

Abstract
OBJECTIVE
Brief systemic hypoxia protects the rodent brain from subsequent ischemic injury, although the protection wanes within days. We hypothesized that the duration of ischemic tolerance could be extended from days to months by repeated intermittent hypoxia of varying magnitude and duration.
METHODS
Infarction volumes following a 60-min transient middle cerebral artery occlusion were determined in adult male mice 2 days through 8 wks after completion of a 2-week repetitive hypoxic preconditioning (RHP) protocol. Separate cohorts were studied for the protective effects of RHP on postischemic and cytokine-induced cerebrovascular inflammation, and for potential deleterious effects of the RHP stimulus itself.
RESULTS
RHP protection against transient focal stroke persisted for 8 weeks. Leukocyte adherence to cortical venules was attenuated in response to stroke, as well as following TNF-α administration, indicating that reductions in postischemic inflammation were not secondary to smaller infarct volumes. RHP reduced post-stroke leukocyte diapedesis concomitant with a long-lasting downregulation of endothelial adhesion molecule mRNAs, and also reduced postischemic blood-brain barrier permeability to endogenous IgG. RHP was without effect on hippocampal CA1 pyramidal cell viability, only transiently elevated hematocrit, and did not affect the magnitude of CBF during and after ischemia.
INTERPRETATION
Taken together, our findings reveal a novel form of epigenetic neurovascular plasticity characterized by a prominent anti-inflammatory phenotype that provides protection against stroke many weeks longer than previously established windows of preconditioning-induced tolerance. Translating these endogenous protective mechanisms into therapeutics could afford sustained periods of cerebroprotection in subpopulations of individuals at identified risk for stroke.
Introduction
New or recurrent stroke is the third-leading cause of death and the leading cause of long-term adult disability in the Western world.1 Despite our growing understanding of stroke-induced injury and recovery, there remains no clinically approved treatment for stroke except time-limited thrombolysis. Many individuals at an identified risk for stroke might instead benefit from therapies that enhance the brain’s resistance to ischemic injury prior to stroke onset. One paradigm for inducing this cerebroprotective phenotype is “preconditioning”, wherein genomic and proteomic reprogramming occurring in response to a non-damaging, noxious stimulus affords transient protection from subsequent injury.2–4 While neuroprotection following preconditioning in preclinical models is robust, the time window for the ischemia-tolerant phenotype persists only a few days2,3 and thus limits clinical applicability.
Previously, we showed that exposing adult mice to a single, brief period of systemic hypoxia induced robust, but time-limited, cerebroprotection following transient stroke.5 Studies in both animals and humans, however, indicate that episodic hypoxia can induce a variety of beneficial and/or injury-reducing changes in brain,6–9 heart,10–12 and other tissues.13 We postulated that repeated exposures to systemic hypoxia might promote a novel, long-lasting ischemia-tolerant phenotype. In this report, we detail studies wherein a repetitive hypoxic preconditioning (RHP) protocol induced sustained neurovascular plasticity that extended the window of tolerance to transient focal stroke to an unprecedented two months following the completion of preconditioning.
Material and Methods
Repetitive Hypoxic Preconditioning
Washington University’s IACUC approved all experimental procedures. Swiss Webster/ND4 male mice (25–35g; 9–12 wks; Harlan) were used in all experiments except for the leukocyte diapedesis and IgG permeability studies, which used male transgenic mice14 with EGFP-expressing myelomonocytic cells (LY-EGFP; courtesy of Dr. Thomas Graf, Albert Einstein College of Medicine). Power analyses estimated sample sizes and mice were randomized into control (no hypoxia) or repetitive hypoxic preconditioning (RHP; Figure 1A) groups. RHP mice were preconditioned in modified home cages with air continuously flushed and monitored (1.5 L/min; Vascular Technologies). Sham preconditioned controls were handled in the same manner, but exposed only to room air and no protection/tolerance was induced by this treatment (data not shown). Some animals were exposed to single hypoxic preconditioning (SHP; 4 h, 8% O2). At all times during hypoxic exposure, animals had access to food and water.
Figure 1.
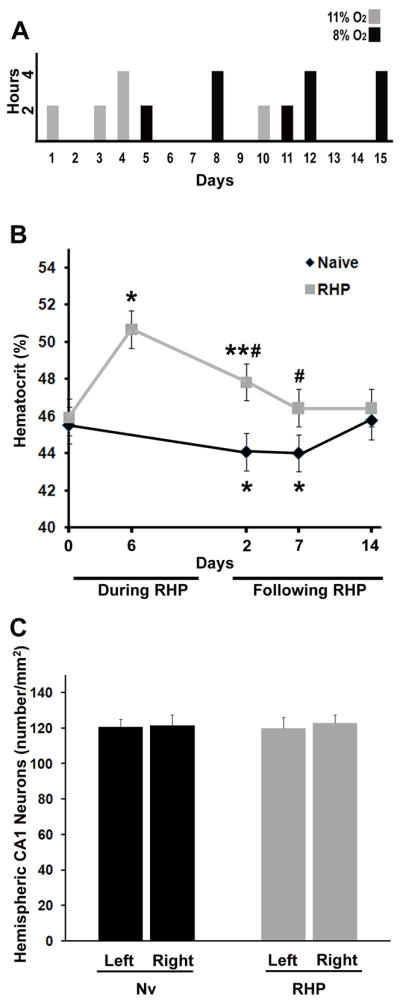
Repetitive hypoxic preconditioning (RHP) model. (A) RHP protocol; animals were exposed to 9 hypoxic exposures over ~2 wks for either 2 or 4 h (y-axis) at either 8% (black bars) or 11% O2 (grey bars). (B) RHP increased hematocrit (y-axis) above baseline (i.e. Day 0) when measured 6 days into, and 2 days following, the RHP protocol (grey line), but returned to baseline by one week (n=18); naïve (Nv) animals (n=16) without prior hypoxia (black line). *p<0.05, **p<0.01 vs. baseline; #p<0.05 vs. corresponding naïve expression at the same time point. (C) Hypoxia was without effect on hippocampal neuronal density when measured 7 days after the completion of RHP (n=5; grey bars). Measures reflect both interhemispheric comparisons (left vs. right) and comparisons to naïve animals with no prior RHP (n=5; black bars).
