

Abstract
Purpose
Since TLR agonists have been well characterized as DC activators, we hypothesized that the admixture of TLR4 agonist into a cellular vector could improve the anti-tumor response in vivo.
Experimental Design
GM-CSF secreting whole cell tumor cell vector (GVAX) was formulated with LPS, a TLR4 agonist, and its intratumoral therapeutic efficacy was tested in three different murine models. We utilized immunohistochemistry, FACS, ELISPOT, and in vivo CTL analysis to assess both local innate immune responses within the tumor tissue as well as the downstream generation of anti-tumor T-cell responses.
Results
Intratumoral treatment of LPS absorbed GVAX showed efficacy in improving an antitumor response in vivo in comparison to GVAX alone. Improved anti-tumor efficacy of this novel admixture was not present in TLR4 signaling impaired mice. In the CT26 model, 40-60% of the mice showed regression of the transplanted tumor. When rechallenged with CT26 tumor cells, these mice proved to be immunized against the tumor. Tumors treated with TLR4 agonist absorbed GVAX showed increased infiltrating CD4 and CD8 T-cells as well as increased numbers of CD86+ cells in the tumor tissue. Draining lymph nodes from the treated mice had enhanced number of activated CD86+, MHCII+, and CD80+ dendritic cells in comparison to GVAX alone and mock treated groups. ELISPOT assay and in vivo CTL assay showed increased numbers of CTLs specific for the AH1 tumor antigen in mice treated with LPS absorbed GVAX.
Conclusions
TLR4 on APCs in the tumor microenvironment may be targeted using cell-based vectors for improved anti-tumor response in vivo.
Introduction
The role of TLR mediated inflammation in the tumor microenvironment is a complex process whose role in carcinogenesis is still unclear. Various tumor models dissecting the downstream MyD88 and NF-kB signaling pathways initially demonstrated procarcinogenic role of TLR signaling (1-3). However, Salcedo, et.al. have demonstrated anti-tumor role of MyD88 signaling while Garrett et.al. have noted no carcinogenic effect of MyD88 signaling in Rag2−/− mice (4,5). TLR signaling in the hematopoietic compartment, however, has been shown to elicit anti-tumor responses, which have translated into multiple clinical trials (6,7). In the context of infection, TLR4 agonists have been shown to render dendritic cell (DC) activation immunogenic whereas lack of TLR4 signaling can lead to tolerance (8). Medzhitov showed phagocytosed microbial antigens can be more efficiently presented in the presence of LPS (9). Cumulative, one valid strategy is to increase TLR stimulation directed towards increasing the number of activated APCs within the tumor microenvironment in vivo (10-12).
The current study focuses on intratumoral injection as a direct means of modifying the tumor microenvironment to enhance both innate and adaptive anti-tumor immunity. In order to minimize potential TLR4 signaling on the tumor cells and to target the antigen presenting cells in the tumor microenvironment, we evaluated intratumoral delivery of cellular vectors loaded with TLR4 agonist. The implication from Medzhitov’s studies is that co-localization of TLR4 agonist and antigen can potentially enhance antitumor response when given as part of a combinatorial cellular vector with tumor antigens (9). An excellent candidate as a cellular vector for localized TLR agonist delivery is GVAX. The paracrine GM-CSF delivery afforded by GVAX cells dramatically increases the numbers of myeloid cells – including DCs, macrophages and granulocytes, at the site of injection (13-15). GVAX has been found to be safe from numerous phase I trials, but was recently found to have uncertain efficacy in a phase III trial for advanced hormone refractory prostate cancer (16) As a cellular vector, however, GVAX may offer a starting platform to immunize the patients with multiple tumor antigens and deliver apoptotic and cellular debris loaded with TLR4 agonists to activate APCs in the tumor microenvironment as part of a combinatorial therapy.
In order to activate the locoregional APCs, LPS was formulated with GVAX cells, and this novel combinatorial regimen was injected intratumorally and studied for anti-tumor efficacy in several murine models.
Methods
Murine Tumor Cell Lines
The SCCFVII/SF head and neck squamous carcinoma, B16-F0 melanoma, and B16-F0 transduced to secrete GM-CSF cell lines were cultured in RPMI 1640 with 10% FCS, penicillin (100 U/ml) and streptomycin (100 U/ml). CT26 was cultured similarly with MEM nonessential amino acids (Sigma), 1 mM sodium pyruvate, and 2 mM of L-glutamine. The bystander B78HI cells transduced with GM-CSF were cultured in the same media as CT26 with the addition of Hygromycin B (1g/L) (Roche).
