

Abstract
Pharmacogenomics is yet to fulfill its promise of manifestly altering clinical medicine. As one example, a predictive test for tardive dyskinesia (an adverse drug reaction consequent to antipsychotic exposure) could greatly improve the clinical treatment of schizophrenia but human studies are equivocal. A complementary approach is the mouse-then-human design in which a valid mouse model is used to identify susceptibility loci which are subsequently tested in human samples. We used inbred mouse strains from the Mouse Phenome Project to estimate the heritability of haloperidol-induced activity and orofacial phenotypes. 159 mice from 27 inbred strains were chronically treated with haloperidol (3 mg/kg/day via subdermal slow-release pellets) and monitored for the development of vacuous chewing movements (VCMs, the mouse analog of TD) and other movement phenotypes derived from open field activity and the inclined screen test. The test battery was assessed at 0, 30, 60, 90, and 120 days in relation to haloperidol exposure. As expected, haloperidol caused marked changes in VCMs, activity in the open field, and EPS. Unexpectedly, factor analysis demonstrated that these measures were imprecise assessments of a latent construct rather than discrete constructs. The heritability of a composite phenotype was ~0.9 after incorporation of the longitudinal nature of the design. Murine VCMs are a face valid animal model of antipsychotic-induced TD and heritability estimates from this study support the feasibility of mapping of susceptibility loci for VCMs.
Keywords: Antipsychotic, haloperidol, adverse drug reaction, tardive dyskinesia, mouse model, vacuous chewing movements
INTRODUCTION
The truth of the proposition that pharmacogenomics is crucial to the future of medicine is usually assumed 1. The logic seems sound: humans vary in propensity for adverse drug reactions and efficacy, and genetic testing could deliver a cardinal feature of individualized medicine via tailoring pharmacotherapy to individual genomes. However, given limited progress with human pharmacogenomic studies, we believe it timely to explore a complementary paradigm 1, mouse-then-human designs. In the first stage, we expose mouse strains to humanlike steady-state drug concentrations under rigorous experimental conditions and carefully and reliably measured outcomes of interest. Control is achieved for genetic background as well as for extraneous environmental effects. These phenotypic data also allow estimation of heritability. As all of these mice have been genotyped using a modern GWAS chip 2, future work will entail genetic mapping as in silico analyses. In the second stage, the human orthologs of genomic regions implicated in mouse are studied in human samples to determine whether the association replicates across species. In this way, the mouse is used to screen the genomic search space in order to identify a small number of candidate regions with high prior probabilities that are then tested in humans. Most of the multiple comparison debt is paid in mouse, and precious human samples are used only for regions that are far more probable than average.
In this report, we investigate a mouse model of human tardive dyskinesia (TD). TD is a clinically important adverse drug reaction whose basis is unknown. Exposure to conventional or typical antipsychotics (prototype haloperidol) causes extrapyramidal syndromes (EPS). Acute EPS occurs in ~40% of subjects and includes akathisia, acute dystonia, and secondary parkinsonism 3. TD is a late-onset form of EPS that develops in ~35% of subjects treated chronically with typical antipsychotics 4 and may be irreversible in about half of subjects 5. TD is characterized by repetitive, involuntary, and purposeless movements primarily of the orofacial region (e.g., chewing and tongue protrusion) 6. There are no heritability estimates for TD. Despite the lack of these crucial data, multiple human candidate gene association studies have been published 7–12. This literature has not converged on strong and replicated associations.
After chronic treatment with typical antipsychotics, rodents show purposeless mouth openings in the vertical plane, or vacuous chewing movements (VCMs) 13. VCMs are a phenotypically and pharmacologically valid animal model of TD that has been used for decades by behavioral pharmacologists 13. The full argument for the validity of VCMs as a rodent model for TD is summarized in Table S1. In Figure S1, we add a new component to this argument. TD can persist in humans for years after exposure to a typical antipsychotic; chronic haloperidol exposure in mouse induces VCMs that persist for 1.5 years (a large fraction of the mouse lifespan) and well after haloperidol levels are undetectable.