Transient and permanent focal cerebral ischemia
A surgeon, blinded to treatment group, anesthetized animals (5% halothane/70% NO2/30%O2) and induced a transient middle cerebral artery occlusion (tMCAo) by intraluminal suture insertion for 60 min.5,15 A >80% reduction in relative cerebral blood flow (CBF; laser Doppler flowmetry; TSI, Inc.), and a 10-min reperfusion >50% CBF baseline, were required for study inclusion. Neurological deficit was measured at 15 min and 24h of reperfusion (Table 2) and scored on a scale of 0–4, with 0 being no observable deficit, and 4 being an inability to walk spontaneously.15 One of the 41 tMCAo-treated mice was excluded due to 5% CBF baseline at 24h. For permanent middle cerebral artery occlusion (pMCAo), a distal occlusion of the MCA was performed via a craniotomy, with electrocoagulation of the surface presentation of the MCA.5,15 Of the 52 animals with pMCAo, 4 animals did not survive 24h, and 1 animal fell outside the 95% confidence interval. Animals were sacrificed 24h following tMCAo/pMCAo and brains were exposed to 2,3,5-triphenyl tetrazolium chloride (TTC) to delineate infarct regions.15,16 A blinded observer quantified infarct volumes, which were corrected for edema based on contralateral hemispheres.5,15 Hemispheric swelling was calculated using total tissue volume of the ipsilateral and contralateral hemispheres.17
Table 2.
Cerebral blood flow and neurological deficits following tMCAo in mice receiving repetitive hypoxic preconditioning (RHP) 2 days to 8 wks earlier
| Analysis | (n) | CBF ischemia* | CBF reperfusion | CBF 24h | NDS 15 min | NDS 24h |
|---|---|---|---|---|---|---|
| Control tMCAo | ||||||
| (9) | 7 ± 1 | 97 ± 6 | 89 ± 10 | 1.7 ± 0.4 | 1.9 ± 0.5 | |
| RHP + 2 day | ||||||
| (8) | 5 ± 0 | 96 ± 4 | 102 ± 3 | 1.8 ± 0.5 | 1.4 ± 0.6 | |
| RHP + 2 wks | ||||||
| (10) | 5 ± 0 | 104 ± 4 | 102 ± 3 | 1.7 ± 0.3 | 1.2 ± 0.3 | |
| RHP + 4 wks | ||||||
| (7) | 6 ± 1 | 105 ± 5 | 106 ± 4 | 1.8 ± 0.5 | 1.6 ± 0.6 | |
| RHP + 8 wks | ||||||
| (6) | 5 ± 1 | 113 ± 5† | 107 ± 4 | 1.8 ± 0.3 | 1.6 ± 0.4 | |
Values are mean (SEM). CBF values are shown as a percent of baseline.; CBF, cerebral blood flow; NDS, Neurological deficit score; tMCAo, transient middle cerebral artery occlusion; RHP, repetitive hypoxic preconditioning;
all groups p<0.05 vs. baseline;
p<0.05 vs. baseline.
Quantification of leukocyte dynamics and BBB integrity
Postischemic leukocyte-endothelial adherence in cortical venules was quantified by epifluorescence videomicroscopy through cranial windows placed over the MCA territory.15 Lymphocytes and neutrophils were fluorescently labeled (0.007% rhodamine6G in 0.01M PBS, intra-arterial) and adherence to the venular endothelium quantified either 24h following 60-min tMCAo in the ipsilesional cortex, or 4h after TNF-α (1.5μg/kg i.p.18; R&D Systems). One animal each from the control (no RHP), 2-wk, and 4-wk groups exhibited values outside of the 95% confidence interval and were excluded from final analysis. In a separate cohort of LY-EGFP mice, brains were removed 24h following tMCAo, flash-frozen, and cryosectioned (10-μm) with coronal sections obtained from the anterior, middle, and posterior portions of MCA territory for stereologic quantification of leukocyte diapedesis in both hemispheres. IgG extravasation into brain parenchyma was quantified in adjacent sections19 (Alexa Fluor 568 goat anti-mouse IgG, 6.7μg/mL; ToPro3; 1:300; Invitrogen), with gain and offset minimized during data acquisition (Olympus Fluoview FV1000 confocal). To determine endothelial mRNA expression, microvessels (including capillaries and small arterioles and venules) were isolated from flash-frozen neocortex via differential centrifugation in sucrose buffer from ipsilesional hemispheres.20 A microvessel-enriched pellet was pooled from two mice, and mRNA expression was normalized against ribosomal 18S.