Mice
Adult (>50 days of age) female C57BL/6, Balb/C, C3H/HeOUJ, and C3H/HeJ mice were purchased from Jackson Laboratory and housed according to the JHH Animal Care and Use Committee. C57BL/6 MyD88−/− and C57BL/6 MyD88−/−TRIF−/− mice were obtained from Dr. Franck Housseau (Johns Hopkins University).
TLR4 Agonist formulation with GVAX
Lipofectamine (120μg/ml), LPS from Escherichia coli 026:B6 (10-50 μg/ml), and LPS-BODIPY (Molecular Probes) were combined into liposome complex. Cells for lipopolysaccharide (LPS) formulation were seeded at a density of 5×105 cells were incubated with the Lipofectamine-LPS complex for 6 hrs, washed 5 times prior to injection. To quantitate LPS incorporated into cells, Limulus Amebocyte Lysate (LAL) assay (Cambrex) was performed as directed by the manufacturer. LPS (23 EU/ng) concentrations ranging from 0 to 3.0 EU/ml were used as standards. Prior to lethal irradiation, LPS absorbed GVAX was cultured to ensure cell growth. Annexin staining verified no evidence of apoptosis after formulation (data not shown).
In vivo vaccine treatment assay
C57BL/6 mice were injected subcutaneously in the right flank with 5×104 B16-F0 cells. Three to five days later, 106 lethally irradiated (150 Gy) B16 GM-CSF (GVAX), 106 lethally irradiated (150 Gy) LPS formulated GVAX or LPS absorbed 293T cells were injected intratumorally. In some cases, the vaccines were injected in the contralateral limb from the tumor-inoculated limb. C3H/HeOUJ mice and Balb/c mice were used with SCCFVII/SF cells and CT26 cells, respectively with comparable methods.
Immunohistochemistry
Frozen CT26 tumor were cut in 10μm thickness and blocked with 1% BSA 30 minutes at RT. Anti-mouse CD45 (eBioscience) and anti-mouse CD4-FITC, CD8-FITC, CD86-FITC antibodies (BD Pharmingen) incubated at 4°C for 1 hour were used. Anti-CD45-Cy3 (Invitrogen) was used as secondary in some cases. DAPI was used as counter stain for 10 minutes. Cells in 10 randomly selected fields at 40x magnification were counted using Nikon Eclipse F800 microscope and developed with Nikon DS-Qi1mc camera. NIS-Element AR 3.0 was the software used for these experiments.
Dendritic cell (DC) activation assay
Spleens and draining lymph nodes from tumor challenged mice were harvested five days post GVAX and TEGVAX treatment. Crushed spleens were digested in media containing DNAse I (Roche) and Liberase Blendzyme 2 (20,000 Mandl U/ml) (Roche). DC-enriched populations were obtained by depleting CD3+ and CD19+, and gated for CD11c+ and B220+. These were evaluated by a multicolored FACS analysis using CD80, CD86, and MHCII antibodies from BD Biosciences.
ELISPOT assay
ELISPOT plates (MultiScreenHTS filter plate, Millipore) were coated with a mouse IFN-γ Ab (MabTech) for 24 hours and 4T1 breast cancer cells were pulsed with 10 μg/ml of AH1 peptide overnight. 106 CD8 cells from spleen and lymph nodes were plated in triplicates to be co-cultured with pulsed or unpulsed 105 4T1 cells or stimulated with 1μM of PMA and 10ng/ml of Ionomycin as positive controls. On day 3, biotinylated anti-mouse IFN-γ Ab (MabTech) and Strepavidin-HRP for ELISPOT (BD) were added. AEC Substrate Reagent Set for ELISPOT (BD) was used to develop spots and analyzed using an ELISPOT Plate Reader (Immunospot).
In vivo CTL assay
A CellTrace CFSE Cell Proliferation Kit (Molecular Probes) was used to test the cytolytic activity of CTLs in CT26 tumor challenged mice treated with or without TEGVAX. Splenocytes were processed and pulsed with either β-gal or AH1 peptide at a concentration of 10ug/ml for 90 minutes. The β-gal population was CFSE labeled low (0.5μM) and the AH1 population was CFSE labeled high (5 μM). After 10 minutes, both CFSE labeled cells were injected into the mice at 107cells/mouse. Twenty-four hours post-injection, splenic cells were harvested and analyzed for detection of ratios of CFSE labeled cells (19). Peptide-specific killing was calculated using the formula: (1-% of CFSE peptide/% of CFSE no peptide) × 100.