The purpose of this paper is to initiate evaluation-of-concept studies whose overarching goal is to investigate whether the MTH design can be used to inform studies of human TD. We show that it is possible to deliver human-like steady state concentrations of haloperidol. As expected, VCMs develop reliably and we show that VCMs can be measured reliably. We then exposed 27 genetically diverse inbred mouse strains to standardized doses of haloperidol in order to calculate heritability and to identify optimal phenotypes for genetic association mapping.
MATERIALS AND METHODS
Ethics Statement
All testing procedures were conducted in strict compliance with the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Research Council, 1996) and approved by the Institutional Animal Care and Use Committee of the University of North Carolina.
Animals
Male mice (aged 8–10 weeks) from 27 inbred strains were obtained from the Jackson Laboratory (Bar Harbor, ME) as part of the Mouse Phenome Project. The 22 classical-derived strains were 129S1/SvImJ, A/J, AKR/J, BALB/cByJ, BTBR T<+> tf/J, C3H/HeJ, C57BL/6J, C57BLKS/J, CBA/J, DBA/2J, DDY, FVB/NJ, KK/HIJ, MA/MY, MRL/MpJ, NOD/LtJ, NON/SHILTJ, NZL, NZO, NZW/LacJ, SJL/J, and SM/J. The five wild-derived strains were CAST/EiJ, MOLF/EiJ, MSM/Ms, PWK, and WSB/EiJ.
Male mice were studied to minimize variation due to sex. Animals were maintained on a 12h light:12h dark schedule with lights on at 0700. The housing room was maintained at 20–24°C with 40–50% relative humidity. Mice were housed in standard 20 cm × 30 cm ventilated polycarbonate cages with laboratory grade Bed-O-Cob bedding. Water and Purina ProLab IsoPro 3000 were available ad libitum. A small section of PVC pipe was present in each cage for enrichment. In pilot work, we observed considerable weight loss due to dehydration in the first few days after exposure to haloperidol. This appeared to be due to acute EPS resulting in an inability to rear and to reach water and chow on the roof of the cage. Thus, for the first few days after exposure to haloperidol, food and water were positioned near the bedding surface. All mice from each strain were group-housed (maximum of five mice per cage) except that BALB/cByJ, CAST/EiJ, and SJL/J mice were separated due to fighting after 7, 10 and 13 weeks of housing.
Antipsychotic exposure
The goal was to achieve a human-like steady-state concentration of haloperidol (10–50 nanomoles/L, nM, or 3.75–19 ng/ml) 14 for a minimum of 30 days. Pilot studies showed that implantable pellets yielded considerably lower coefficients of variation in steady-state haloperidol concentrations in comparison to injections, implantable mini-pumps, or haloperidol in drinking water (Supplemental Methods, Table S2). Dose-ranging pilot studies in C57BL/6J mice indicated that 3.0 mg/kg/day (60 day release tablets) yielded plasma haloperidol concentrations in the 10–50 nM range. Haloperidol pellets (Innovative Research of America; Sarasota, FL) 15 were implanted subcutaneously with a trocar under two minutes of isoflurane anesthesia (pilot studies indicated that anesthesia minimized handling stress and pain). Pellets of incremental dosages were implanted to compensate for varying body weights. Mice were followed for 120 days in total in order to examine changes in VCMs when haloperidol concentrations waned. Blood plasma was collected via tail nick for drug concentration assays after 30, 60, 90 and 120 days of exposure to haloperidol. Haloperidol assays were performed using mass spectrometry by the Analytical Psychopharmacology Laboratory at the Nathan Kline Institute for Psychiatric Research (Orangeburg, NY).