Hematocrit and hippocampal CA1 cell density determinations
Hematocrit (Hct) was measured in RHP-treated and control cohorts at the start, during, and following the completion of RHP. A 75-μl blood sample was obtained from the retro-orbital sinus and animals were monitored until ambulatory. Viability of CA1 pyramidal neurons was assessed in another cohort 7 days after the completion of RHP. Coronal sections (16-μm) were obtained beginning 1 mm posterior to Bregma, and Nissl-stained prior to post-fixation. A blinded observer manually quantified total viable pyramidal cells in 3 non-adjacent sections over a 0.1-mm length of the CA1 sector of the dorsal hippocampus.21
Statistical analyses
Data are presented as mean ± standard error of the mean (SEM). Statistical comparisons were evaluated using one-way ANOVA for physiologic parameters (blood pH, PaCO2, PaO2, MABP, and blood glucose), infarct volumes, leukocyte adherence, and leukocyte extravasation. Mann-Whitney rank sum test was used for CA1 neuronal density and neurologic deficit scores, and repeated measures ANOVA for serial measures of hematocrit and body weight. Significance was determined as p<0.05.
Results
Establishing a safe and efficacious RHP protocol
We previously demonstrated that a single hypoxic stimulus established ischemic tolerance in both the mouse brain5 and retina22 for 1–3 days. We later found that, in the retina, repeating the hypoxic stimulus six times over a 2-wk period extended the window of retinal ischemic tolerance to 4 wks.23 However, this same RHP stimulus was not sufficient to reduce cortical injury caused by tMCAo (data not shown), which we interpreted as a reflection of the need for tissue-dependent titration of the preconditioning stimulus. Indeed, a smaller dose of lipopolysaccharide can precondition the retina24 from ischemic injury relative to that used for stroke preconditioning25. In addition, neuronal plasticity is enhanced by irregularly presented stimuli of varying magnitude, which serves as the molecular basis for learning, memory and drug addiction/withdrawal.26 Based on these findings, we manipulated the frequency, duration, and intensity of the hypoxic stimulus train (Figure 1A) until we achieved a new RHP protocol that induced a long-lasting cerebral tolerance to focal stroke.
We then examined whether the RHP stimulus elicited changes in hematocrit (Hct)27 that might, in turn, affect oxygen delivery and other microcirculatory hemodynamics (Fig. 1B). RHP promoted a transient, stimulatory effect on erythropoiesis. Six days into the RHP protocol, Hct was increased by 11% (p<0.05), and remained above baseline (a 5% increase; p<0.01) 2 days after the last RHP stimulus. Within one week, however, Hct had returned to within baseline levels. Animals in the control group actually exhibited decreased Hct values over this same time (p<0.01), but we interpret the 1.5% decrease as physiologically insignificant.
Given that longer exposures (> 5 h) to a magnitude of hypoxia (8% O2) utilized in some of our RHP exposures can result in significant hippocampal neuronal death,27 we also sought to determine whether the durations and intensities of the repeated hypoxia exposures of RHP affected the viability of CA1 pyramidal cells, known to be the most vulnerable cerebral cell population to global hypoxia-ischemia. When compared to matched naïve animals, however, no significant differences in CA1 density were noted in RHP-treated mice, and no significant inter-hemispheric differences in CA1 density were found in either group at 7 days following the completion of RHP (Fig. 1C).
RHP leads to a long-lasting cerebroprotective phenotype
When mice were exposed to our RHP stimulus, and then subjected to tMCAo 2 days through 8 wks later, infarct volumes were significantly reduced at all times (Fig. 2A–C). Although the most robust reduction occurred when tMCAo was induced 2 wks after RHP (a 66% infarct-sparing effect; p<0.0001), in mice wherein RHP ended 8 wks prior to tMCAo there remained a 48% reduction (p<0.05) in infarct volume. RHP-treated animals also exhibited reduced hemispheric swelling ipsilateral to the stroke. In the representative TTC-stained coronal sections, note that the region of infarct remaining in all animals with prior RHP was localized to subcortical structures and the pattern of TTC staining was more heterogeneous, with a less delineated border, relative to that in nonpreconditioned controls. We cannot rule out the possibility that the long-lasting ischemic tolerance was indirectly the result of a RHP-induced delay in the rate of infarct maturation. While unlikely, future studies will be needed to address this caveat more closely.
Figure 2.

Repetitive hypoxic preconditioning (RHP) promotes long-lasting neurovascular protection. (A and B) RHP reduced (A) infarct volume, and (B) hemispheric swelling following tMCAo in animals with RHP completed 2 days though 8 wks prior to tMCAo (n=6–10/group) versus control animals with tMCAo but no prior RHP (n=9). Individual values (black-filled circles), mean (black horizontal bars), and values (open circles) that fell outside of the 95% confidence interval (gray boxes) are given. (C) Representative TTC-stained coronal sections from groups shown in (A) of the infarct volume closest to the mean. (D) RHP 2 wks prior to pMCAo reduced infarct volume (n=10) compared to control animals with pMCAo but no prior RHP (n=16). SHP was protective only when pMCAo followed SHP at1 day (n=13), but not 2 wks (n=9). (E) Corresponding hemispheric swelling in mice subjected to pMCAo. *p<0.05, **p<0.01, ***p<0.001 vs. Control.
In our previous study, a single hypoxic stimulus did not impart ischemic tolerance against pMCAo,5 although another lab, using a more robust hypoxic stimulus, was successful in this regard.28 In the present investigation, an increased magnitude and duration of hypoxia was also effective, promoting a 32% reduction in infarct volume when pMCAo followed 1 day after the modified SHP stimulus (p<0.05; Fig. 2D,E), but not when pMCAo was induced 2 wks after SHP. As with tMCAo, RHP afforded sustained protection against pMCAo-induced injury, with reductions in infarct volume (p<0.05) and infarct-associated hemispheric swelling in mice subjected to pMCAo 2 wks after RHP.