Statistical Analysis
We used paired t-test to calculate two-tailed p value to estimate statistical significance of differences between two treatment groups using Excel software. Kaplan-Meier curves were generated using GraftPad Prism software and analyzed with log rank test. Statistically significant p values are labeled in the figures and the legends with asterisks.
Results
TLR4 Agonist formulation with GVAX
In order to enhance locoregional innate immune cell activation as well as minimize systemic toxicity of TLR4 stimulation and minimize TLR4 stimulation on tumor cells, LPS was formulated into GVAX (TEGVAX – TLR agonist enhanced GVAX). We used a commercially available vector – Lipofectamine – to optimize absorption of LPS into GVAX cells prior to lethal irradiation. LPS-BODIPY flurophore was used to show that 25μg/ml of LPS for 6 hours resulted in 99.4% of the cells to be labeled (Figure 1). Limulus Amebocyte Lysate (LAL) assay (Cambrex) was then used to demonstrate that 4.73+/− 0.2 ng of LPS was absorbed into 5 × 105 cells using the lipofectamine method that optimized LPS formulation (data not shown). For each of the in vivo murine tumor experiments, aliquots of TEGVAX were tested using LAL assay as well as murine GM-CSF ELISA assay to ensure a comparable amount of LPS and GM-CSF in the TEGVAX formulation. The typical GM-CSF secreted ranged from 50-200 ng/ml/106 cells/24 hours.
Figure 1.

Formulation of TLR4 agonist into GVAX. To optimize the absorption of LPS, the concentrations of Lipofectamine and LPS-BODIPY fluorophore conjugate were varied as noted. Cells were washed 5 times, and LPS was undetectable in the final wash using the LAL assay. Optimal absorption was at 6 hr incubation with 40μg/ml of Lipofectamine and 25μg/ml of LPS per 1×105 cells. These conditions were used for quantitation of LPS on a per cell basis.
TEGVAX can induce in vivo anti-tumor response in multiple murine models
We initially tested the efficacy of intratumoral TEGVAX injection in a B16 murine model whereby TEGVAX was delivered intratumorally 3 to 5 days after tumor inoculation in a therapeutic model. One rationale for intratumoral injection was to ensure that locoregional APCs that circulate between the tumor and the draining lymph nodes were targeted. These time points of treatment relative to initial tumor implantation were selected because we previously demonstrated that anergic and tolerant tumor specific T-cells were present as early as 3 days after B16 injection (17). As shown in Figure 2, intratumoral TEGVAX administration decreased the growth rate of the B16 tumor in comparison to intratumoral injection of GVAX alone, which translated to survival curve differences (Figure 2A). However, all the mice treated with TEGVAX eventually developed large tumors. We injected equimolar amount of LPS intratumorally as in the TEGVAX treatment group and these mice were no different than mice treated with GVAX or PBS (Supplemental Figure 1). Equimolar LPS injected with GVAX separately without formulation also did not demonstrate an antitumor response, suggesting that the specific formulation of LPS with a cellular vector is critical for its anti-tumor response (Supplemental Figure 1).
Figure 2.

TEGVAX can induce an anti-tumor response in vivo. (A) B16 inoculated C57BL/6, (B) SCCFVII/SF inoculated C3H/HeOUJ, and (C) CT26 inoculated BALB/c mice were treated with appropriate PBS, GVAX, or TEGVAX intratumorally typically from 3-5 days after the tumor injection. In vivo tumor progression in the TEGVAX group in all the murine models studied was statistically slower than those in the GVAX group (*P<0.05) (left panels). 10-20 mice per group were used, and all the experiments replicated 3 times. For the B16 and the SCCFVII/SF models, there were no differences between GVAX treated group and the PBS treated group (Supplemental Figure 1 and data not shown). In the CT26 model, 40-60% of the mice in the TEGVAX group experienced a regression of their palpable tumor to undetectable levels by 3 weeks. Equimolar LPS formulated into 293T cells also showed no difference in growth rate. Mice showing regression of tumor were rechallenged with CT26 tumor (2×106) and no tumor growth were noted. In all three murine models, the reduced tumor growth rate correlated with enhanced survival for mice in the TEGVAX group compared to those in the GVAX group (**P<0.01) (right panels).
To test if TEGVAX can also demonstrate an anti-tumor response in other murine models, SCCFVII/SF cells were subcutaneously injected into the flanks of syngeneic C3H/HeOUJ mice with a wildtype TLR4 (Figure 2B) with comparable results as the B16 model.