Scoring VCMs
High-resolution digital videotapes of orofacial behavior were made by modifying the method of Tomiyama et al. 16. Each mouse was briefly anesthetized using isoflurane and placed in a restrictor device (a plastic collar that lightly restrained the mouse around the neck) attached to a horizontal platform (Figure S2A). Collars were composed of two semicircular elements: a lower trough fixed to the platform and an upper restrainer that completed light enclosure of the neck. Both the diameter of the collar and its height above the platform were adjustable according to body size in order to allow a comfortable posture to be maintained. This restrictor device allowed a high-resolution digital camcorder (JVC Everio GZ-MG360BU; Wayne, NJ) to be focused onto the orofacial region from below with minimal disturbance to facial movements. Each mouse was placed in the restrictor device for 25 minutes, and the final 15 minutes were scored for orofacial movement phenotypes: tongue protrusions, overt chewing movements, subtle chewing movements and jaw tremors. Tongue protrusions and chewing movements were measured as counts of individual events and tremors as total duration (seconds). A chewing movement was defined as a single mouth opening in the vertical plane not directed toward physical material, not occurring during a period of struggling, and not coincident with tongue protrusion. Overt chewing movements were distinguished from subtle chewing movements if the interior of the oral cavity could be seen. Each video recording was randomly assigned to one of three raters and scored using the Observer video analysis system (Noldus, The Netherlands). Crucially, raters were blinded to all study data including strain and study time point. Inter-rater reliability among the three raters was high (intra-class correlation of 0.92), as determined by individual scoring of 32 randomly-assigned and blinded video recordings. All raters were initially trained by an experienced rater in a three-step process: viewing of a training tape with clips from several different strains of mice demonstrating each movement phenotype; comparative scoring of a training set composed of 36 video clips (one haloperidol and one placebo treated mouse from 18 different strains); and scoring of training tapes continued until the new rater reached consensus criterion. To monitor drift, the same training tape was periodically and blindly scored. Orofacial observations were made on days 0, 30, 60, 90 and 120 relative to drug treatment (day 1).
Open field activity
Extrapyramidal side effects may appear as general motor deficits in mice, therefore spontaneous locomotor activity in the open field 17 was measured for 1 hour using a photocell-equipped automated open field apparatus (Versamax system, Accuscan Instruments, Columbus, OH; 40 cm wide × 40 cm long × 30 cm high). Four phenotypes were extracted from these activity data: total distance traveled (cm), vertical activity (number of beam breaks), stereotypy (repeated breaking of the same beam; number of beam breaks), and time spent in the central region of the chamber (percent of total time; central 20 cm × 20 cm). Activity chambers were inside sound-attenuating boxes equipped with houselights and fans. Open field activity was measured on days 0, 30, 60, 90 and 120 relative to drug treatment (day 1).
Extrapyramidal side effects (EPS)
The inclined screen test 18 (Figure S1B) was used as an index of Parkinsonian rigidity and sedation. Mice were placed on a wire mesh screen inclined at 45° and the latency to move all four paws was recorded (to a maximum of 300 seconds). EPS was measured on days 0, 30, 60, 90 and 120 relative to drug treatment (day 1).
Statistical Analyses
Linear mixed effects models were used to decompose phenotype variances for the calculation of heritability and inter-rater reliability as well as to assess the significance of covariate fixed effects (R 2.6.0 and Stata 9.2). Mixed models are generally considered superior to competing analytical approaches for variance decomposition of longitudinal data because they can accommodate missing observations and irregular assessment spacing, and enable modeling of random effects and within-subject residual covariance structures 19.
