective measure to millions of US soldiers in the Pacific region during World War II.
Some of the more serious side effects of quinacrine are mild in comparison to other anti-cancer chemotherapeutics and most of the conditions can be easily reversed after treatment cessation or dose reduction. Many of quinacrine's side effects develop gradually, starting from minor lesions in the case dermatitis or a slight decrease in blood counts in the development of anemia, and have been found to be completely and easily reversible, if quinacrine use is discontinued at this early stage [21,49,64,276]. Indeed some of the side effects exhibited due to quinacrine treatment can be used in the clinical setting to confirm proper dosing of the drug in the treatment of cancer patients. The yellow discoloration of the skin due to the accumulation of the bright yellow compound would indicate to the clinician that the drug has reached the equilibrium and as in the case of squamous cell carcinomas, has potentially reached areas where tumor has developed.
Furthermore, the polypharmacology of quinacrine make it an attractive drug in the use of different cancer types. In addition, as inflammation is now being considered the seventh hallmark of cancer [277], quinacrine's anti-inflammatory effects would seem to increase its potential utility as a anti-cancer drug. As more research is being conducted into quinacrine's mechanisms of action, investigators have begun to realize that its interactions extend beyond mere DNA binding and effects on nuclear proteins. Quinacrine has thus been shown to bind and inhibit proteins involved in multidrug resistance, to disrupt the arachidonic acid pathway, as well as affecting the p53, NF-κB and AKT pathway. Its effects on multiple key signaling pathways, implicated in the malignant progression of numerous cancer types, make quinacrine an exciting candidate as a chemotherapeutic agent in new types of combination treatments. Continued research into the mechanisms of this drug is clearly warranted as it may be used in addition to established therapeutic regimes in hopes of ultimately reducing toxic side effects of drugs, such as DNA damaging agents, currently used in the clinic.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors contributed to the writing of this manuscript and approved the final version.
Contributor Information
Reza Ehsanian, Email: rezae@stanford.edu.
Carter Van Waes, Email: vanwaesc@nidcd.nih.gov.
Stephan M Feller , Email: stephan.feller@imm.ox.ac.uk.
Acknowledgements
The authors would like to thank Zhong Chen, Jay Friedman, and Liesl Nottingham for their input. This work was partially supported by the HeadsUp and CRUK charities. Reza Ehsanian was supported by an NIH-Oxford Fellowship, a Stanford Medscholars Fellowship and HHMI Scholars Fellowship. RE and CVW were supported by NIDCD intramural project ZIA-DC-000016.
References
- Browning CH, Cohen JB, Gaunt R, Gulbransen R. Relationships between antiseptic action and chemical consitution with special reference to compounds of the pyridine, quinoline, acridine and phenazine series. Proceedings of the Royal Society. 1922;93:329–366. doi: 10.1098/rspb.1922.0025. [DOI] [Google Scholar]
- Browning CH. Synthetic dyes as antiseptics and chemotherapeutic agents. Nature. 1922;109:750–751. [Google Scholar]
- Wainwright M. Acridine-a neglected antibacterial chromophore. J Antimicrob Chemother. 2001;47:1–13. doi: 10.1093/jac/47.1.1. [DOI] [PubMed] [Google Scholar]
- Greenwood D. Conflicts of interest: the genesis of synthetic antimalarial agents in peace and war. J Antimicrob Chemother. 1995;36:857–872. doi: 10.1093/jac/36.5.857. [DOI] [PubMed] [Google Scholar]
- Office of the Surgeon General UCln. The drug treament of malaria, suppressive and clinical. J Am Med Assoc. 1943;123:205–208. [Google Scholar]
- Goodman LS, Gilman A, (Eds.) Goodman and Gilman's The Pharmacological Basis of Therapeutics. 2. New Yourk: Macmillian; 1954. [Google Scholar]
- Joint Report of the Armored Medical Research Laboratory and Commission on Tropical Diseases AEB, Preventative Medicine Service, Office of the Surgen General, US Army. Plasma quinacrine concentration as a function of dosage and enviornment. Arch Intern Med. 1946;78:64–107. doi: 10.1001/archinte.1946.00220010074007. [DOI] [PubMed] [Google Scholar]
- Shannon JA, Earle DP, Brodie BB, Taggart JV, Berliner RW, Service TRSotR. The pharmacological basis for the rational use of Atabrine in the treatment of malaria. J Pharmacol Exp Ther. 1944;81:307–330. [Google Scholar]
- Jailer JW. Fluorescent microscopic study of the physiological distribution of Atabrine. Science. 1945;102:258–259. doi: 10.1126/science.102.2645.258. [DOI] [PubMed] [Google Scholar]
- Cancel AM, Dillberger JE, Kelly CM, Bolte HF, Creasy DM, Sokal DC. A lifetime cancer bioassay of quinacrine administered into the uterine horns of female rats. Regul Toxicol Pharmacol. 2009. [DOI] [PubMed]
- Babb RR. Giardiasis. Taming this pervasive parasitic infection. Postgrad Med. 1995;98:155–158. [PubMed] [Google Scholar]
- Mandell GL, Bennett JE, Dolin R. In: Principles and practice of infectious diseases. 4. Mandell GL, Bennett JE, Dolin R, editor. New York: Churchill Livingstone; 1995. Principles and practice of infectious diseases; pp. 4–69. [Google Scholar]
- Lerman SJ, Walker RA. Treatment of giardiasis: literature review and recommendations. Clin Pediatr (Phila) 1982;21:409–414. doi: 10.1177/000992288202100704. [DOI] [PubMed] [Google Scholar]
- Pappas PW, Leiby DA. Competitive, uncompetitive, and mixed inhibitors of the alkaline phosphatase activity associated with the isolated brush border membrane of the tapeworm Hymenolepis diminuta. J Cell Biochem. 1989;40:239–248. doi: 10.1002/jcb.240400212. [DOI] [PubMed] [Google Scholar]
- Cruz AC. Treatment of human taeniasis in the Philippines: a review. Southeast Asian J Trop Med Public Health. 1991;22(Suppl):271–274. [PubMed] [Google Scholar]
- Koul PA, Wahid A, Bhat MH, Wani JI, Sofi BA. Mepacrine therapy in niclosamide resistant taeniasis. J Assoc Physicians India. 2000;48:402–403. [PubMed] [Google Scholar]
- Feldmann R, Salomon D, Saurat JH. The association of the two antimalarials chloroquine and quinacrine for treatment-resistant chronic and subacute cutaneous lupus erythematosus. Dermatology. 1994;189:425–427. doi: 10.1159/000246899. [DOI] [PubMed] [Google Scholar]
- Werth V. Current treatment of cutaneous lupus erythematosus. Dermatol Online J. 2001;7:2. [PubMed] [Google Scholar]
- Kalia S, Dutz JP. New concepts in antimalarial use and mode of action in dermatology. Dermatol Ther. 2007;20:160–174. doi: 10.1111/j.1529-8019.2007.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana I, Sala R, Bazzani C, Ceribelli A, Zane C, Cattaneo R, Tincani A, Calzavara-Pinton PG, Franceschini F. Treatment of lupus skin involvement with quinacrine and hydroxychloroquine. Lupus. 2009;18:735–739. doi: 10.1177/0961203308101714. [DOI] [PubMed] [Google Scholar]
- Wallace DJ. The use of quinacrine (Atabrine) in rheumatic diseases: a reexamination. Semin Arthritis Rheum. 1989;18:282–296. doi: 10.1016/0049-0172(89)90050-4. [DOI] [PubMed] [Google Scholar]
- Lipsker D, Piette JC, Cacoub P, Godeau P, Frances C. Chloroquine-quinacrine association in resistant cutaneous lupus. Dermatology. 1995;190:257–258. doi: 10.1159/000246710. [DOI] [PubMed] [Google Scholar]
- Rynes RI. Antimalarial drugs in the treatment of rheumatological diseases. Br J Rheumatol. 1997;36:799–805. doi: 10.1093/rheumatology/36.7.799. [DOI] [PubMed] [Google Scholar]