Lastly, intratumoral administration of TEGVAX was also tested in the CT26 colon carcinoma model, where it also showed an anti-tumor response (Figure 2C). For CT26, 40-60% of the mice treated with TEGVAX actually demonstrated regression of tumor. Mice whose tumor regressed completely after intratumoral TEGVAX administration rejected subsequent challenges with CT26, indicating that the initial intratumoral treatment resulted in systemic immunization. For the CT26 model, we also tested LPS formulated with lethally irradiated 293 cells in equimolar amounts as TEGVAX. This experiment was done to determine whether co-localization of LPS and tumor antigen in TEGVAX was important for the anti-tumor response. LPS formulated with cells not expressing an identical set of tumor antigens as the treated tumor did not elicit an anti-tumor response in the CT26 model (Figure 2).
Anti-tumor response to TEGVAX is MyD88-TRIF and TLR4 dependent
In order to verify that the in vivo anti-tumor response noted above were due to the TLR4 signaling, B16 tumor was treated with TEGVAX and GVAX in mice with MyD88−/− as well as MyD88−/−TRIF−/− genotypes (Figure 3A). Small differences noted early between the GVAX group and PBS groups eventually disappeared at later time points in MyD88−/− mice. MyD88 and TRIF are essential intracellular mediators of TLR4 signaling, and the complete absence of these downstream TLR4 signaling mediators also abrogated the enhanced in vivo anti-tumor response noted in wild type mice. For the SCCFVII model, the experiment was performed in C3H/HeJ mice that lack functional TLR4, and, once again, the antitumor effect of TEGVAX was abrogated (Figure 3B).
Figure 3.

Improved in vivo anti-tumor response noted for TEGVAX is MyD88 dependent and TLR4 dependent. (A) Using the B16 model in the MyD88−/− mice and MyD88−/−TRIF−/− mice, the in vivo tumor growth rate was not delayed by the vaccine treatment performed intratumorally 3-5 days after tumor inoculation. (B) For the SCCFVII model, the experiment was performed in C3H/HeJ that lack TLR4, and for these mice, TEGVAX treatment did not demonstrate the anti-tumor response. Experiments using C3H/HeOUJ mice replicated results from Figure 2 (*P<0.05). All these experiments were replicated three times with 10 mice per group.
TEGVAX induces enhanced T-cell infiltration and APC maturation in the tumor
In order to examine the potential mechanism of the in vivo anti-tumor responses from TEGVAX treatment, the tumor tissue was harvested and analyzed for lymphocytic infiltrate. As shown in Figure 4, tumor treated with TEGVAX had quantitatively increased CD4 and CD8 infiltration in comparison to the control and the GVAX treated tumors. Moreover, given our hypothesis that TLR4 agonist stimulates the locoregional APCs to mature, we probed the tissue with αCD86 and noted quantitative enhancement of CD86+ cells in the tumor treated with TEGVAX. We found no significant differences in the infiltration of F4+ macrophages between the treatment groups (data not shown).
Figure 4.

TEGVAX treatment increases the lymphocytic and antigen presenting cell infiltration into the tumor microenvironment. Immunohistochemical staining was used to detect the expression of CD4+, CD8+, CD86+ and CD45+ cells in CT26 tumor treated with control, GVAX, or TEGVAX. The yellow cells represent the co-localized staining of CD4 or CD8 with CD45 in the first two columns. The arrow points to the non-aggregating, diffusely infiltrating lymphocytes in the tumor tissue. The third column represents CD86 conjugate staining alone. These cells were formally quantified in 10 randomly selected fields per slide at 40X magnification. Statistical differences were obtained in the CD4, CD8, CD86 and CD45 cells between the GVAX and TEGVAX groups (P<0.01). These experiments were replicated at least 3 times.
TEGVAX augments dendritic cell activation in the locally draining lymph nodes
The immunostaining data from Figure 4 were consistent with the hypothesis that TLR4 agonist absorbed cell vaccines can increase the number of activated locoregional DCs. Given that the LPS absorbed into GVAX is probably phagocytosed into the infiltrating DCs as cellular debris and micelles with tumor antigens, we predicted that there would be increased number of activated locoregional DC population with TEGVAX treatment. In order to test this, we purified dendritic cells from the draining lymph nodes from tumor bearing mice treated with either PBS, GVAX, or TEGVAX and gated for the conventional DC population with B220 and CD11c. Multiparametric staining of DC activation marker CD86, CD80, and MHCII from these gated cells shows that DCs from the TEGVAX treated group has greater population of activated phenotype (Figure 5).
Figure 5.