Representative physiologic data (Table 1) showed no consistent between-group differences following transient focal stroke or TNF-α in RHP-treated animals relative to shams or controls, suggesting that the neurovascular protection does not manifest secondary to changes in these physiologic parameters. Moreover, the lack of consistent between-group differences for the magnitude of CBF reduction, early (10 min) or late (24h) in reperfusion, as well as for the early postischemic neurologic deficit scores, strongly suggest that a less severe ischemic insult did not occur in RHP-treated mice (Table 2). The lack of statistical significance for improved neurologic deficit scores in the RHP-treated mice likely reflects the relatively modest degree of sensitivity in the semi-quantitative neurological deficit scoring system we used relative to other, more extensive neuro-behavioral scoring systems.29,30
Table 1.
Physiologic parameters of repetitive hypoxic preconditioning (RHP)
| (n) | pH | PaCO2(mm Hg) | PaO2(mmHg) | MABP(mmHg) | Glucose(mg/dL) | |
|---|---|---|---|---|---|---|
| Sham Surgery | ||||||
| (7) | 7.27 ± 0.02 | 35 ± 2 | 1 05 ± 9 | 57 ± 2 | 352 ± 47 | |
| Control transient MCAo | ||||||
| (5) | 7.25 ± 0.01* | 45 ± 2 | 78 ± 9 | 65 ± 11 | 244 ± 39 | |
| RHP + transient MCAo | ||||||
| 2 days | (6) | 7.23 ± 0.03 | 45 ± 3 | 90 ± 5 | 82 ± 10 | 300 ± 46 |
| 2 wks | (6) | 7.32 ± 0.02 | 36 ± 3 | 102 ± 4 | 64 ± 5 | 259 ± 26 |
| 4 wks | (5) | 7.13 ± 0.08† | 36 ± 9 | 110 ± 14† | 70 ± 19 | 259 ± 26 |
| Control TNFα | ||||||
| (9) | 7.34 ± 0.02* | 29 ± 2 | 90 ± 4 | 61 ± 2 | 273 ± 29 | |
| RHP + TNFα | ||||||
| 2 days | (5) | 7.3 ± 0.04 | 33 ± 4 | 105 ± 10 | 62 ± 6 | 322 ± 31 |
| 2 wks | (6) | 7.30 ± 0.02 | 32 ± 2 | 86 ± 9 | 56 ± 2 | 224 ± 33 |
| 4 wks | (4) | 7.26 ± 0.04† | 35 ± 7 | 91 ± 5 | 75 ± 10*† | 295 ± 55 |
| Control permanent MCAo | ||||||
| (9) | 7.28 ± 0.02 | 43 ± 6* | 105 ± 5 | 79 ± 2* | 158 ± 19* | |
| SHP + permanent MCAo | ||||||
| 1 day | (9) | 7.32 ± 0.02 | 38 ± 1 | 100 ± 6 | 80 ± 4* | 133 ± 25* |
| 2 wks | (5) | 7.29 ± 0.04 | 47 ± 6* | 100 ± 11 | 72 ± 9 | 173 ± 17* |
| RHP + permanent MCAo | ||||||
| 2 wks | (6) | 7.35 ± 0.02*† | 44 ± 2* | 100 ± 6 | 75 ± 5* | 217 ± 30* |
All values (mean ± SEM) were measured 24h after MCAo or 4h after TNF-α; PaCO2, arterial PCO2; PaO2, arterial PO2; MABP, mean arterial blood pressure; tMCAo, transient middle cerebral artery occlusion; RHP, repetitive hypoxic preconditioning; SHP, single hypoxic preconditioning.
p<0.05 vs. sham;
p<0.05 vs. control
RHP reduces vascular inflammation following tMCAo or systemic TNFα
Post-stroke leukocyte recruitment requires the endothelial upregulation of selectins and integrins,31 which is attenuated in preconditioned animals.32,33 To determine if RHP reduces endothelial selectin and integrin expression, we quantified message expression in cortical microvessels isolated from 24-h infarcted hemispheres (Fig. 3A). P-selectin mRNA levels were increased 121-fold over naïve (p<0.01), while E-selectin mRNA increased 144-fold (p<0.05) in response to tMCAo. Prior RHP reduced the post-ischemic upregulation of both selectins by as much as 88%. VCAM-1 mRNA exhibited minimal post-ischemic upregulation in control animals, possibly due to the high basal expression levels for VCAM-1 message in cortical microvessels.31 Nevertheless, VCAM-1 expression was downregulated at both 2 (p<0.05 vs. control tMCAo) and 4 wks following RHP. In addition to E-selectin, P-selectin, and VCAM-1, we analyzed the post-stroke microvessel mRNA expression of ICAM-1 and several chemokines/chemokine receptors implicated in leukocyte recruitment (e.g., CCL2, CCR2, CXCL2, CXCR2, CXCL12, CXCR4). A nonsignificant (p=0.067) trend for increased ICAM-1 mRNA expression was noted in control tMCAo mice relative to RHP-treated mice (data not shown). For the aforementioned chemokines, no consistent, statistically significant differences in the postischemic expression pattern were evidenced in untreated vs. RHP-treated animals (data not shown).
Figure 3.