- Denny WA, Baguley BC, Cain BF, Waring MJ. In: Molecular apects of anticancer drug action. Neidle S, Waring MJ, editor. London: Macmillin; 1983. Antitumor acridines; pp. 1–34. [Google Scholar]
- Hiller RI. A study of quinacrine dihyochloride in the human breast in vitro and in vivo. Am J Surg. 1970;119:317–321. doi: 10.1016/0002-9610(70)90058-9. [DOI] [PubMed] [Google Scholar]
- Taylor SA, Hooton NS, Macarthur AM. Quinacrine in the management of malignant pleural effusion. Br J Surg. 1977;64:52–53. doi: 10.1002/bjs.1800640113. [DOI] [PubMed] [Google Scholar]
- Koldsland S, Svennevig JL, Lehne G, Johnson E. Chemical pleurodesis in malignant pleural effusions: a randomised prospective study of mepacrine versus bleomycin. Thorax. 1993;48:790–793. doi: 10.1136/thx.48.8.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrenius V, Ukale V, Widstrom O, Kallenius G, Svenson SB. Quinacrine-induced pleural inflammation in malignant pleurisy: relation between drainage time of pleural fluid and local interleukin-1 beta levels. Respiration. 1993;60:366–372. doi: 10.1159/000196237. [DOI] [PubMed] [Google Scholar]
- Janzing HM, Derom A, Derom E, Eeckhout C, Derom F, Rosseel MT. Intrapleural quinacrine instillation for recurrent pneumothorax or persistent air leak. Ann Thorac Surg. 1993;55:368–371. doi: 10.1016/0003-4975(93)90999-X. [DOI] [PubMed] [Google Scholar]
- Larrieu AJ, Tyers GF, Williams EH, O'Neill MJ, Derrick JR. Intrapleural instillation of quinacrine for treatment of recurrent spontaneous pneumothorax. Ann Thorac Surg. 1979;28:146–150. doi: 10.1016/S0003-4975(10)63772-1. [DOI] [PubMed] [Google Scholar]
- Karnofsky DA. Cancer Chemotherapeutic Agents. CA Cancer J Clin. 1964;14:67–72. doi: 10.3322/canjclin.14.2.67. [DOI] [PubMed] [Google Scholar]
- Bhatt RV, Aparicio A, Laufe LE, Parmley T, King TM. Quinacrine-induced pathologic changes in the fallopian tube. Fertil Steril. 1980;33:666–667. [PubMed] [Google Scholar]
- Hieu DT, Tan TT, Tan DN, Nguyet PT, Than P, Vinh DQ. 31,781 cases of non-surgical female sterilisation with quinacrine pellets in Vietnam. Lancet. 1993;342:213–217. doi: 10.1016/0140-6736(93)92302-A. [DOI] [PubMed] [Google Scholar]
- Kessel E. 100,000 quinacrine sterilizations. Adv Contracept. 1996;12:69–76. doi: 10.1007/BF01849629. [DOI] [PubMed] [Google Scholar]
- Zipper J, Trujillo V. 25 years of quinacrine sterilization experience in Chile: review of 2,592 cases. Int J Gynaecol Obstet. 2003;83(Suppl 2):S23–29. [PubMed] [Google Scholar]
- Physicians' Desk Reference 199347Montavile, NJ: Thomas PDR; 21584946 [Google Scholar]
- Ottolenghi-Lodigiani F. Treatment of chronic lupus erythematosus, local intradermal infiltration with an acridine preparation. Hautarzt. 1955;6:24–27. [PubMed] [Google Scholar]
- Thies W. Recent experience in the treatment of erythematodes chronicus discoides with atebrin and resochin, with special reference to local and combined therapeutic methods. Hautarzt. 1955;6:227–232. [PubMed] [Google Scholar]
- Bayly TC, Kisner DL, Sybert A, Macdonald JS, Tsou E, Schein PS. Tetracycline and quinacrine in the control of malignant pleural effusions. A randomized trial. Cancer. 1978;41:1188–1192. doi: 10.1002/1097-0142(197803)41:3<1188::AID-CNCR2820410357>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Campbell WC. The chemotherapy of parasitic infections. J Parasitol. 1986;72:45–61. doi: 10.2307/3281795. [DOI] [PubMed] [Google Scholar]
- Bjorkman S, Elisson LO, Gabrielsson J. Pharmacokinetics of quinacrine after intrapleural instillation in rabbits and man. J Pharm Pharmacol. 1989;41:160–163. doi: 10.1111/j.2042-7158.1989.tb06421.x. [DOI] [PubMed] [Google Scholar]
- Laufe LE, Sokal DC, Cole LP, Shoupe D, Schenken RS. Phase I prehysterectomy studies of the transcervical administration of quinacrine pellets. Contraception. 1996;54:181–186. doi: 10.1016/S0010-7824(96)00174-6. [DOI] [PubMed] [Google Scholar]
- Looareesuwan S, Phillips RE, Edwards G, Rodick CL, Chanthavanich P, Supanaranond W, Warrell DA. Mepacrine accumulation during treatment of chloroquine-resistant falciparum malaria. Ann Trop Med Parasitol. 1988;82:107–112. doi: 10.1080/00034983.1988.11812216. [DOI] [PubMed] [Google Scholar]
- Ackerman NB, Haldorsen DK, Wallace DL, Madsen AJ, McFee AS. Aminoacridine uptake by experimental tumors. JAMA. 1965;191:103–104. doi: 10.1001/jama.1965.03080020031008. [DOI] [PubMed] [Google Scholar]
- Ackerman NB, Shemesh A. Localization of aminoacridine fluorescence in lung tumors of rats. JAMA. 1964;187:832–833. doi: 10.1001/jama.1964.03060240040009. [DOI] [PubMed] [Google Scholar]
- Anghileri LJ. Uptake of iodine-131-labelled atabrine by Ehrlich ascites tumour and by sarcoma S-180 BALB. Nature. 1966;211:878. doi: 10.1038/211878a0. [DOI] [PubMed] [Google Scholar]
- Gibb W, Isenberg DA, Snaith ML. Mepacrine induced hepatitis. Ann Rheum Dis. 1985;44:861–862. doi: 10.1136/ard.44.12.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SJ, Montgomery S, Brearley R, Galloway A. A case of primary peritonitis possibly related to mepacrine. J R Coll Surg Edinb. 1987;32:321–322. [PubMed] [Google Scholar]
- Zuehlke RL, Lillis PJ, Tice A. Antimalarial therapy for lupus erythematosus: an apparent advantage of quinacrine. Int J Dermatol. 1981;20:57–61. doi: 10.1111/j.1365-4362.1981.tb05295.x. [DOI] [PubMed] [Google Scholar]
- Carr RE, Henkind P, Rothfield N, Siegel IM. Ocular toxicity of antimalarial drugs. Long-term follow-up. Am J Ophthalmol. 1968;66:738–744. doi: 10.1016/0002-9394(68)91300-7. [DOI] [PubMed] [Google Scholar]
- Ansdell VE, Common JD. Corneal changes induced by mepacrine. J Trop Med Hyg. 1979;82:206–207. [PubMed] [Google Scholar]
- Chamberlain WP, Boles DJ. Edema of cornea precipitated by quinacrine (Atabrine) Arch Ophthalmol. 1946;35:120–134. doi: 10.1001/archopht.1946.00890200125005. [DOI] [PubMed] [Google Scholar]
- Engel GL. Quinacrine effects on the central nervous system. JAMA. 1966;197:515. doi: 10.1001/jama.197.6.515. [DOI] [PubMed] [Google Scholar]
- Lindenmayer JP, Vargas P. Toxic psychosis following use of quinacrine. J Clin Psychiatry. 1981;42:162–164. [PubMed] [Google Scholar]
- Ward WQ, Walter-Ryan WG, Shehi GM. Toxic psychosis: a complication of antimalarial therapy. J Am Acad Dermatol. 1985;12:863–865. doi: 10.1016/S0190-9622(85)70109-0. [DOI] [PubMed] [Google Scholar]
- Evans RL, Khalid S, Kinney JL. Antimalarial psychosis revisited. Arch Dermatol. 1984;120:765–767. doi: 10.1001/archderm.120.6.765. [DOI] [PubMed] [Google Scholar]
- Gaskill HS, Fitz-Hugh T. Toxic psychosis following Atabrine. Bull US Army Med Dept. 1945;86:63–69. [Google Scholar]
- Lidz T, Kahn RL. Toxicity of quinacrine (Atabrine) for the cental nervous system. Arch Neurol. 1946;56:284–289. [PubMed] [Google Scholar]
- Custer RP. Aplastic anemia in soldiers treated with Atabrine (quinacrine) Am J Med Sci. 1946;212:211–224. [PubMed] [Google Scholar]
- Fishman AP, Kinsman JM. Hypoplastic anemia due to atabrine. Blood. 1949;4:970–976. [PubMed] [Google Scholar]
- Freedman A, Bach F. Mepacrine and rheumatoid arthritis. Lancet. 1952;2:321. doi: 10.1016/s0140-6736(52)92481-1. [DOI] [PubMed] [Google Scholar]
- Parmer LG, Sawitsky A. Fatal aplastic anemia following quinacrine therapy in chronic discoid lupus erythematosus. J Am Med Assoc. 1953;153:1172–1174. doi: 10.1001/jama.1953.02940300030009a. [DOI] [PubMed] [Google Scholar]