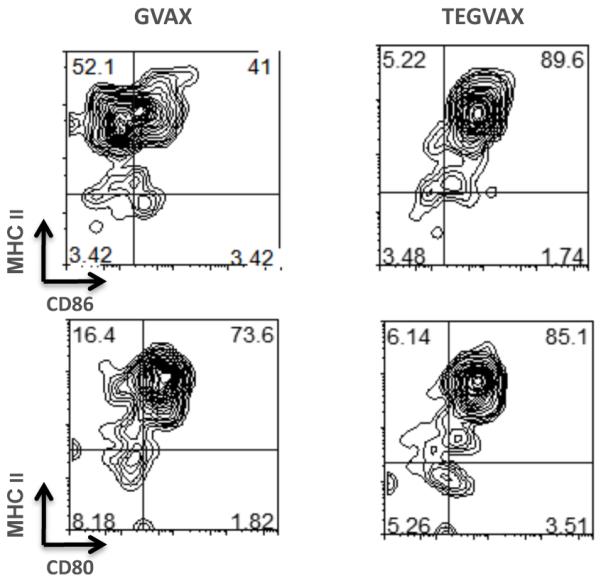
TEGVAX treatment increases activated dendritic cells (DC) in the draining lymph nodes (DLN). 10-20 CT26 tumor bearing BALB/c mice were treated with PBS (not shown), GVAX, or TEGVAX peritumorally 3 days after the tumor injection. DCs were isolated from the DLN 5-8 days after treatment by enzymatic digestion. B220-CD11c+ conventional DC cells were gated and CD86, MHCII, and CD80 staining were analyzed as shown. TEGVAX had an increased number of CD86+MHCII+ as well as CD80+MHCII+ DCs in the DLN in comparison to GVAX treated group. These experiments were replicated more than 3 times. The results from these other experiments are depicted in Supplementary Figure 3.
Tumor specific cytotoxic T-cells are expanded in mice treated with TEGVAX
CT26 model is associated with a well-characterized immunodominant CT26 antigen, AH1, that can facilitate quantitation of tumor specific cytotoxic T-cells. To test if the in vivo anti-tumor responses for TEGVAX that activates locoregional dendritic cells can increase the downstream population of tumor specific cytotoxic effector cells, ELISPOT assays were performed (18). T-cells from the draining lymph node and spleen were harvested and IFN-γ producing cytotoxic T-cells screened in the presence of MHC class I Ld restricted AH1 peptides pulsed with APC in the ELISPOT assay. While minimal AH1 specific T-cells were detected on days 3-5 after treatment with TEGVAX (data not shown), by day 8, there were statistically significant AH1 specific T-cells in the TEGVAX group in both the draining lymph nodes and the spleen as shown in Figure 6A. In vivo CTL assays also demonstrated enhanced cytotoxic T-cell priming for the AH1 peptide in the TEGVAX treated group in comparison to the control groups (Figure 6B). The “average killing” value calculated between TEGVAX and GVAX only reached statistical significance at P<0.07, but each of the experiments showed consistent trend for enhanced number of AH1 specific T-cells in the TEGVAX treated groups. In vivo CTL assays with p15E specific T-cell from the B16 model also demonstrated enhanced p15E-specific T-cells in the TEGVAX group (data not shown).
Figure 6.

TEGVAX treatment increases the number of tumor specific CD8+ T-cells. (A) CT26 tumor bearing BALB/c mice were treated with PBS, GVAX, or TEGVAX, and 5 days later CD8+ cells were isolated and purified from the spleen and lymph nodes. ELISPOT assays were performed using 4T1 cells as APCs and AH1 peptide, and IFN-γ levels were measured. The number of AH1 specific IFN-γ producing T-cells were statistically greater in the TEGVAX group compared to GVAX group in both the spleen and the draining LN (P<0.01). (B) In vivo CTL assays were used to measure cell killing by AH1 specific CTLs in CT26 tumor bearing mice treated with or without TEGVAX (see Methods section for calculation of average cell killing). The average cell killing, which measure AH1 specific CTLs, was higher in the TEGVAX group compared to the untreated groups (P<0.07). These experiments were replicated 3 times.
Discussion
In this report, we tested the efficacy of intratumoral injection of TEGVAX, a novel TLR4 agonist formulated GVAX, whereby we were able to absorb TLR4 agonist into GVAX cells. We demonstrated that TLR4 agonist absorbed GVAX has significantly improved anti-tumor response in three different therapeutic murine models, including SCCFVII/SF, Bl6, and CT26. For the CT26 model, TEGVAX injected intratumorally was able to prevent tumor growth in 40-60% of the mice, while GVAX or LPS injection alone had no such effect.