Repetitive hypoxic preconditioning (RHP) reduces postischemic leukocyte adherence. (A) In homogenates of isolated capillary endothelium, transient middle cerebral artery occlusion (tMCAo) increased E-selectin and P-selectin mRNA expression (n=11) relative to shams (n=8), but in RHP-treated mice, the extent of this postischemic upregulation was reduced. In RHP-treated mice, postischemic VCAM-1 mRNA expression was significantly downregulated. (B and D) Leukocyte adherence to cortical venular endothelium increased (B) following tMCAo (n=5), or (D) after TNF-α i.p. (n=9), relative to adherence in in sham-operated mice (n=7). RHP 2 wks (n=8) or 4 wks (n=4) prior significantly reduced both ischemia-induced and TNF-induced adherence, an effect also shown in representative video photomicrographs of cortical venules following (C) tMCAo or (E) TNF-α (2 wks, n=6; 4 wks, n=5). Scale bar = 100μm. *p<0.05, **p<0.01, ***p<0.001 vs. naïve or sham, or for 2 wks vs. 4 wks following RHP; †p<0.05, ††p<0.01, †††p<0.001 vs. control tMCAo or control TNF-α.
To confirm that postischemic reductions in adhesion molecule mRNA expression similarly affected the leukocyte recruitment phenotype, we measured leukocyte adherence in cortical venules following tMCAo. We found a 64-fold increase (p<0.0001) in leukocyte adherence to surface venular endothelium within the MCA territory in ischemic mice without RHP. The extent of adherence was reduced by 68% (p<0.0001) in mice with RHP completed 2 wks prior to stroke, and by 32% (p<0.05) in mice with RHP completed 4 wks prior to stroke (Figs. 3B,C). To verify that this anti-inflammatory phenotype was not simply secondary to a smaller volume of infarcted subcortical tissue, a second series of experiments evaluated leukocyte-endothelial adherence caused by systemic administration of TNF-α. TNF-α promotes leukocyte adherence to cortical venules,18,34 and in nonpreconditioned mice, induced a 52-fold increase in leukocyte-endothelial adherence (p<0.0001). This response was reduced by 88% in mice with RHP completed 2 wks prior to TNF-α (p<0.0001), to levels not statistically distinguishable from sham controls (Fig. 3D,E). In mice with RHP completed 4 wks prior to TNF-α, adherence was reduced by 51% (p<0.01). These findings confirm that RHP promotes a sustained and generalized anti-inflammatory phenotype at the blood-cerebral endothelial interface.
RHP reduces leukocyte diapedesis and preserves BBB integrity
We also determined whether the attenuation of postischemic leukocyte adherence in RHP-treated mice was concomitantly associated with a reduction in leukocyte infiltration into brain parenchyma.35 Relative to the contralateral, non-ischemic hemisphere, leukocyte diapedesis was elevated 26-fold 24h after tMCAo (Fig. 4A). RHP 2 wks or 4 wks prior to stroke reduced leukocyte diapedesis by 44% (p<0.05) or 75% (p<0.001), respectively. Leukocyte diapedesis is one factor associated with BBB disruption following stroke.15 Thus, we also quantified the endogenous extravasation of IgG, a highly sensitive marker for loss of postischemic BBB integrity in areas of infarction.36 In nonpreconditioned mice, BBB permeability to IgG increased more than 2-fold (p<0.01) in the ischemic hemisphere (Fig. 4B). In contrast, in mice with RHP completed 2 wks or 4 wks prior to stroke, ipsilateral IgG extravasation was robustly reduced in both instances (p<0.0001), to levels indistinguishable from those in the nonischemic, contralateral hemisphere.
Figure 4.

Repetitive hypoxic preconditioning (RHP) maintains postischemic barrier integrity in LY-EGFP mice. (A,D) Leukocyte diapedesis is increased following transient middle cerebral artery occlusion (tMCAo) in ipsilateral (solid bar) vs. contralateral (hatched bar) hemispheres (n=6; fluorescent leukocytes shown in lower panels by arrowhead from the representative area denoted by a black box in C). RHP, 2 and 4 wks prior to tMCAo, significantly reduced leukocyte diapedesis (n=4,5). (B,E,F) Increased ipsilateral IgG extravasation in controls (solid black) is completely abrogated in animals with RHP. Representative extravasated immunoglobulin G (IgG; red), nuclei (counterstained with ToPro3; blue), and diapedesed leukocytes (arrowheads) shown in (E) the ipsilesional hemisphere from area denoted by a black box in C, and (F) the corresponding contralesional hemisphere from area denoted by the grey square in C. (C) Adjacent cresyl violet stained sections, with an increased magnification (from the black box) of one of the five areas used in quantification. *p<0.05, **p<0.01, ***p<0.001 vs. contralateral nonischemic hemisphere; †p<0.05, ††p<0.01, †††p<0.001 vs. nonpreconditioned group. Scale bar = 50 μm.
Discussion
Although windows of ischemic tolerance lasting to one week have occasionally been reported,6,37 cerebral protection induced by a single preconditioning stimulus typically fades within a few days. Chronic hypoxia can also lead to a host of “acclimatization” responses, but these adaptations also wane shortly after returning to a normoxic environment.38 We postulated that an intermediate approach, using a patterned series of intermittently presented hypoxic challenges, might induce a period of tolerance that was sustained well after the treatment series was completed. We were encouraged by the findings that repetitive hypoxia, interspersed with periods of normoxia, elicits beneficial effects in the CNS and other tissues under baseline conditions8,10 and following ischemia,6,9,11,12 and induces novel long-term facilitation in respiratory motoneurons.39 Moreover, repetitive stimuli are known to trigger persistent neuronal plasticity,26 and both cerebrovascular and neuronal plasticity are induced following intermittent interventions such as exercise.40 We found that the stochastic, intermittent hypoxic exposures that define our RHP stimulus were quite efficacious in increasing cellular ischemic resistance in a uniquely protracted fashion, and, as such, reflect a fundamentally new type of extended neurovascular plasticity.