- Paton MD, Riddell MJ, Strong JA. Aplastic anaemia following mepacrine treatment of lupus erythematosus. Lancet. 1955;268:281–282. doi: 10.1016/s0140-6736(55)90109-4. [DOI] [PubMed] [Google Scholar]
- Biro L, Leone N. Aplastic anemia induced by quinacrine. Arch Dermatol. 1965;92:574–576. doi: 10.1001/archderm.1965.01600170090017. [DOI] [PubMed] [Google Scholar]
- Schmid I, Anasetti C, Petersen FB, Storb R. Marrow transplantation for severe aplastic anemia associated with exposure to quinacrine. Blut. 1990;61:52–54. doi: 10.1007/BF02076699. [DOI] [PubMed] [Google Scholar]
- Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, Thomas D, Walker S, Webb T, Wroe S, Darbyshire J. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8:334–344. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer F. Quinacrine hydrochloride drug eruption (tropical lichenoid dermatitis). Its early and late sequelae and its malignant potential: a review. J Am Acad Dermatol. 1981;4:239–248. doi: 10.1016/S0190-9622(81)70025-2. [DOI] [PubMed] [Google Scholar]
- Lerman MA, Karimbux N, Guze KA, Woo SB. Pigmentation of the hard palate. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:8–12. doi: 10.1016/j.tripleo.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Lippard VW, Kauer G. Pigmentation of the palate and subungual tissue associated with suppressive quinacrine hydrochloride therapy. Am J Tropic Med. 1945;25:469–471. doi: 10.4269/ajtmh.1945.s1-25.469. [DOI] [PubMed] [Google Scholar]
- Kleinegger CL, Hammond HL, Finkelstein MW. Oral mucosal hyperpigmentation secondary to antimalarial drug therapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:189–194. doi: 10.1067/moe.2000.106340. [DOI] [PubMed] [Google Scholar]
- Egorin MJ, Trump DL, Wainwright CW. Quinacrine ochronosis and rheumatoid arthritis. JAMA. 1976;236:385–386. doi: 10.1001/jama.236.4.385. [DOI] [PubMed] [Google Scholar]
- Leigh IM, Kennedy CT, Ramsey JD, Henderson WJ. Mepacrine pigmentation in systemic lupus erythematosus. New data from an ultrastructural, biochemical and analytical electron microscopic investigation. Br J Dermatol. 1979;101:147–153. doi: 10.1111/j.1365-2133.1979.tb05599.x. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Lichtenstein PK, Farrell MK. Quinacrine hydrochloride-induced yellow discoloration of the skin in children. Pediatrics. 1982;69:232–233. [PubMed] [Google Scholar]
- Callaway JL. Late sequelae of quinacrine dermatitis, a new premalignant entity. J Am Acad Dermatol. 1979;1:456. doi: 10.1016/S0190-9622(79)80072-9. [DOI] [PubMed] [Google Scholar]
- Bauer F. Late sequelae of atabrine dermatitis--a new pre-malignant entity. Australas J Dermatol. 1978;19:9–12. doi: 10.1111/j.1440-0960.1978.tb00178.x. [DOI] [PubMed] [Google Scholar]
- Tuffanelli DL. Quinacrine ochronosis. JAMA. 1976;236:2491. [PubMed] [Google Scholar]
- Dabancens A, Sokal DC, Pruyas M, Rivera M, Zipper J. Prevalence and standardized incidence rates of preclinical cervical pathology among 1,061 women sterilized with transcervical quinacrine hydrochloride pellets. Fertil Steril. 1995;64:444–446. [PubMed] [Google Scholar]
- Sokal DC, Zipper J, Guzman-Serani R, Aldrich TE. Cancer risk among women sterilized with transcervical quinacrine hydrochloride pellets, 1977 to 1991. Fertil Steril. 1995;64:325–334. [PubMed] [Google Scholar]
- Sokal DC, Dabancens A, Guzman-Serani R, Zipper J. Cancer risk among women sterilized with transcervical quinacrine in Chile: an update through 1996. Fertil Steril. 2000;74:169–171. doi: 10.1016/S0015-0282(00)00581-1. [DOI] [PubMed] [Google Scholar]
- Castelli M, Baggio G, Ruberto AI, Tampieri A, Tartoni PL, Rossi T, Bossa MR, Galatulas I. Influence of antimalarials chloroquine, quinine, primaquine and mepacrine on the evolution of Ehrlich ascites tumour. Anticancer Res. 1996;16:2673–2675. [PubMed] [Google Scholar]
- Dutta P, Karmali R, Pinto JT, Rivlin RS. Enhanced growth of mammary adenocarcinoma in rats by chloroquine and quinacrine. Cancer Lett. 1994;76:113–119. doi: 10.1016/0304-3835(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Dabancens A, Zipper J, Guerrero A. Quinacrine and copper, compounds with anticonceptive and antineoplastic activity. Contraception. 1994;50:243–251. doi: 10.1016/0010-7824(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Zipper JA, Dabancens AO, Guerrero AC. Opposing effects of quinacrine and chloroquine on the development of TA3 transplanted tumors in mice. Biol Res. 1995;28:227–230. [PubMed] [Google Scholar]
- Zipper J, Dabancens A, Guerrero A, Trujillo V. Quinacrine revised. Hum Reprod Update. 1995;1:324–342. doi: 10.1093/humupd/1.4.324. [DOI] [PubMed] [Google Scholar]
- Siegel H, Mushett CW. Structural changes following administration of quinacrine hydrochloride. Arch Path. 1944;38 [PubMed] [Google Scholar]
- Mushett CW, Siegel H. Hematological changes following the administration of large doses of quinacrine hydrochloride. Blood. 1946;1:537–547. [PubMed] [Google Scholar]
- Keeler R, Richardson H, Watson AJ. Enteromegaly and steatorrhea in the rat following intraperitoneal quinacrine (atrabrine) Lab Invest. 1966;15:1253–1262. [PubMed] [Google Scholar]
- Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16:127–136. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Sivachenko A, Kalinin A, Yuryev A. Pathway analysis for design of promiscuous drugs and selective drug mixtures. Curr Drug Discov Technol. 2006;3:269–277. doi: 10.2174/157016306780368117. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Boer DR, Canals A, Coll M. DNA-binding drugs caught in action: the latest 3D pictures of drug-DNA complexes. Dalton Trans. 2009. pp. 399–414. [DOI] [PubMed]
- Pindur U, Jansen M, Lemster T. Advances in DNA-ligands with groove binding, intercalating and/or alkylating activity: chemistry, DNA-binding and biology. Curr Med Chem. 2005;12:2805–2847. doi: 10.2174/092986705774454698. [DOI] [PubMed] [Google Scholar]
- Strekowski L, Wilson B. Noncovalent interactions with DNA: an overview. Mutat Res. 2007;623:3–13. doi: 10.1016/j.mrfmmm.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Lerman LS. Structural considerations in the interaction of DNA and acridines. J Mol Biol. 1961;3:18–30. doi: 10.1016/S0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- Waring MJ. DNA-binding characteristics of acridinylmethanesulphonanilide drugs: comparison with antitumour properties. Eur J Cancer. 1976;12:995–1001. doi: 10.1016/0014-2964(76)90066-9. [DOI] [PubMed] [Google Scholar]
- Lerman LS. The structure of the DNA-acridine complex. Proc Natl Acad Sci USA. 1963;49:94–102. doi: 10.1073/pnas.49.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanoglu M, Ayne G. Voltammetric studies of the interaction of quinacrine with DNA. Anal Bioanal Chem. 2004;380:658–663. doi: 10.1007/s00216-004-2797-5. [DOI] [PubMed] [Google Scholar]
- Doglia SM, Albinsson B, Hiort C, Norden B, Graslund A. Quinacrine: Spectroscopic properties and interactions with polynucleotides. Biopolymers. 1993;33:1431–1442. doi: 10.1002/bip.360330913. [DOI] [Google Scholar]
- Doglia S, Graslund A, Ehrenberg A. Specific interactions between quinacrine and self-complementary deoxydinucleotides. Anticancer Res. 1986;6:1363–1368. [PubMed] [Google Scholar]
- Lober G. The fluorescence of dye-nucleic acid complexes. J Luminescence. 1981;22:221–265. doi: 10.1016/0022-2313(81)90022-3. [DOI] [Google Scholar]
- Rivas L, Murza A, Sanchez-Cortes S, Garcia-Ramos JV. Interaction of antimalarial drug quinacrine with nucleic acids of variable sequence studied by spectroscopic methods. J Biomol Struct Dyn. 2000;18:371–383. doi: 10.1080/07391102.2000.10506674. [DOI] [PubMed] [Google Scholar]