Heritability was calculated using intra-class correlation coefficients. First, we present heritability estimates for the raw data as a baseline to which the processed data can be compared. Second, heritability estimates for change scores were estimated (i.e., √timet – √time0). Given that many phenotypes had non-zero values prior to exposure, change scores isolate the phenotypic component attributable to haloperidol exposure. Third, a major weakness of the two prior methods of estimating heritability is inefficiency. Longitudinal analyses are appropriate to our study design and incorporate all available data to refine the heritability measures by studying over-time response trajectories induced by haloperidol for each mouse (as opposed to data from time points in isolation). The heritabilities of the over-time trajectories in haloperidol-induced movement disorder phenotypes were assessed using an extension of the mixed model for behavioral genetic analysis (see Figure S3 for idealized method illustration) 20. For each phenotype, the optimal functional form of the over-time trajectory was determined by fitting a series of mixed models in which the trajectory was specified as a linear trend, a plateau (linear change until timet, and flat thereafter), or a lag effect (no phenotype change until timet, and flat thereafter), with models fit for all possible plateau/lag points. After determining optimal trajectory functional forms by comparing model fit statistics, three level mixed models (assessments nested within individual mice nested within strains) were fit that decomposed trajectory variance into components due to mouse-specific and strain-specific differences. Trajectory heritabilities were calculated as the ratio of strain-level variance to strain-level + mouse-level variance. Fourth, there are strong intercorrelations between the nine dependent variables (Table S4). If each observed phenotype is considered as an imperfect measurement of an unobserved latent factor, it may be possible to discover unexpected connections across phenotypes and to derive improved estimates of more fundamental traits. Thus, we applied factor analysis to examine the factor structure of the mouse-specific response trajectories (i.e., the random slope coefficients) (MPlus 5.21) 21–23. Orthogonal iterative principal factor exploratory models were fit followed by an examination of various rotations. The variance of the latent trajectory factors underlying the individual response phenotype trajectories was decomposed into strain- and mouse-level components, and heritabilities of the latent trajectory factors were calculated as the ratio of strain-level variance to strain-level + mouse-level variance.
RESULTS
Mice & haloperidol delivery
Haloperidol tablets were initially implanted in 156 mice from 27 inbred strains with the goal of obtaining a human-like steady-state concentration of haloperidol (10–50 nM) for a minimum of 30 days. A pilot dose-ranging study suggested that this could be achieved using 3.0 mg/kg/day for a 60-day tablet (Figure S1). Four of five MOLF/EiJ mice died within one day of haloperidol exposure and all five mice were excluded (mortality was likely due to temperature dysregulation or dehydration due to acute EPS). Subsequently, five MOLF/EiJ mice were successfully run through the protocol when the implanted haloperidol dose was split over successive days. In addition, all four BTBR mice developed a dermatological problem, which may be related to characteristic coat abnormalities in this strain, two months after drug treatment and were sacrificed. Eight additional BTBR mice were subsequently run through the unmodified protocol successfully. Thus, the sample for this report consists of 159 mice from 27 strains. Complete data for the key dependent variables (VCMs) were available for three time-points (0, 30, and 60 days) for 157 mice (98.7%) and for five time-points (0, 30, 60, 90, and 120 days) for 148 mice (93.1%).
Table S3 summarizes the number of mice used in this experiment, attrition over time, and the numbers of mice that achieved the minimum desired haloperidol concentrations at each time-point. The numbers of mice with steady-state plasma haloperidol concentrations ≥10 nM were 156/159 at 30 days, 151/158 at 60 days, 73/149 at 90 days, and 9/148 at 120 days, consistent with the function of the 60 day release tablets. Haloperidol concentrations in the three mice under the 10 nM threshold at 30 days were 9.4, 9.6, and 9.9 nM. Figure 1 shows full data for plasma haloperidol concentrations at 30, 60, 90, and 120 days. Although there is considerable variation across strains, haloperidol levels were almost always tightly clustered within each strain-time point with the possible exception of the heaviest strain (the mean baseline weight for NZL mice was four times the lightest strain). The medians (inter-quartile ranges) of the coefficients of variation for haloperidol concentrations across strains were 0.19 (0.15–0.25) at 30 days and 0.19 (0.13–0.25) at 60 days. These data are consistent with the pilot data in Table S2 and indicate that the delivery method almost always achieved our goal of exposure to sustained human-like haloperidol steady-state concentrations.
Figure 1. Haloperidol plasma concentrations (in nM) per strain at four time points.