Titration is essential to the effectiveness of all preconditioning stimuli. The stimulus must be ‘strong’ enough to elicit an adaptive response, but not so strong as to cause injury itself or worsen ischemic outcomes.2,3,27 Moreover, given that all stimuli are defined by their frequency, magnitude, and duration, achieving the optimal titration of a repetitive stimulus is not necessarily straightforward in either concept or practice. For example, a much more severe intermittent hypoxic challenge (15 h/day of ~10% O2 for 4 wks) than that defining our RHP stimulus failed to protect when administered 3 wks prior to transient focal stroke in rats.6 Moreover, accumulating evidence shows that high frequency intermittent hypoxia – as occurs with sleep apnea – increases the risk for stroke.41,42 With respect to ensuring that the magnitude and frequency of our RHP stimulus was without adverse effects, we documented normal CA1 pyramidal cell viability, indicating an intermediate hypoxic stimulus that protected without harm. The mild, transitory increase in hematocrit during RHP, also observed by others in response to intermittent hypoxia,43 does not bear on our results, either positively or negatively, since protection persists 4 and 8 wks after RHP, well after the time when hematocrit values returned to baseline.
Often overlooked in studies of standard preconditioning-induced tolerance is the notion that the robust protection is likely to also include survival-promoting mechanisms acting directly in astrocytes, microglia, oligodendroglia, and endothelial cells. Reductions in leukocyte-endothelial adherence, diapedesis, and IgG extravasation specifically reflect anti-inflammatory events occurring directly at the level of the BBB that may also contribute to the reductions in infarct volume.. TNFα-induced inflammation in the nonischemic brain was also attenuated in RHP-treated mice indicating that, with respect to the early stages of postischemic leukocyte recruitment, a generalized anti-inflammatory phenotype is established at the blood-cerebral endothelial cell interface. This cerebrovascular protection is mediated by reductions in endothelial adhesion molecule gene transcription, although diminished leukocyte-derived elastases and matrix metalloproteinases,15,44 as well as preservation of tight junctions,45 may also participate. Mechanisms underlying the loss of BBB integrity following stroke include both leukocyte-independent and leukocyte-dependent injury of endothelium and basal lamina, as well as extracellular matrix disruption.31,46,47 Our data do not address the extent to which the documented reductions in postischemic leukocyte-endothelial adherence and diapedesis causally result in a more intact BBB. We can only conclude the RHP phenotype includes a significant improvement in postischemic BBB integrity concomitant with reduced vascular inflammation.
The multi-factorial mechanistic basis underlying the neurovascular plasticity that affords long-lasting protection documented in our study will require further investigation to elucidate. The sustained stabilization of transcription factors (e.g., DeltaFosB in response to repeated stress)48 may contribute to the evolution of tolerance during the weeks following preconditioning. Whether protection is hypoxia-inducible factor (HIF)-dependent, especially considering the unique mechanisms that regulate HIF activity under conditions of single vs. intermittent vs. chronic hypoxia,38,49–51 should be addressed. The finding that stabilizing HIF transcription by repeated administration of a prolyl hydroxylase inhibitor enhanced hippocampal memory one month after treatment was completed52 and may reflect a similar HIF-driven plasticity.
In summary, we have demonstrated that a relatively short period of repetitive hypoxia, varying in both intensity and duration, extends the ‘window’ of the ischemia-tolerant phenotype from several days to many weeks. These findings reflect a novel form of induced neurovascular plasticity that enhances the resistance of the neurovascular unit to both nonperfused and reperfused ischemic brain injury in a uniquely protracted fashion. Intermittent bouts of systemic hypoxia – similar in both intensity and duration to that comprising our RHP protocol – provide a host of cytoprotective effects in animal models of cerebral and myocardial ischemia6,8–12 and are well-tolerated by humans, improving both endurance and exercise efficiency.10,13 Ultimately, however, repeated stimulation by nonhypoxic, pharmacological stimuli may be chosen for inducing survival-promoting plasticity in the human CNS,53 in line with our recent demonstration that a repetitive, 2-wk dosing regimen of the FDA-approved drug, deferroxamine, protects the retina from ischemic injury for one month.54 This sustained manipulation of endogenous mechanisms of protection through repeated exposures to preconditioning stimuli may also confer long-term tolerance to many other acute and chronic neurodegenerative and autoimmune CNS diseases in identified, at-risk populations.