By adding a potent TLR4 agonist into the tumor microenvironment via a cellular vector with our TEGVAX reagent, we demonstrated that the growth rate of the inoculated tumor is significantly blunted. We believe that it is not only the addition of TLR4 agonist, but also its novel delivery absorbed into lethally irradiated vaccine cells with tumor antigens as vectors. Equimolar LPS injection alone did not have an in vivo response. Injection of equimolar LPS directly into the tumor separately from GVAX as well as control experiments using LPS absorbed into 293T cells also did not produce the anti-tumor response in our mice models (Figure 2 and Supplementary Figure 1). The effect of TLR4 agonist in the tumor microenvironment is a complex process that has produced conflicting in vivo responses. Recent reports showed that TLR4 expressed on HNSCC cell lines could promote carcinogenesis (3). However, expression of TLR4 in HNSCC primary tumor is heterogeneous and it is unclear in whether TLR4 signaling is a critical carcinogenic signaling in vivo. Others have shown that TLR4 stimulation can break CD8+ T-cell tolerance and eradicate established tumors (19,20). MyD88 signaling, which is downstream to TLR4, has been shown to be procarcinogenic in some animal models (5). However, our results are consistent with a recently presented hypothesis developed by Medzhitov and others that TLR4 agonists absorbed into micelles can induce an up regulation of antigen presenting machinery in the phagolysosome by the APCs (9).
With TEGVAX, our strategy was to dress syngeneic cells as “foreign” cells with strong danger signals that can activate APCs in the tumor microenvironment. LPS formulated into cellular vaccines may be preferentially targeted towards the locoregional APCs such as dendritic cells. We hypothesized that apoptotic cellular debris with TLR4 agonist from the injected TEGVAX cells would be phagocytosed by the local APCs such that TLR4 signaling on the tumor cells would be minimized. Hence this would explain the consistent lack of in vivo anti-tumor response in the mouse groups treated with LPS alone or with LPS and GVAX without the absorption formulation. When we examined the draining LN, TEGVAX treatment was associated with an increased number of activated DCs as defined by high expression of CD86+, CD80+, and MHC class II+ cells in comparison to GVAX treated groups as shown in Figure 5. GVAX treated groups also had increased number of activated DCs but less than TEGVAX treated DCs, which is consistent with the immunohistochemical data from Figure 4. GM-CSF from GVAX can also activate DC cells that explain the increased number of activated DC in GVAX group in comparison to the control group treated with PBS alone. The lack of anti-tumor response in GVAX treatment group may stem from possible upregulation of myeloid derived suppressor cells (MDSC) that has been associated with GM-CSF signaling in the tumor microenvironment(21). Regardless, TEGVAX treated groups had greater number of activated DCs in comparison to the GVAX or the PBS treated groups to potentially induce tumor specific CTL response in our experimental models.
Activated APCs can prime tumor specific T-cells as downstream effectors, so we examined the tumor tissue from each of the treated and the control groups for CD4 and CD8 T-cell infiltration using immunohistochemistry. Quantitatively, TEGVAX treated tumors demonstrated greater infiltration of both CD4 and CD8 T-cells in comparison to GVAX or PBS treated tumors. Whether the increase in CD4+ cells is also associated with any changes in Treg or Th17 cells are still being investigated. Regardless, there appeared to be a positive correlation between T-cell infiltration and the overall tumor response in vivo. We stained for NK cells in the tumor tissue, but did not see any qualitative differences (data not shown).
We quantitated the tumor specific Ld restricted AH1 specific IFN-γ secreting CTLs from each of the control and treated groups in the CT26 model, and both ELISPOT and in vivo CTL assay demonstrated that TEGVAX treated mice had increased number of AH1 specific CTLs. These results strongly suggest that one downstream consequence of TEGVAX treatment is the up regulation of anti-tumor cytotoxic IFN-γ producing T-cells in both the draining lymph nodes and the spleen that can slow the rate of the tumor growth. Consistent with this mechanistic picture is an experiment whereby TEGVAX injected on the contralateral limb from the tumor growth site had comparable in vivo anti-tumor response as intratumoral injection as shown in Supplementary Figure 2. Both intratumoral and systemic treatments were lymphocyte dependent as demonstrated by the abrogation of these effects in Rag2−/− mice. While intratumoral injection can elicit both innate and adaptive immune response, injection of TEGVAX on the contralateral limb predominantly increases the priming of naïve T-cells into vaccine specific T-cells.