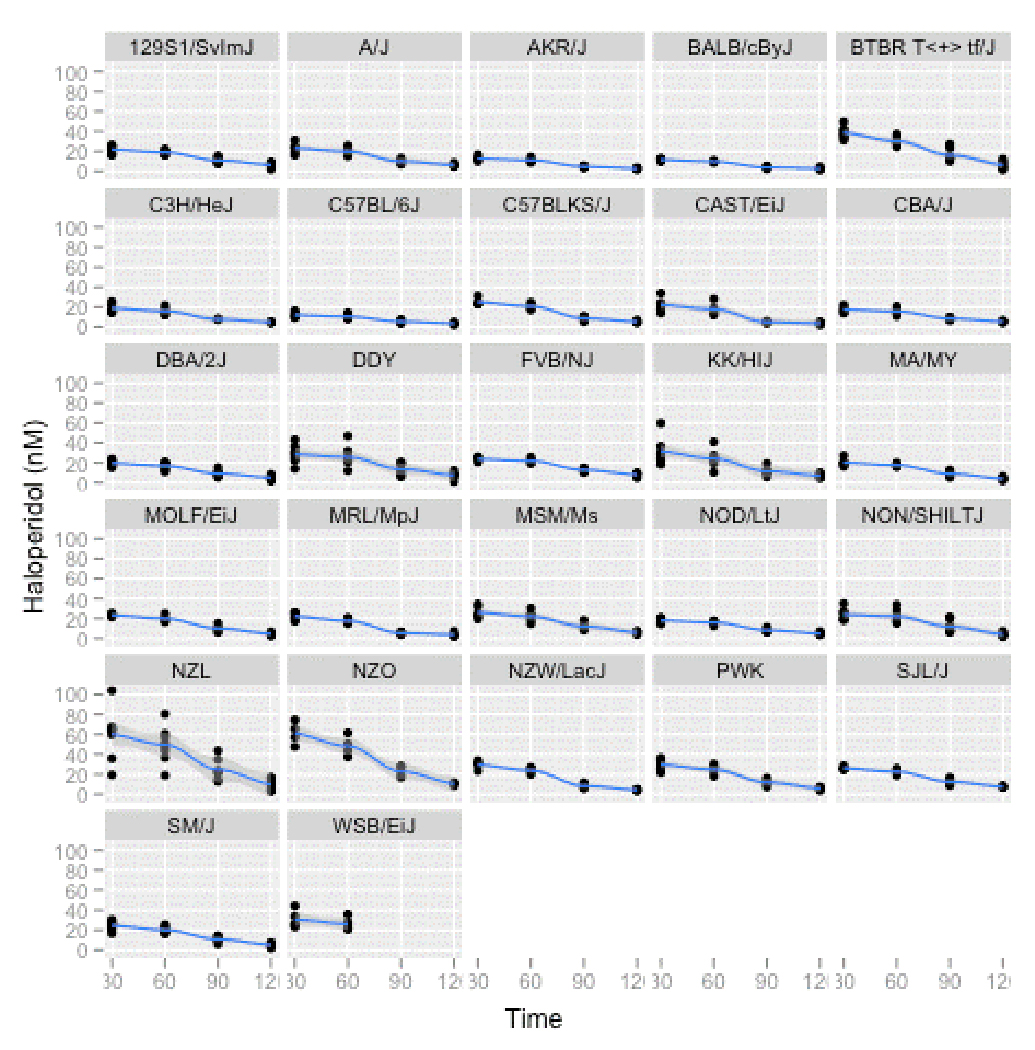
All data points are shown for all strains with a non-linear smoothing function added.
We investigated the impact of confounding variables on the substantial strain variation in haloperidol concentrations (Supplemental Methods). Linear regression modeling was used to investigate the effects of mouse strain, pellet dosage, and body weight on plasma haloperidol concentration at 30 days. Mouse strain was the predominant effect (P ~ 10−16). The heritability of log10(haloperidol) was 0.70 at 30 days and 0.67 at 60 days during the period when drug was being released by the pellets (heritabilities were lesser as haloperidol concentrations declined, 0.61 at 90 days and 0.27 at 120 days). These data strongly suggest that the observed variation in haloperidol concentrations is due to strain effects (a proxy for genetic differences between inbred lines).