Acknowledgments
The authors thank T.S. Park for his early support of this line of investigation. Invaluable technical contributions were made by Ernesto R. Gonzales, Ronald S. Perez, Aarti R. Shah, Jennifer Perfater, Yolanda M. Rangel, and Ivo A.C.W. Tiebosch. We recognize NIH RO1 HL79278 (JMG), NIH P01 NS32636 (JMG), NIH P30 NS057105 (Alafi Neuroimaging Core, Washington University), American Heart Association (AMS), and Spastic Paralysis Research Foundation of the Illinois-Eastern Iowa District of Kiwanis International for generously funding this research. Dr. Stowe was also supported by The Hope Center for Neurological Disorders, Washington University School of Medicine.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 4.Yenari M, Kitagawa K, Lyden P, Perez-Pinzon M. Metabolic downregulation: a key to successful neuroprotection? Stroke. 2008;39:2910–2917. doi: 10.1161/STROKEAHA.108.514471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller BA, Perez RS, Shah AR, et al. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport. 2001;12:1663–1669. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- 6.Lin AM, Dung SW, Chen CF, et al. Hypoxic preconditioning prevents cortical infarction by transient focal ischemia-reperfusion. Ann N Y Acad Sci. 2003;993:168–178. doi: 10.1111/j.1749-6632.2003.tb07527.x. [DOI] [PubMed] [Google Scholar]
- 7.Rybnikova E, Mironova V, Pivina S, et al. Antidepressant-like effects of mild hypoxia preconditioning in the learned helplessness model in rats. Neurosci Lett. 2007;417:234–239. doi: 10.1016/j.neulet.2007.02.048. [DOI] [PubMed] [Google Scholar]
- 8.Zhu XH, Yan HC, Zhang J, et al. Intermittent hypoxia promotes hippocampal neurogenesis and produces antidepressant-like effects in adult rats. J Neurosci. 2010;30:12653–12663. doi: 10.1523/JNEUROSCI.6414-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rybnikova E, Vataeva L, Tyulkova E, et al. Mild hypoxia preconditioning prevents impairment of passive avoidance learning and suppression of brain NGFI-A expression induced by severe hypoxia. Behav Brain Res. 2005;160:107–114. doi: 10.1016/j.bbr.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 10.Katayama K, Matsuo H, Ishida K, et al. Intermittent hypoxia improves endurance performance and submaximal exercise efficiency. High Alt Med Biol. 2003;4:291–304. doi: 10.1089/152702903769192250. [DOI] [PubMed] [Google Scholar]
- 11.Fitzpatrick CM, Shi Y, Hutchins WC, et al. Cardioprotection in chronically hypoxic rabbits persists on exposure to normoxia: role of NOS and KATP channels. Am J Physiol Heart Circ Physiol. 2005;288:H62–68. doi: 10.1152/ajpheart.00701.2004. [DOI] [PubMed] [Google Scholar]
- 12.Zhu WZ, Xie Y, Chen L, et al. Intermittent high altitude hypoxia inhibits opening of mitochondrial permeability transition pores against reperfusion injury. J Mol Cell Cardiol. 2006;40:96–106. doi: 10.1016/j.yjmcc.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 13.Wiesner S, Haufe S, Engeli S, et al. Influences of normobaric hypoxia training on physical fitness and metabolic risk markers in overweight to obese subjects. Obesity (Silver Spring) 2010;18:116–120. doi: 10.1038/oby.2009.193. [DOI] [PubMed] [Google Scholar]
- 14.Faust N, Varas F, Kelly LM, et al. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- 15.Stowe AM, Adair-Kirk TL, Gonzales ER, et al. Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis. 2009;35:82–90. doi: 10.1016/j.nbd.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swanson RA, Morton MT, Tsao-Wu G, et al. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 17.Gerriets T, Stolz E, Walberer M, et al. Noninvasive quantification of brain edema and the space-occupying effect in rat stroke models using magnetic resonance imaging. Stroke. 2004;35:566–571. doi: 10.1161/01.STR.0000113692.38574.57. [DOI] [PubMed] [Google Scholar]
- 18.Altay T, McLaughlin B, Wu JY, et al. Slit modulates cerebrovascular inflammation and mediates neuroprotection against global cerebral ischemia. Exp Neurol. 2007;207:186–194. doi: 10.1016/j.expneurol.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo EH, Pan Y, Matsumoto K, Kowall NW. Blood-brain barrier disruption in experimental focal ischemia: comparison between in vivo MRI and immunocytochemistry. Magn Reson Imaging. 1994;12:403–411. doi: 10.1016/0730-725x(94)92533-x. [DOI] [PubMed] [Google Scholar]
- 20.Gerhart DZ, Broderius MA, Drewes LR. Cultured human and canine endothelial cells from brain microvessels. Brain Res Bull. 1988;21:785–793. doi: 10.1016/0361-9230(88)90047-0. [DOI] [PubMed] [Google Scholar]
- 21.Altay T, Gonzales ER, Park TS, Gidday JM. Cerebrovascular inflammation after brief episodic hypoxia: modulation by neuronal and endothelial nitric oxide synthase. J Appl Physiol. 2004;96:1223–1230. doi: 10.1152/japplphysiol.00798.2003. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Y, Ohlemiller KK, McMahan BK, Gidday JM. Mouse models of retinal ischemic tolerance. Invest Ophthalmol Vis Sci. 2002;43:1903–1911. [PubMed] [Google Scholar]
- 23.Zhu Y, Zhang Y, Ojwang BA, et al. Long-term tolerance to retinal ischemia by repetitive hypoxic preconditioning: role of HIF-1alpha and heme oxygenase-1. Invest Ophthalmol Vis Sci. 2007;48:1735–1743. doi: 10.1167/iovs.06-1037. [DOI] [PubMed] [Google Scholar]
- 24.Franco PJ, Fernandez DC, Sande PH, et al. Effect of bacterial lipopolysaccharide on ischemic damage in the rat retina. Invest Ophthalmol Vis Sci. 2008;49:4604–4612. doi: 10.1167/iovs.08-2054. [DOI] [PubMed] [Google Scholar]