In terms of the architecture of the tumor microenvironment, TEGVAX treated tumor had broad infiltration of CD86+ cells. While both PBS and GVAX treated tumor had localized aggregates of lymphocytes and APCs, TEGVAX treated tumors had diffuse infiltration of CD4+, CD8+, and CD86+ cells (see arrow in Figure 4). These ectopic lymphoid aggregates have been described in pancreatic cancer specimens from patients treated with GVAX(22), (personal communication Drs. Elizabeth Jaffee and Lei Zhang). The significance of these aggregates is unclear, but there appears to be a negative correlation between these aggregates and the tumor response in vivo.
Given that one limitation of GVAX is limited number of activated APCs, development of TEGVAX is a step towards an efficacious combinatorial immunotherapeutic reagent for advanced cancer patients. GVAX has been found to be safe in multiple phase I trials, and preclinical in vivo efficacy of TEGVAX, as demonstrated in this report, warrants further investigation. Of greatest concern is the use of TLR4 agonist therapy in cancer patients. Goto et.al. had previously injected 10μg of LPS into cancer patients with tolerable side effects that were not greater than WHO grade 3 (23). Extrapolating from previous trials with GVAX in cancer patients where 108-9 GVAX cells were used, this would amount to micrograms of LPS injected submucosally or subdermally in human subjects as currently formulated in TEGVAX. In order to minimize the exposure of LPS, we are currently formulating TEGVAX with a lower amount of LPS on a per cell basis to titrate the minimal amount of LPS required for in vivo response in murine models. We are also currently developing TEGVAX with nontoxic lipid A mimetics.
Much like the vaccines for infectious agents, the importance of TLR4 adjuvants has been acknowledged by the fact that the multiple clinical cancer vaccines have been typically mixed with TLR4 agonists. The MAGE-A3 ASCI lung cancer vaccine for NSC lung cancer as well as Cervarix for cervical cancer contains monophosphoryl lipid A (MPL), a TLR4 agonist (7). BCG, another TLR4 agonist, was found to be important for low volume melanoma disease (24). Our report, however, points out the importance of TLR4 agonist formulation in terms of in vivo efficacy. Simple mixing of cellular vaccine and adjuvants may be a suboptimal method to integrate the power of danger signals into the combinatorial vaccine. Future studies will also compare different TLR4 agonists that have been tested in patients with comparable formulations.
In conclusion, we report a novel formulation of GVAX tumor vaccines with improved anti-tumor efficacy with intratumoral injection in several murine models. Intratumoral injection of vaccine is potentially feasible in select cases of accessible tumors such as head and neck and skin malignancies. Moreover, intratumoral injection has important translational implications for neoadjuvant therapy prior to surgery in patients with a high risk of relapse. Intratumoral injection prior to resection could generate systemic anti-tumor immune responses that could eliminate the micrometastasis undetectable at the time of surgery, which are the sources of tumor relapse.
Translational Relevance.
Use of TLR agonists as adjuvants for advanced malignancies in patients is still controversial. One limitation is that TLR signaling can have mixed results in the tumor microenvironment. In order to target TLR4 signaling in antigen presenting cells within the tumor microenvironment, we have formulated TLR4 agonist with a safe cellular vaccine, GVAX, which is currently undergoing clinical trials for multiple solid tumors. The following preclinical studies, therefore, provide scientific rationale for potential modification of a well-studied tumor vaccine platform to improve its clinical response in solid tumors amenable for intratumoral injections such as head and neck tumors.
Supplementary Material
Supplementary Figure 1: GVAX with LPS injected separately (without TEGVAX co-formulation) do not induce an anti-tumor response. B16 bearing mice were treated intratumorally with either GVAX, LPS, or with a mixture of LPS and GVAX without liposomal formulation. Equimolar amounts of LPS as used in Figure 2 were injected intratumorally in these experiments.
Supplementary Figure 2: TEGVAX can also induce a systemic anti-tumor response that is dependent on normal lymphocytes. Using the B16 tumor model, mice were treated with either TEGVAX intratumorally or TEGVAX injected into the contralateral limb from the site of tumor inoculation. Both TEGVAX groups showed similar in vivo tumor growth rate (Supp 2A). These experiments were also performed in Rag2−/− mice, which showed that the anti-tumor response is lymphocyte dependent (Supp 2B).
Supplementary Figure 3: TEGVAX consistently increases DC activation profile markers. These are three examples of DC activation profile from three independent experiments. Conventional DC were gated as CD11chighB220− and stained for CD86.
Acknowledgements
Grant Support: NIH K23-DE018464-02 and Triological Society / American College of Surgeon Career Developmental Award (YJK).