The effects of haloperidol on VCMs, EPS, and activity
The central aim of this study was to investigate the relationship between genetic background and susceptibility to haloperidol-induced VCMs and other EPS phenotypes across 27 inbred mouse strains. Figure 2 summarizes nine dependent variables over all strains at baseline and after 30, 60, 90, and 120 days of exposure to haloperidol. Five points are noteworthy. First, the wide error bars in Figure 2 (even on a square root scale) suggest that, on average, there is considerable variation in all phenotypes across strains. Individual strain and time-point data for these nine variables indicate that between strain variation was considerably greater than within strain variation at most time points (Figure S4). Second, exposure to haloperidol had profound average effects. For example, comparing baseline with day 30, there was a 7-fold decrease in vertical activity, 263-fold increase in EPS, and 8-fold increase in overt chewing movements. Third, the baseline activity measures showed considerable variability across strains (consistent with attempts to map genetic loci for open-field activity) 24, 25. The average effect of haloperidol was maximal at day 30 or 60. Average values moved toward baseline on days 90 and 120 but did not return to baseline suggesting that the behavioral effects of haloperidol persist even when plasma concentrations are low (Table 2). Fourth, there was little average variability in EPS at baseline and the strong effects of haloperidol exposure were still present at day 120. Fifth, perhaps unlike the activity and EPS measures, the average effect of haloperidol on the four VCM measures were notably persistent through day 120 without a trend toward baseline values.
Figure 2. Summary of dependent variables (activity, EPS, and VCMs) over all strains.
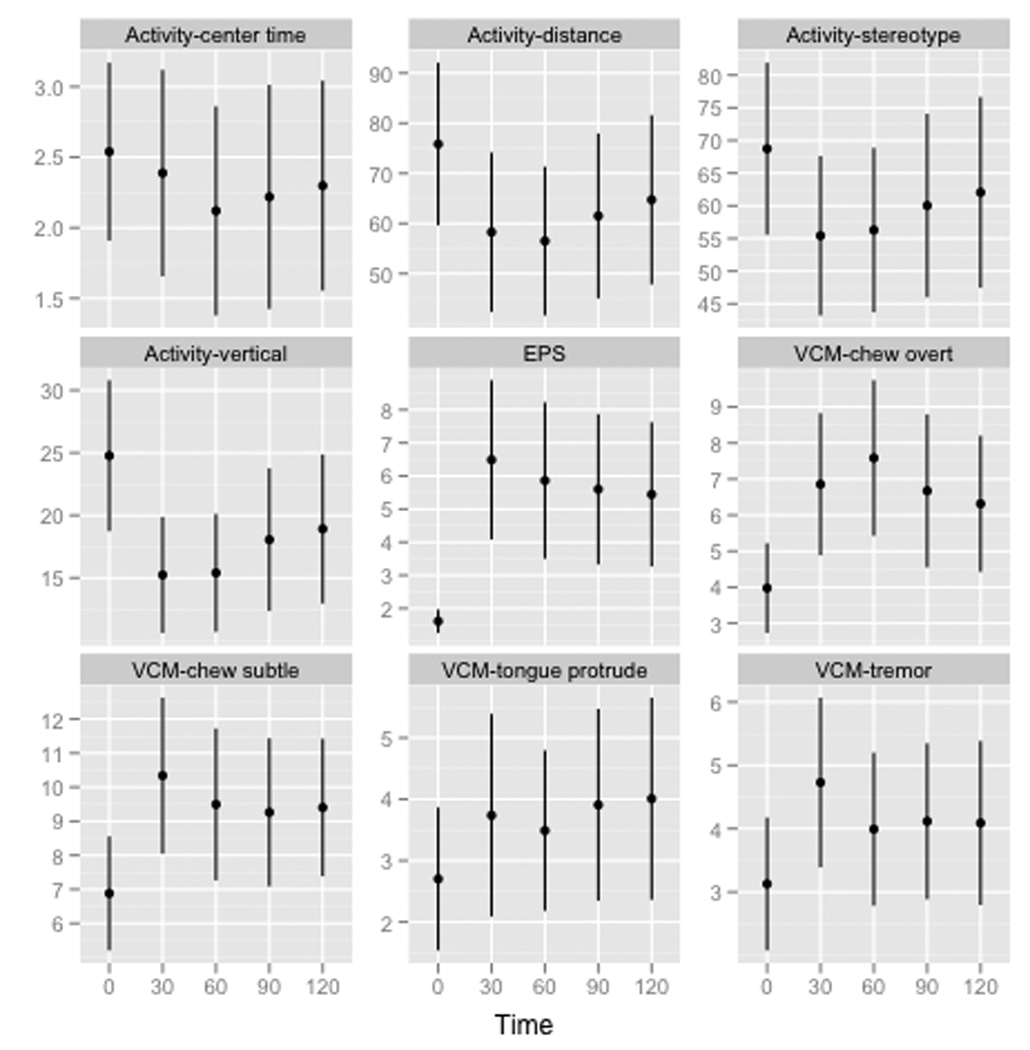
Each plot shows data for one dependent variable for five time points (baseline and 30, 60, 90, and 120 days). Shown are means and SEM error bars for over all strains (square root transformation).