- Weisblum B, De Haseth PL. Quinacrine, a chromosome stain specific for deoxyadenylate-deoxythymidylaterich regions in DNA. Proc Natl Acad Sci USA. 1972;69:629–632. doi: 10.1073/pnas.69.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastasi M, Yip RW, Seligy VL, Szabo AG, Williams RE. Exciton-like splitting in acridine dye-nucleic acid complexes. Nature. 1974;249:248–250. doi: 10.1038/249248a0. [DOI] [PubMed] [Google Scholar]
- Gorenstein DG, Lai K. 31P NMR spectra of ethidium, quinacrine, and daunomycin complexes with poly(adenylic acid).poly(uridylic acid) RNA duplex and calf thymus DNA. Biochemistry. 1989;28:2804–2812. doi: 10.1021/bi00433a010. [DOI] [PubMed] [Google Scholar]
- Selander RK. Interaction of quinacrine mustard with mononucleotides and polynucleotides. Biochem J. 1973;131:749–755. doi: 10.1042/bj1310749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoni A, Cubeddu R, De Silvestri S, Laporta P. Time-resolved fluorescence spectrum of quinacrine mustard bound to synthetic polynucleotides. Chemical Physics Letters. 1981;80:323–326. doi: 10.1016/0009-2614(81)80118-2. [DOI] [Google Scholar]
- Constant JF, Laugaa P, Roques BP, Lhomme J. Heterodimeric molecules including nucleic acid bases and 9-aminoacridine. Spectroscopic studies, conformations, and interactions with DNA. Biochemistry. 1988;27:3997–4003. doi: 10.1021/bi00411a016. [DOI] [PubMed] [Google Scholar]
- Snyder RD, Arnone MR. Putative identification of functional interactions between DNA intercalating agents and topoisomerase II using the V79 in vitro micronucleus assay. Mutat Res. 2002;503:21–35. doi: 10.1016/s0027-5107(02)00028-3. [DOI] [PubMed] [Google Scholar]
- O'Brien RL, Olenick JG, Hahn FE. Reactions of quinine, chloroquine, and quinacrine with DNA and their effects on the DNA and RNA polymerase reactions. Proc Natl Acad Sci USA. 1966;55:1511–1517. doi: 10.1073/pnas.55.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn FE, O'Brien RL, Ciak J, Allison JL, Olenick JG. Studies on modes of action of chloroquine, quinacrine, and quinine and on chloroquine resistance. Mil Med. 1966;131(Suppl):1071–1089. [PubMed] [Google Scholar]
- Kagemoto A, Kunihiro A, Baba Y. Thermodynamic studies on interactions between DNA and dye. Thermochimica Acta. 1994;242:65–75. [Google Scholar]
- Haerd T, Fan P, Magde D, Kearns DR. On the flexibility of DNA: time-resolved fluorescence polarization of intercalated quinacrine and 9-amino-6-chloro-2-methoxyacridine. The Journal of Physical Chemistry. 2002;93:4338–4345. [Google Scholar]
- Sumner AT. Mechanisms of quinacrine binding and fluorescence in nuclei and chromosomes. Histochemistry. 1986;84:566–574. doi: 10.1007/BF00482993. [DOI] [PubMed] [Google Scholar]
- Baldini G, Doglia S, Dolci S, Sassi G. Fluorescence-determined preferential binding of quinacrine to DNA. Biophys J. 1981;36:465–477. doi: 10.1016/S0006-3495(81)84746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xamena N, Creus A, Velazquez A, Marcos R. Testing of chloroquine and quinacrine for mutagenicity in Drosophila melanogaster. Mutat Res. 1985;158:177–180. doi: 10.1016/0165-1218(85)90081-3. [DOI] [PubMed] [Google Scholar]
- Denny WA. DNA-intercalating ligands as anti-cancer drugs: prospects for future design. Anticancer Drug Des. 1989;4:241–263. [PubMed] [Google Scholar]
- Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- Le Pecq JB, Nguyen Dat X, Gosse C, Paoletti C. A new antitumoral agent: 9-hydroxyellipticine. Possibility of a rational design of anticancerous drugs in the series of DNA intercalating drugs. Proc Natl Acad Sci USA. 1974;71:5078–5082. doi: 10.1073/pnas.71.12.5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JA, Reszka K, Zuo ET, Wilson WD, Morgan AR, Lown JW. Characteristics of the interaction of anthrapyrazole anticancer agents with deoxyribonucleic acids: structural requirements for DNA binding, intercalation, and photosensitization. Mol Pharmacol. 1988;33:265–271. [PubMed] [Google Scholar]
- Valentini L, Nicolella V, Vannini E, Menozzi M, Penco S, Arcamone F. Association of anthracycline derivatives with DNA: a fluorescence study. Farmaco Sci. 1985;40:377–390. [PubMed] [Google Scholar]
- Mergny JL, Mailliet P, Lavelle F, Riou JF, Laoui A, Helene C. The development of telomerase inhibitors: the G-quartet approach. Anticancer Drug Des. 1999;14:327–339. [PubMed] [Google Scholar]
- Harrison RJ, Gowan SM, Kelland LR, Neidle S. Human telomerase inhibition by substituted acridine derivatives. Bioorg Med Chem Lett. 1999;9:2463–2468. doi: 10.1016/S0960-894X(99)00394-7. [DOI] [PubMed] [Google Scholar]
- Perry PJ, Jenkins TC. Recent advances in the development of telomerase inhibitors for the treatment of cancer. Expert Opin Investig Drugs. 1999;8:1981–2008. doi: 10.1517/13543784.8.12.1981. [DOI] [PubMed] [Google Scholar]
- Redman JE, Granadino-Roldan JM, Schouten JA, Ladame S, Reszka AP, Neidle S, Balasubramanian S. Recognition and discrimination of DNA quadruplexes by acridine-peptide conjugates. Org Biomol Chem. 2009;7:76–84. doi: 10.1039/b814682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read MA, Wood AA, Harrison JR, Gowan SM, Kelland LR, Dosanjh HS, Neidle S. Molecular modeling studies on G-quadruplex complexes of telomerase inhibitors: structure-activity relationships. J Med Chem. 1999;42:4538–4546. doi: 10.1021/jm990287e. [DOI] [PubMed] [Google Scholar]
- Corbett AH, Osheroff N. When good enzymes go bad: conversion of topoisomerase II to a cellular toxin by antineoplastic drugs. Chem Res Toxicol. 1993;6:585–597. doi: 10.1021/tx00035a001. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Osheroff N. Stabilization of the topoisomerase II-DNA cleavage complex by antineoplastic drugs: inhibition of enzyme-mediated DNA religation by 4'-(9-acridinylamino)methanesulfon-m-anisidide. Biochemistry. 1990;29:2511–2515. doi: 10.1021/bi00462a012. [DOI] [PubMed] [Google Scholar]
- Su TL. Development of DNA topoisomerase II-mediated anticancer agents, 3-(9-acridinylamino)-5-hydroxymethylanilines (AHMAs) and related compounds. Curr Med Chem. 2002;9:1677–1688. doi: 10.2174/0929867023369231. [DOI] [PubMed] [Google Scholar]
- Denny WA, Baguley BC. Dual topoisomerase I/II inhibitors in cancer therapy. Curr Top Med Chem. 2003;3:339–353. doi: 10.2174/1568026033452555. [DOI] [PubMed] [Google Scholar]
- Denny WA. Acridine derivatives as chemotherapeutic agents. Curr Med Chem. 2002;9:1655–1665. doi: 10.2174/0929867023369277. [DOI] [PubMed] [Google Scholar]
- Buys CHCM. Telomeres, telomerase, and cancer. N Engl J Med. 2000;342:1282–1283. doi: 10.1056/NEJM200004273421710. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, Manns MP, Rudolph KL. Telomeres, telomerase and cancer: an endless search to target the ends. Cell Cycle. 2004;3:1138–1150. [PubMed] [Google Scholar]
- Robinson MO. Telomerase and cancer. Genet Eng (N Y) 2000;22:209–222. doi: 10.1007/978-1-4615-4199-8_12. [DOI] [PubMed] [Google Scholar]
- Wong HM, Payet L, Huppert JL. Function and targeting of G-quadruplexes. Curr Opin Mol Ther. 2009;11:146–155. [PubMed] [Google Scholar]
- Tan JH, Gu LQ, Wu JY. Design of selective G-quadruplex ligands as potential anticancer agents. Mini Rev Med Chem. 2008;8:1163–1178. doi: 10.2174/138955708785909880. [DOI] [PubMed] [Google Scholar]
- Hsiang YH, Wu HY, Liu LF. Topoisomerases: novel therapeutic targets in cancer chemotherapy. Biochem Pharmacol. 1988;37:1801–1802. doi: 10.1016/0006-2952(88)90453-4. [DOI] [PubMed] [Google Scholar]