References
- 1.Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schreiber RD, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105(2):652–6. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–4. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 3.Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, et al. Triggering of toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69(7):3105–13. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J Exp Med. 2010;207(8):1625–36. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16(3):208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murad YM, Clay TM, Lyerly HK, Morse MA. CPG-7909 (PF-3512676, ProMune): Toll-like receptor-9 agonist in cancer therapy. Expert Opin Biol Ther. 2007;7(8):1257–66. doi: 10.1517/14712598.7.8.1257. [DOI] [PubMed] [Google Scholar]
- 7.Dubensky TW, Jr, Reed SG. Adjuvants for cancer vaccines. Semin Immunol. 2010;22(3):155–61. doi: 10.1016/j.smim.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299(5609):1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 9.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(7085):808–12. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 10.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi: 10.1016/S0065-2776(06)90001-7. [DOI] [PubMed] [Google Scholar]
- 12.Long CM, van Laarhoven HW, Bulte JW, Levitsky HI. Magnetovaccination as a novel method to assess and quantify dendritic cell tumor antigen capture and delivery to lymph nodes. Cancer Res. 2009;69(7):3180–7. doi: 10.1158/0008-5472.CAN-08-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–43. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eager R, Nemunaitis J. GM-CSF gene-transduced tumor vaccines. Mol Ther. 2005;12(1):18–27. doi: 10.1016/j.ymthe.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Couch M, Saunders JK, O’Malley BW, Jr, Pardoll D, Jaffee E. Genetically engineered tumor cell vaccine in a head and neck cancer model. Laryngoscope. 2003;113(3):552–6. doi: 10.1097/00005537-200303000-00029. [DOI] [PubMed] [Google Scholar]
- 16.Lassi K, Dawson NA. Emerging therapies in castrate-resistant prostate cancer. Curr Opin Oncol. 2009;21(3):260–5. doi: 10.1097/CCO.0b013e32832a1868. [DOI] [PubMed] [Google Scholar]
- 17.Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95(3):1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain A, Slansky JE, Matey LC, Allen HE, Pardoll DM, Schulick RD. Synergistic effect of a granulocyte-macrophage colony-stimulating factor-transduced tumor vaccine and systemic interleukin-2 in the treatment of murine colorectal cancer hepatic metastases. Ann Surg Oncol. 2003;10(7):810–20. doi: 10.1245/aso.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5(5):508–15. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 20.Paulos CM, Kaiser A, Wrzesinski C, Hinrichs CS, Cassard L, Boni A, et al. Toll-like receptors in tumor immunotherapy. Clin Cancer Res. 2007;13(18 Pt 1):5280–9. doi: 10.1158/1078-0432.CCR-07-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64(17):6337–43. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 22.Bombardieri M, Barone F, Humby F, Kelly S, McGurk M, Morgan P, et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in sjogren’s syndrome. J Immunol. 2007;179(7):4929–38. doi: 10.4049/jimmunol.179.7.4929. [DOI] [PubMed] [Google Scholar]
- 23.Goto S, Sakai S, Kera J, Suma Y, Soma GI, Takeuchi S. Intradermal administration of lipopolysaccharide in treatment of human cancer. Cancer Immunol Immunother. 1996;42(4):255–61. doi: 10.1007/s002620050279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiFronzo LA, Gupta RK, Essner R, Foshag LJ, O’Day SJ, Wanek LA, Stern SL, Morton DL. Enhanced humoral immune response correlates with improved disease-free and overall survival in AJCC stage II melanoma patients receiving adjuvant polyvalent vaccine. J Clin Oncol. 2002;20(15):3242–8. doi: 10.1200/JCO.2002.01.065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: GVAX with LPS injected separately (without TEGVAX co-formulation) do not induce an anti-tumor response. B16 bearing mice were treated intratumorally with either GVAX, LPS, or with a mixture of LPS and GVAX without liposomal formulation. Equimolar amounts of LPS as used in Figure 2 were injected intratumorally in these experiments.
Supplementary Figure 2: TEGVAX can also induce a systemic anti-tumor response that is dependent on normal lymphocytes. Using the B16 tumor model, mice were treated with either TEGVAX intratumorally or TEGVAX injected into the contralateral limb from the site of tumor inoculation. Both TEGVAX groups showed similar in vivo tumor growth rate (Supp 2A). These experiments were also performed in Rag2−/− mice, which showed that the anti-tumor response is lymphocyte dependent (Supp 2B).
Supplementary Figure 3: TEGVAX consistently increases DC activation profile markers. These are three examples of DC activation profile from three independent experiments. Conventional DC were gated as CD11chighB220− and stained for CD86.