We next investigated the effects of confounding variables on the core dependent variables. We fit 45 regression models with the dependent variable each phenotype at each time point (9 phenotypes × 5 time points). The predictors were drug dose, weight at that time point, haloperidol concentration at that time point (log10, omitted for time0), and strain. P-values for the coefficients for dose, weight, and haloperidol concentration were always > 0.001 (Bonferroni correction) whereas P-values for the coefficients for strain were 10−8–10−37 (the single exception was P=0.01 for strain for EPS at baseline). Thus, strain effects were the main determinant of phenotypic variation with no evident covariate effects. Strain effects also accounted for considerable proportions of variation (R2 > 0.6 for most activity and VCM measures and >0.7 for EPS after day 0).
Heritability estimates
The study of 27 inbred mouse strains allowed calculation of heritability for VCMs, EPS, and activity measures. We estimated heritability for these phenotypic data in four progressive ways. Figure 3a shows heritability estimates at five time points for the nine phenotypes (square root transformations of raw data). Strong genetic effects are evident for virtually all phenotypes. To isolate the impact of exposure to haloperidol, Figure 3b depicts the heritabilities of haloperidol-induced change scores (√timet – √time0). Heritabilities are considerably lower but generally > 0.4. Change scores can be unreliable and reflect error variance 26, particularly if the variables in the change score calculation are correlated. For example, the correlations of time0 with time30 measures were high for the activity (~0.6) and VCM measures (~0.5) but near zero for EPS (−0.07). For all correlated measures, the heritability of change scores is markedly lower but not for the uncorrelated EPS measure. Thus, the estimates in Figure 3b appear to be artifactually low. Figure 3c provides an improved estimation of phenotype-level heritability via analysis of change trajectories. The heritability of seven phenotypes exceeded 75% (all except center time and stereotopy).
Figure 3. Heritability estimates.