- Dominick PK, Keppler BR, Legassie JD, Moon IK, Jarstfer MB. Nucleic acid-binding ligands identify new mechanisms to inhibit telomerase. Bioorg Med Chem Lett. 2004;14:3467–3471. doi: 10.1016/j.bmcl.2004.04.055. [DOI] [PubMed] [Google Scholar]
- Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- Werbovetz KA, Lehnert EK, Macdonald TL, Pearson RD. Cytotoxicity of acridine compounds for Leishmania promastigotes in vitro. Antimicrob Agents Chemother. 1992;36:495–497. doi: 10.1128/aac.36.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer SW, Schmidt G, Sorensen M, Sehested M, Jensen PB. Inhibitors of topoisomerase II as pH-dependent modulators of etoposide-mediated cytotoxicity. Clin Cancer Res. 1999;5:2899–2907. [PubMed] [Google Scholar]
- Popanda O, Thielmann HW. The function of DNA topoisomerases in UV-induced DNA excision repair: studies with specific inhibitors in permeabilized human fibroblasts. Carcinogenesis. 1992;13:2321–2328. doi: 10.1093/carcin/13.12.2321. [DOI] [PubMed] [Google Scholar]
- Sideropoulos AS, Specht SM, Jones MT. Feasibility of testing DNA repair inhibitors for mutagenicity by a simple method. Mutation Research/Environmental Mutagenesis and Related Subjects. 1980;74:95–105. doi: 10.1016/0165-1161(80)90235-6. [DOI] [PubMed] [Google Scholar]
- Fuks Z, Smith KC. Effect of quinacrine on x-ray sensitivity and the repair of damaged DNA in Escherichia coli K-12. Radiat Res. 1971;48:63–73. doi: 10.2307/3573395. [DOI] [PubMed] [Google Scholar]
- Skare JA, Wong TK. Lack of specific inhibition of DNA repair in WI-38 human diploid fibroblasts by sodium saccharin. Cancer Letters. 1985;26:191–200. doi: 10.1016/0304-3835(85)90026-6. [DOI] [PubMed] [Google Scholar]
- Thielmann HW, Popanda O, Gersbach H, Gilberg F. Various inhibitors of DNA topoisomerases diminish repair-specific DNA incision in UV-irradiated human fibroblasts. Carcinogenesis. 1993;14:2341–2351. doi: 10.1093/carcin/14.11.2341. [DOI] [PubMed] [Google Scholar]
- Popanda O, Thielmann HW. The function of DNA polymerases in DNA repair synthesis of ultraviolet-irradiated human fibroblasts. Biochim Biophys Acta. 1992;1129:155–160. doi: 10.1016/0167-4781(92)90480-n. [DOI] [PubMed] [Google Scholar]
- Voiculetz N, Smith KC, Kaplan HS. Effect of Quinacrine on survival and DNA repair in X-irradiated chinese hamster cells. Cancer Res. 1974;34:1038–1044. [PubMed] [Google Scholar]
- Downes CS, Johnson RT. DNA topoisomerases and DNA repair. Bioessays. 1988;8:179–184. doi: 10.1002/bies.950080602. [DOI] [PubMed] [Google Scholar]
- Lieberman MW. Alterations in chromatin structure during DNA excision repair. Basic Life Sci. 1982;20:303–314. doi: 10.1007/978-1-4613-3476-7_20. [DOI] [PubMed] [Google Scholar]
- Arnold GE, Dunker AK, Smerdon MJ. Limited nucleosome migration can completely randomize DNA repair patches in intact human cells. J Mol Biol. 1987;196:433–436. doi: 10.1016/0022-2836(87)90703-0. [DOI] [PubMed] [Google Scholar]
- Sidik K, Smerdon MJ. Rearrangement of nucleosome structure during excision repair in xeroderma pigmentosum (group A) human fibroblasts. Carcinogenesis. 1987;8:733–736. doi: 10.1093/carcin/8.5.733. [DOI] [PubMed] [Google Scholar]
- Schneider E, Darkin SJ, Lawson PA, Ching LM, Ralph RK, Baguley BC. Cell line selectivity and DNA breakage properties of the antitumour agent N-[2-(dimethylamino)ethyl]acridine-4-carboxamide: role of DNA topoisomerase II. Eur J Cancer Clin Oncol. 1988;24:1783–1790. doi: 10.1016/0277-5379(88)90082-X. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. Yeast genes involved in DNA-repair processes: new looks on old faces. Mol Microbiol. 1991;5:2303–2310. doi: 10.1111/j.1365-2958.1991.tb02074.x. [DOI] [PubMed] [Google Scholar]
- Van Dyke K, Lantz C, Szustkiewicz C. Quinacrine: mechanisms of antimalarial action. Science. 1970;169:492–493. doi: 10.1126/science.169.3944.492. [DOI] [PubMed] [Google Scholar]
- Chou SC, Ramanathan S. Quinacrine: site of inhibition of synchronized cell division in Tetrahymena. Life Sci. 1968;7:1053–1062. doi: 10.1016/0024-3205(68)90142-2. [DOI] [PubMed] [Google Scholar]
- Fox G, Popanda O, Edler L, Thielmann HW. Preferential inhibition of DNA polymerases alpha, delta, and epsilon from Novikoff hepatoma cells by inhibitors of cell proliferation. J Cancer Res Clin Oncol. 1996;122:78–94. doi: 10.1007/BF01226265. [DOI] [PubMed] [Google Scholar]
- Wang TS, Wong SW, Korn D. Human DNA polymerase alpha: predicted functional domains and relationships with viral DNA polymerases. FASEB J. 1989;3:14–21. doi: 10.1096/fasebj.3.1.2642867. [DOI] [PubMed] [Google Scholar]
- Wang TS. Eukaryotic DNA polymerases. Annu Rev Biochem. 1991;60:513–552. doi: 10.1146/annurev.bi.60.070191.002501. [DOI] [PubMed] [Google Scholar]
- Hirschman SZ, Garfinkel E. Inhibition of hepatitis B DNA polymerase by intercalating agents. Nature. 1978;271:681–683. doi: 10.1038/271681a0. [DOI] [PubMed] [Google Scholar]
- Hess G, Arnold W, Moller B, Gahl GM, Meyerzum Buschenfelde KH. Inhibition of hepatitis B virus specific DNA polymerase by intercalating agents. Med Microbiol Immunol. 1980;168:25–34. doi: 10.1007/BF02121649. [DOI] [PubMed] [Google Scholar]
- Goda K, Bacso Z, Szabo G. Multidrug resistance through the spectacle of P-glycoprotein. Curr Cancer Drug Targets. 2009;9:281–297. doi: 10.2174/156800909788166493. [DOI] [PubMed] [Google Scholar]
- Lehne G. P-glycoprotein as a drug target in the treatment of multidrug resistant cancer. Curr Drug Targets. 2000;1:85–99. doi: 10.2174/1389450003349443. [DOI] [PubMed] [Google Scholar]
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–4602. [PubMed] [Google Scholar]
- Singh P, Kaur J, Kaur P, Kaur S. Search for MDR modulators: design, syntheses and evaluations of N-substituted acridones for interactions with p-glycoprotein and Mg2+ Bioorg Med Chem. 2009;17:2423–2427. doi: 10.1016/j.bmc.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Mayur YC, Peters GJ, Prasad VV, Lemo C, Sathish NK. Design of new drug molecules to be used in reversing multidrug resistance in cancer cells. Curr Cancer Drug Targets. 2009;9:298–306. doi: 10.2174/156800909788166619. [DOI] [PubMed] [Google Scholar]
- Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- Traunecker HC, Stevens MC, Kerr DJ, Ferry DR. The acridonecarboxamide GF120918 potently reverses P-glycoprotein-mediated resistance in human sarcoma MES-Dx5 cells. Br J Cancer. 1999;81:942–951. doi: 10.1038/sj.bjc.6690791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- Liang GW, Lu WL, Wu JW, Zhao JH, Hong HY, Long C, Li T, Zhang YT, Zhang H, Wang JC, Zhang X, Zhang Q. Enhanced therapeutic effects on the multi-drug resistant human leukemia cells in vitro and xenograft in mice using the stealthy liposomal vincristine plus quinacrine. Fundam Clin Pharmacol. 2008;22:429–437. doi: 10.1111/j.1472-8206.2008.00613.x. [DOI] [PubMed] [Google Scholar]
- Inaba M, Maruyama E. Reversal of resistance to vincristine in P388 leukemia by various polycyclic clinical drugs, with a special emphasis on quinacrine. Cancer Res. 1988;48:2064–2067. [PubMed] [Google Scholar]
- Beck WT, Cirtain MC, Glover CJ, Felsted RL, Safa AR. Effects of indole alkaloids on multidrug resistance and labeling of P-glycoprotein by a photoaffinity analog of vinblastine. Biochem Biophys Res Commun. 1988;153:959–966. doi: 10.1016/S0006-291X(88)81321-4. [DOI] [PubMed] [Google Scholar]
- Zamora JM, Pearce HL, Beck WT. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells. Mol Pharmacol. 1988;33:454–462. [PubMed] [Google Scholar]