- 25.Marsh B, Stevens SL, Packard AE, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- 27.Prass K, Scharff A, Ruscher K, et al. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke. 2003;34:1981–1986. doi: 10.1161/01.STR.0000080381.76409.B2. [DOI] [PubMed] [Google Scholar]
- 28.Bernaudin M, Nedelec AS, Divoux D, et al. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab. 2002;22:393–403. doi: 10.1097/00004647-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Ashioti M, Beech JS, Lowe AS, et al. Multi-modal characterisation of the neocortical clip model of focal cerebral ischaemia by MRI, behaviour and immunohistochemistry. Brain Res. 2007;1145:177–189. doi: 10.1016/j.brainres.2007.01.111. [DOI] [PubMed] [Google Scholar]
- 30.Hunter AJ, Hatcher J, Virley D, et al. Functional assessments in mice and rats after focal stroke. Neuropharmacology. 2000;39:806–816. doi: 10.1016/s0028-3908(99)00262-2. [DOI] [PubMed] [Google Scholar]
- 31.Gavins F, Yilmaz G, Granger DN. The evolving paradigm for blood cell-endothelial cell interactions in the cerebral microcirculation. Microcirculation. 2007;14:667–681. doi: 10.1080/10739680701404903. [DOI] [PubMed] [Google Scholar]
- 32.Bowen KK, Naylor M, Vemuganti R. Prevention of inflammation is a mechanism of preconditioning-induced neuroprotection against focal cerebral ischemia. Neurochem Int. 2006;49:127–135. doi: 10.1016/j.neuint.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Hoyte LC, Brooks KJ, Nagel S, et al. Molecular magnetic resonance imaging of acute vascular cell adhesion molecule-1 expression in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010 doi: 10.1038/jcbfm.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carvalho-Tavares J, Hickey MJ, Hutchison J, et al. A role for platelets and endothelial selectins in tumor necrosis factor-alpha-induced leukocyte recruitment in the brain microvasculature. Circ Res. 2000;87:1141–1148. doi: 10.1161/01.res.87.12.1141. [DOI] [PubMed] [Google Scholar]
- 35.Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 36.Chen B, Friedman B, Cheng Q, et al. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke. 2009;40:e666–674. doi: 10.1161/STROKEAHA.109.551341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barone FC, White RF, Spera PA, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke. 1998;29:1937–1950. doi: 10.1161/01.str.29.9.1937. [DOI] [PubMed] [Google Scholar]
- 38.Chavez JC, Agani F, Pichiule P, LaManna JC. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol. 2000;89:1937–1942. doi: 10.1152/jappl.2000.89.5.1937. [DOI] [PubMed] [Google Scholar]
- 39.Wilkerson JE, Mitchell GS. Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp Neurol. 2009;217:116–123. doi: 10.1016/j.expneurol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swain RA, Harris AB, Wiener EC, et al. Prolonged exercise induces angiogenesis and increases cerebral blood volume in primary motor cortex of the rat. Neuroscience. 2003;117:1037–1046. doi: 10.1016/s0306-4522(02)00664-4. [DOI] [PubMed] [Google Scholar]
- 41.Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med. 2010;182:269–277. doi: 10.1164/rccm.200911-1746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353:2034–2041. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 43.Siques P, Brito J, Leon-Velarde F, et al. Time course of cardiovascular and hematological responses in rats exposed to chronic intermittent hypobaric hypoxia (4600 m) High Alt Med Biol. 2006;7:72–80. doi: 10.1089/ham.2006.7.72. [DOI] [PubMed] [Google Scholar]
- 44.Gidday JM, Gasche YG, Copin JC, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 45.An P, Xue YX. Effects of preconditioning on tight junction and cell adhesion of cerebral endothelial cells. Brain Res. 2009;1272:81–88. doi: 10.1016/j.brainres.2009.03.031. [DOI] [PubMed] [Google Scholar]
- 46.Mayhan WG. Leukocyte adherence contributes to disruption of the blood-brain barrier during activation of mast cells. Brain Res. 2000;869:112–120. doi: 10.1016/s0006-8993(00)02376-3. [DOI] [PubMed] [Google Scholar]
- 47.Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. doi: 10.1016/j.surneu.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 48.McClung CA, Ulery PG, Perrotti LI, et al. DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res. 2004;132:146–154. doi: 10.1016/j.molbrainres.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 49.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 50.Prabhakar NR, Kumar GK, Nanduri J. Intermittent hypoxia-mediated plasticity of acute O2 sensing requires altered red-ox regulation by HIF-1 and HIF-2. Ann N Y Acad Sci. 2009;1177:162–168. doi: 10.1111/j.1749-6632.2009.05034.x. [DOI] [PubMed] [Google Scholar]
- 51.Koh MY, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. doi: 10.1016/j.tibs.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Adamcio B, Sperling S, Hagemeyer N, et al. Hypoxia inducible factor stabilization leads to lasting improvement of hippocampal memory in healthy mice. Behav Brain Res. 2009;208:80–84. doi: 10.1016/j.bbr.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 53.Gidday JM. Pharmacologic preconditioning: Translating the promise. Transl Stroke Res. 2010;1:29–30. doi: 10.1007/s12975-010-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu Y, Zhang L, Gidday JM. Deferroxamine preconditioning promotes long-lasting retinal ischemic tolerance. J Ocul Pharmacol Ther. 2008;24:527–535. doi: 10.1089/jop.2008.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]