- Boscoboinik D, Epand RM. Increased cellular internalization of amphiphiles in a multidrug-resistant CHO cell line. Biochim Biophys Acta. 1989;1014:53–56. doi: 10.1016/0167-4889(89)90239-5. [DOI] [PubMed] [Google Scholar]
- de Souza PL, Castillo M, Myers CE. Enhancement of paclitaxel activity against hormone-refractory prostate cancer cells in vitro and in vivo by quinacrine. Br J Cancer. 1997;75:1593–1600. doi: 10.1038/bjc.1997.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- Mann JR, DuBois RN. Cyclooxygenase-2 and gastrointestinal cancer. Cancer J. 2004;10:145–152. doi: 10.1097/00130404-200405000-00001. [DOI] [PubMed] [Google Scholar]
- Kurie JM, Dubois RN. Prostaglandin E synthase: another enzyme in the cyclooxygenase pathway driving epithelial cancer? Clin Cancer Res. 2001;7:2608–2610. [PubMed] [Google Scholar]
- Bunn PA Jr, Keith RL. The future of cyclooxygenase-2 inhibitors and other inhibitors of the eicosanoid signal pathway in the prevention and therapy of lung cancer. Clin Lung Cancer. 2002;3:271–277. doi: 10.3816/CLC.2002.n.012. discussion 278. [DOI] [PubMed] [Google Scholar]
- Thun MJ, Blackard B. Pharmacologic effects of NSAIDs and implications for the risks and benefits of long-term prophylactic use of aspirin to prevent cancer. Recent Results Cancer Res. 2009;181:215–221. doi: 10.1007/978-3-540-69297-3_20. [DOI] [PubMed] [Google Scholar]
- Patel MI, Kurek C, Dong Q. The arachidonic acid pathway and its role in prostate cancer development and progression. J Urol. 2008;179:1668–1675. doi: 10.1016/j.juro.2007.12.037. [DOI] [PubMed] [Google Scholar]
- Keith RL, Miller YE. Lung cancer: genetics of risk and advances in chemoprevention. Curr Opin Pulm Med. 2005;11:265–271. doi: 10.1097/01.mcp.0000166493.77412.2d. [DOI] [PubMed] [Google Scholar]
- Jimenez P, Garcia A, Santander S, Piazuelo E. Prevention of cancer in the upper gastrointestinal tract with COX-inhibition. Still an option? Curr Pharm Des. 2007;13:2261–2273. doi: 10.2174/138161207781368800. [DOI] [PubMed] [Google Scholar]
- Mehta S, Johnson IT, Rhodes M. Systematic review: the chemoprevention of oesophageal adenocarcinoma. Aliment Pharmacol Ther. 2005;22:759–768. doi: 10.1111/j.1365-2036.2005.02667.x. [DOI] [PubMed] [Google Scholar]
- Fosslien E. Molecular pathology of cyclooxygenase-2 in neoplasia. Ann Clin Lab Sci. 2000;30:3–21. [PubMed] [Google Scholar]
- Tokumoto H, Croxtall JD, Choudhury Q, Flower RJ. Phospholipase A2-induced stimulation of A549 lung adenocarcinoma cell line proliferation. Biochim Biophys Acta. 1993;1169:236–242. doi: 10.1016/0005-2760(93)90246-6. [DOI] [PubMed] [Google Scholar]
- Hanada K, Kinoshita E, Itoh M, Hirata M, Kajiyama G, Sugiyama M. Human pancreatic phospholipase A2 stimulates the growth of human pancreatic cancer cell line. FEBS Lett. 1995;373:85–87. doi: 10.1016/0014-5793(95)01005-Y. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Yamashita J, Ogawa M. Overexpression of group II phospholipase A2 in human breast cancer tissues is closely associated with their malignant potency. Br J Cancer. 1994;69:1166–1170. doi: 10.1038/bjc.1994.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata K, Egami H, Kiyohara H, Oshima S, Kurizaki T, Ogawa M. Expression of group-II phospholipase A2 in malignant and non-malignant human gastric mucosa. Br J Cancer. 1993;68:103–111. doi: 10.1038/bjc.1993.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain MK, Yu BZ, Rogers J, Ranadive GN, Berg OG. Interfacial catalysis by phospholipase A2: dissociation constants for calcium, substrate, products, and competitive inhibitors. Biochemistry. 1991;30:7306–7317. doi: 10.1021/bi00243a036. [DOI] [PubMed] [Google Scholar]
- Loffler BM, Bohn E, Hesse B, Kunze H. Effects of antimalarial drugs on phospholipase A and lysophospholipase activities in plasma membrane, mitochondrial, microsomal and cytosolic subcellular fractions of rat liver. Biochim Biophys Acta. 1985;835:448–455. doi: 10.1016/0005-2760(85)90114-6. [DOI] [PubMed] [Google Scholar]
- Schiess K, Kaszkin M, Jordan P, Seidler L, Kinzel V. Mobilization of diacylglycerol in intact HeLa cells by exogenous phospholipase C from Cl. perfringens is accompanied by release of fatty acids including arachidonic acid. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1992;1137:82–94. doi: 10.1016/0167-4889(92)90104-J. [DOI] [PubMed] [Google Scholar]
- Ahmed A, Cameron IT, Ferriani RA, Smith SK. Activation of phospholipase A2 and phospholipase C by endothelin-1 in human endometrium. J Endocrinol. 1992;135:383–390. doi: 10.1677/joe.0.1350383. [DOI] [PubMed] [Google Scholar]
- Horrobin DF, Manku MS, Karmazyn M, Ally AI, Morgan RO, Karmali RA. Quinacrine is a prostaglandin antagonist. Biochem Biophys Res Commun. 1977;76:1188–1193. doi: 10.1016/0006-291X(77)90981-0. [DOI] [PubMed] [Google Scholar]
- Dise CA, Burch JW, Goodman DB. Direct interaction of mepacrine with erythrocyte and platelet membrane phospholipid. J Biol Chem. 1982;257:4701–4704. [PubMed] [Google Scholar]
- Zidovetzki R, Sherman IW, Maguire PA, De Boeck H. A nuclear magnetic resonance study of the interactions of the antimalarials chloroquine, quinacrine, quinine and mefloquine with lipids extracted from normal human erythrocytes. Mol Biochem Parasitol. 1990;38:33–39. doi: 10.1016/0166-6851(90)90202-W. [DOI] [PubMed] [Google Scholar]
- Abdel-Latif AA, Smith JP, Akhtar RA. Studies on the mechanism of alteration by propranolol and mepacrine of the metabolism of phosphoinositides and other glycerolipids in the rabbit iris muscle. Biochemical Pharmacology. 1983;32:3815–3821. doi: 10.1016/0006-2952(83)90154-5. [DOI] [PubMed] [Google Scholar]
- Mustonen P, Lehtonen JYA, Kinnunen PKJ. Binding of quinacrine to acidic phospholipids and pancreatic phospholipase A2. Effects on the catalytic activity of the enzyme. Biochemistry. 1998;37:12051–12057. doi: 10.1021/bi980430q. [DOI] [PubMed] [Google Scholar]
- Yamada K, Okano Y, Miura K, Nozawa Y. A major role for phospholipase A2 in antigen-induced arachidonic acid release in rat mast cells. Biochem J. 1987;247:95–99. doi: 10.1042/bj2470095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugge E, Gamst TM, Hegstad AC, Andreasen T, Ytrehus K. Mepacrine protects the isolated rat heart during hypoxia and reoxygenation--but not by inhibition of phospholipase A2. Basic Res Cardiol. 1997;92:17–24. doi: 10.1007/BF00803753. [DOI] [PubMed] [Google Scholar]
- Beckman JK, Borowitz SM, Burr IM. The role of phospholipase A activity in rat liver microsomal lipid peroxidation. J Biol Chem. 1987;262:1479–1484. [PubMed] [Google Scholar]
- Evans PM, Lanham DF. Effects of inhibitors of arachidonic acid metabolism on intercellular adhesion of SV40-3T3 cells. Cell Biol Int Rep. 1986;10:693–698. doi: 10.1016/0309-1651(86)90126-8. [DOI] [PubMed] [Google Scholar]
- Hurst NP, French JK, Bell AL, Nuki G, O'Donnell ML, Betts WH, Cleland LG. Differential effects of mepacrine, chloroquine and hydroxychloroquine on superoxide anion generation, phospholipid methylation and arachidonic acid release by human blood monocytes. Biochem Pharmacol. 1986;35:3083–3089. doi: 10.1016/0006-2952(86)90390-4. [DOI] [PubMed] [Google Scholar]
- Churchill PC, Churchill MC, McDonald FD. Quinacrine antagonizes the effects of Na, K-ATPase inhibitors on renal prostaglandin E2 release but not their effects on renin secretion. Life Sci. 1985;36:277–282. doi: 10.1016/0024-3205(85)90070-0. [DOI] [PubMed] [Google Scholar]
- Erman A, Azuri R, Raz A. Prostaglandin biosynthesis in rabbit kidney: mepacrine inhibits renomedullary cyclooxygenase. Biochem Pharmacol. 1984;33:79–82. doi: 10.1016/0006-2952(84)90372-1. [DOI] [PubMed] [Google Scholar]
- Raz A. Mepacrine blockade of arachidonate-induced washed platelet aggregation: relationship to mepacrine inhibition of platelet cyclooxygenase. Thromb Haemost. 1983;50:784–786. [PubMed] [Google Scholar]
- Hofmann SL, Prescott SM, Majerus PW. The effects of mepacrine and p-bromophenacyl bromide on arachidonic acid release in human platelets. Arch Biochem Biophys. 1982;215:237–244. doi: 10.1016/0003-9861(82)90300-9. [DOI] [PubMed] [Google Scholar]
- Lot TY, Bennett T. Comparison of the effects of chloroquine quinacrine and quinidine on autonomic neuroeffector mechanisms. Med Biol. 1982;60:307–315. [PubMed] [Google Scholar]
- Authi KS, Traynor JR. Stimulation of polymorphonuclear leucocyte phospholipase A2 activity by chloroquine and mepacrine. J Pharm Pharmacol. 1982;34:736–738. doi: 10.1111/j.2042-7158.1982.tb06212.x. [DOI] [PubMed] [Google Scholar]
- Flynn JT. Inhibition of complement-mediated hepatic thromboxane production by mepacrine, a phospholipase inhibitor. Prostaglandins. 1987;33:287–299. doi: 10.1016/0090-6980(87)90013-X. [DOI] [PubMed] [Google Scholar]
- Louw L, Claassen J. Rationale for adjuvant fatty acid therapy to prevent radiotherapy failure and tumor recurrence during early laryngeal squamous cell carcinoma. Prostaglandins Leukot Essent Fatty Acids. 2008;78:21–26. doi: 10.1016/j.plefa.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Itoh S, Matsui K, Furuta I, Takano Y. Immunohistochemical study on overexpression of cyclooxygenase-2 in squamous cell carcinoma of the oral cavity: its importance as a prognostic predictor. Oral Oncol. 2003;39:829–835. doi: 10.1016/S1368-8375(03)00105-2. [DOI] [PubMed] [Google Scholar]
- Chen WC, McBride WH, Chen SM, Lee KF, Hwang TZ, Jung SM, Shau H, Liao SK, Hong JH, Chen MF. Prediction of poor survival by cyclooxygenase-2 in patients with T4 nasopharyngeal cancer treated by radiation therapy: clinical and in vitro studies. Head Neck. 2005;27:503–512. doi: 10.1002/hed.20178. [DOI] [PubMed] [Google Scholar]
- Chang BW, Kim DH, Kowalski DP, Burleson JA, Son YH, Wilson LD, Haffty BG. Prognostic significance of cyclooxygenase-2 in oropharyngeal squamous cell carcinoma. Clin Cancer Res. 2004;10:1678–1684. doi: 10.1158/1078-0432.CCR-03-0354. [DOI] [PubMed] [Google Scholar]
- Peng JP, Su CY, Chang HC, Chai CY, Hung WC. Overexpression of cyclo-oxygenase 2 in squamous cell carcinoma of the hypopharynx. Hum Pathol. 2002;33:100–104. doi: 10.1053/hupa.2002.30187. [DOI] [PubMed] [Google Scholar]
- Terakado N, Shintani S, Yano J, Chunnan L, Mihara M, Nakashiro K, Hamakawa H. Overexpression of cyclooxygenase-2 is associated with radioresistance in oral squamous cell carcinoma. Oral Oncol. 2004;40:383–389. doi: 10.1016/j.oraloncology.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Gallo O, Masini E, Bianchi B, Bruschini L, Paglierani M, Franchi A. Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Hum Pathol. 2002;33:708–714. doi: 10.1053/hupa.2002.125376. [DOI] [PubMed] [Google Scholar]
- Cho EI, Kowalski DP, Sasaki CT, Haffty BG. Tissue microarray analysis reveals prognostic significance of COX-2 expression for local relapse in T1-2N0 larynx cancer treated with primary radiation therapy. Laryngoscope. 2004;114:2001–2008. doi: 10.1097/01.mlg.0000147936.67379.e7. [DOI] [PubMed] [Google Scholar]
- Li N, Sood S, Wang S, Fang M, Wang P, Sun Z, Yang CS, Chen X. Overexpression of 5-lipoxygenase and cyclooxygenase 2 in hamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clin Cancer Res. 2005;11:2089–2096. doi: 10.1158/1078-0432.CCR-04-1684. [DOI] [PubMed] [Google Scholar]
- Feng L, Wang Z. Chemopreventive effect of celecoxib in oral precancers and cancers. Laryngoscope. 2006;116:1842–1845. doi: 10.1097/01.mlg.0000233778.41927.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata M, Kodani I, Osaki M, Araki K, Adachi H, Ryoke K, Ito H. Cyclo-oxygenase-1 and -2 expression in human oral mucosa, dysplasias and squamous cell carcinomas and their pathological significance. Oral Oncol. 2005;41:304–312. doi: 10.1016/j.oraloncology.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL, Dannenberg AJ. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999;59:991–994. [PubMed] [Google Scholar]
- Blackwell GJ, Duncombe WG, Flower RJ, Parsons MF, Vane JR. The distribution and metabolism of arachidonic acid in rabbit platelets during aggregation and its modification by drugs. Br J Pharmacol. 1977;59:353–366. doi: 10.1111/j.1476-5381.1977.tb07500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Lamberti M, Zacharek A, Li L, Szekeres K, Tang K, Chen Y, Honn KV. Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun. 2000;267:245–251. doi: 10.1006/bbrc.1999.1840. [DOI] [PubMed] [Google Scholar]
- Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008;118:18–35. doi: 10.1016/j.pharmthera.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Wei J, Yan W, Li X, Ding Y, Tai HH. Thromboxane receptor alpha mediates tumor growth and angiogenesis via induction of vascular endothelial growth factor expression in human lung cancer cells. Lung Cancer. 2009. [DOI] [PubMed]
- Oshima H, Oguma K, Du YC, Oshima M. Prostaglandin E2, Wnt, and BMP in gastric tumor mouse models. Cancer Sci. 2009;100:1779–1785. doi: 10.1111/j.1349-7006.2009.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei D, Murakami M, Nakatani Y, Ishikawa Y, Ishii T, Kudo I. Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J Biol Chem. 2003;278:19396–19405. doi: 10.1074/jbc.M213290200. [DOI] [PubMed] [Google Scholar]
- Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K, Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001;7:3971–3976. [PubMed] [Google Scholar]
- Fulton A, Miller F, Weise A, Wei WZ. Prospects of controlling breast cancer metastasis by immune intervention. Breast Dis. 2006;26:115–127. doi: 10.3233/bd-2007-26110. [DOI] [PubMed] [Google Scholar]
- Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001;7:2669–2674. [PubMed] [Google Scholar]
- Jabbour HN, Milne SA, Williams AR, Anderson RA, Boddy SC. Expression of COX-2 and PGE synthase and synthesis of PGE(2)in endometrial adenocarcinoma: a possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. Br J Cancer. 2001;85:1023–1031. doi: 10.1054/bjoc.2001.2033. [DOI] [PubMed] [Google Scholar]
- Yu L, Wu WK, Li ZJ, Li HT, Wu YC, Cho CH. Prostaglandin E(2) promotes cell proliferation via protein kinase C/extracellular signal regulated kinase pathway-dependent induction of c-Myc expression in human esophageal squamous cell carcinoma cells. Int J Cancer. 2009;125:2540–2546. doi: 10.1002/ijc.24607. [DOI] [PubMed] [Google Scholar]
- Camacho M, Leon X, Fernandez-Figueras MT, Quer M, Vila L. Prostaglandin E(2) pathway in head and neck squamous cell carcinoma. Head Neck. 2008;30:1175–1181. doi: 10.1002/hed.20850. [DOI] [PubMed] [Google Scholar]
- Jung TT, Berlinger NT, Juhn SK. Prostaglandins in squamous cell carcinoma of the head and neck: a preliminary study. Laryngoscope. 1985;95:307–312. doi: 10.1288/00005537-198503000-00014. [DOI] [PubMed] [Google Scholar]
- Cohen EG, Almahmeed T, Du B, Golijanin D, Boyle JO, Soslow RA, Subbaramaiah K, Dannenberg AJ. Microsomal prostaglandin E synthase-1 is overexpressed in head and neck squamous cell carcinoma. Clin Cancer Res. 2003;9:3425–3430. [PubMed] [Google Scholar]
- Kawata R, Hyo S, Maeda T, Urade Y, Takenaka H. Simultaneous expression of cyclooxygenase-2 and microsomal prostaglandin E synthase in squamous cell carcinoma of the larynx. Acta Otolaryngol. 2006;126:627–632. doi: 10.1080/00016480500452541. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tergaonkar V, Perkins ND. p53 and NF-kappaB crosstalk: IKKalpha tips the balance. Mol Cell. 2007;26:158–159. doi: 10.1016/j.molcel.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26:75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33–34. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- Delhase M, Li N, Karin M. Kinase regulation in inflammatory response. Nature. 2000;406:367–368. doi: 10.1038/35019154. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Friedman J, Nottingham L, Duggal P, Pernas FG, Yan B, Yang XP, Chen Z, Van Waes C. Deficient TP53 expression, function, and cisplatin sensitivity are restored by quinacrine in head and neck cancer. Clin Cancer Res. 2007;13:6568–6578. doi: 10.1158/1078-0432.CCR-07-1591. [DOI] [PubMed] [Google Scholar]
- Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D, Lvovskiy D, Webb TR, Stark GR, Gudkov AV. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proc Natl Acad Sci USA. 2005;102:17448–17453. doi: 10.1073/pnas.0508888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ho WC, Dicker DT, MacKinnon C, Winkler JD, Marmorstein R, El-Deiry WS. Acridine derivatives activate p53 and induce tumor cell death through Bax. Cancer Biol Ther. 2005;4:893–898. doi: 10.4161/cbt.4.8.2134. [DOI] [PubMed] [Google Scholar]
- Gurova K. New hopes from old drugs: revisiting DNA-binding small molecules as anticancer agents. Future Oncol. 2009;5:1685–1704. doi: 10.2217/fon.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbachev AV, Gasparian AV, Gurova KV, Gudkov AV, Fairchild RL. Quinacrine inhibits the epidermal dendritic cell migration initiating T cell-mediated skin inflammation. Eur J Immunol. 2007;37:2257–2267. doi: 10.1002/eji.200636708. [DOI] [PubMed] [Google Scholar]
- Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- Read MA, Whitley MZ, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–512. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Klein CL, van Kooten TG, Kirkpatrick CJ. Mechanisms of cell activation by heavy metal ions. J Biomed Mater Res. 1998;42:443–452. doi: 10.1002/(SICI)1097-4636(19981205)42:3<443::AID-JBM14>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Konig W, Schonfeld W, Raulf M, Koller M, Knoller J, Scheffer J, Brom J. The neutrophil and leukotrienes--role in health and disease. Eicosanoids. 1990;3:1–22. [PubMed] [Google Scholar]
- Stuhlmeier KM, Kao JJ, Bach FH. Arachidonic acid influences proinflammatory gene induction by stabilizing the inhibitor-kappaBalpha/nuclear factor-kappaB (NF-kappaB) complex, thus suppressing the nuclear translocation of NF-kappaB. J Biol Chem. 1997;272:24679–24683. doi: 10.1074/jbc.272.39.24679. [DOI] [PubMed] [Google Scholar]
- Thommesen L, Sjursen W, Gasvik K, Hanssen W, Brekke OL, Skattebol L, Holmeide AK, Espevik T, Johansen B, Laegreid A. Selective inhibitors of cytosolic or secretory phospholipase A2 block TNF-induced activation of transcription factor nuclear factor-kappa B and expression of ICAM-1. J Immunol. 1998;161:3421–3430. [PubMed] [Google Scholar]
- Palmetshofer A, Robson SC, Nehls V. Lysophosphatidic acid activates nuclear factor kappa B and induces proinflammatory gene expression in endothelial cells. Thromb Haemost. 1999;82:1532–1537. [PubMed] [Google Scholar]
- Pupe A, Degreef H, Garmyn M. Induction of tumor necrosis factor-alpha by UVB: a role for reactive oxygen intermediates and eicosanoids. Photochem Photobiol. 2003;78:68–74. doi: 10.1562/0031-8655(2003)078<0068:IOTNFB>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Pupe A, Moison R, De Haes P, van Henegouwen GB, Rhodes L, Degreef H, Garmyn M. Eicosapentaenoic acid, a n-3 polyunsaturated fatty acid differentially modulates TNF-alpha, IL-1alpha, IL-6 and PGE2 expression in UVB-irradiated human keratinocytes. J Invest Dermatol. 2002;118:692–698. doi: 10.1046/j.1523-1747.2002.01615.x. [DOI] [PubMed] [Google Scholar]
- Heller A, Koch T, Schmeck J, van Ackern K. Lipid mediators in inflammatory disorders. Drugs. 1998;55:487–496. doi: 10.2165/00003495-199855040-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gresham A, Morrison A, Pentland AP. Oxidative stress mediates synthesis of cytosolic phospholipase A2 after UVB injury. Biochim Biophys Acta. 1996;1299:23–33. doi: 10.1016/0005-2760(95)00166-2. [DOI] [PubMed] [Google Scholar]
- Kurose I, Saito H, Miura S, Ebinuma H, Higuchi H, Watanabe N, Zeki S, Nakamura T, Takaishi M, Ishii H. CD18/ICAM-1-dependent oxidative NF-kappaB activation leading to nitric oxide production in rat Kupffer cells cocultured with syngeneic hepatoma cells. J Clin Invest. 1997;99:867–878. doi: 10.1172/JCI119251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. Quinacrine acts like an acetylcholine receptor antagonist rather than like a phospholipase A2 inhibitor in a passive avoidance task in the chick. Neurobiol Learn Mem. 1995;63:206–208. doi: 10.1006/nlme.1995.1022. [DOI] [PubMed] [Google Scholar]
- Stuhlmeier KM. Effects of quinacrine on endothelial cell morphology and transcription factor-DNA interactions. Biochim Biophys Acta. 2001;1524:57–65. doi: 10.1016/s0304-4165(00)00140-9. [DOI] [PubMed] [Google Scholar]
- Fabbri S, Prontera C, Broggini M, D'Incalci M. Differential inhibition of the DNA binding of transcription factors NF kappa B and OTF-1 by nitrogen mustard and quinacrine mustard: transcriptional implications. Carcinogenesis. 1993;14:1963–1967. doi: 10.1093/carcin/14.9.1963. [DOI] [PubMed] [Google Scholar]
- Na SI, Lee MY, Heo JS, Han HJ. Hydrogen peroxide increases [3H]-2-deoxyglucose uptake via MAPKs, cPLA2, and NF-kappaB signaling pathways in mouse embryonic stem cells. Cell Physiol Biochem. 2007;20:1007–1018. doi: 10.1159/000110541. [DOI] [PubMed] [Google Scholar]
- Guo C, Gasparian AV, Zhuang Z, Bosykh DA, Komar AA, Gudkov AV, Gurova KV. 9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-kappaB and p53 pathways. Oncogene. 2009;28:1151–1161. doi: 10.1038/onc.2008.460. [DOI] [PubMed] [Google Scholar]
- Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem. 2002;277:5484–5489. doi: 10.1074/jbc.M108302200. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene. 2005;24:6719–6728. doi: 10.1038/sj.onc.1208825. [DOI] [PubMed] [Google Scholar]
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- Jung KJ, Dasgupta A, Huang K, Jeong SJ, Pise-Masison C, Gurova KV, Brady JN. Small-molecule inhibitor which reactivates p53 in human T-cell leukemia virus type 1-transformed cells. J Virol. 2008;82:8537–8547. doi: 10.1128/JVI.00690-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan S, Carraway RE. Involvement of arachidonic acid metabolism and EGF receptor in neurotensin-induced prostate cancer PC3 cell growth. Regul Pept. 2006;133:105–114. doi: 10.1016/j.regpep.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Heimpel H, Heit W. Drug-induced aplastic anaemia: clinical aspects. Clin Haematol. 1980;9:641–662. [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–140. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- Dey A, Tergaonkar V, Lane DP. Double-edged swords as cancer therapeutics: simultaneously targeting p53 and NF-kappaB pathways. Nat Rev Drug Discov. 2008;7:1031–1040. doi: 10.1038/nrd2759. [DOI] [PubMed] [Google Scholar]