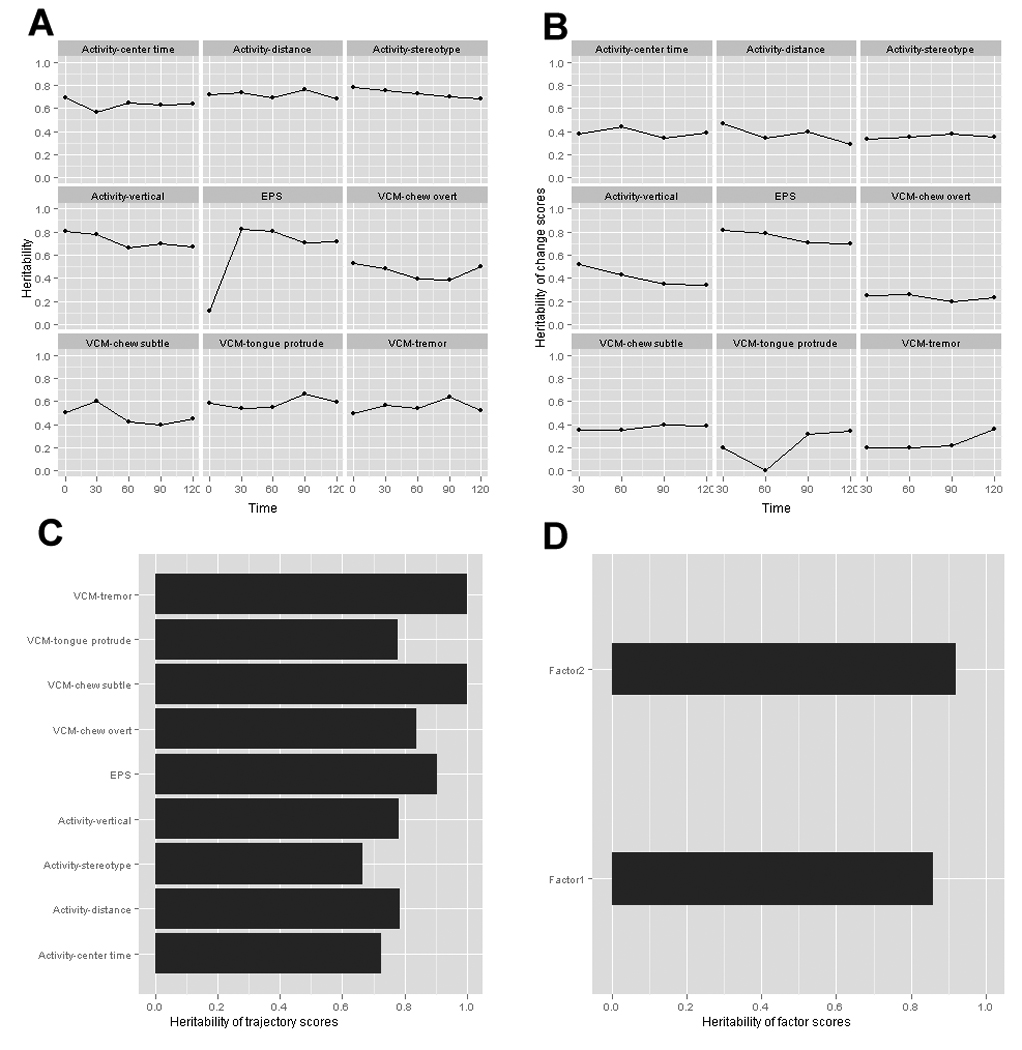
A) Heritability estimates for each dependent variable (square root transformation) at five time points (baseline and 30, 60, 90, and 120 days). B) Heritability estimates of change in phenotype from baseline (√timet – √time0). C) Heritability estimates for response trajectories of change scores. D) Heritability of factor analysis of response trajectories.
Factor analysis was then applied to the change score trajectories. As shown in Table 1, the first two factors had eigenvalues > 1 and together explained 75% of the shared variance among the VCM, activity, and EPS trajectories. These factors are roughly consonant with a priori expectations, with Factor 1 explaining most of the variance in the locomotor activity response trajectories and Factor 2 primarily explaining the orofacial movement response trajectories. However, the factor loadings were not completely specific as some items loaded onto both factors. The heritabilities were very high for both factors, with strain differences explaining 96% of variance in the orofacial movement latent trajectory factor 2, and 86% of variance in the locomotor activity latent trajectory factor 1 (Figure 3D). Strain means for factors 1 and 2 are depicted in Figure S5.
Table 1.
Factor analysis of haloperidol-induced movement phenotype trajectories.
| Factor | Eigenvalue | Proportion | Cum. | Trajectory | Factor 1 | Factor 2 |
|---|---|---|---|---|---|---|
| 1 | 3.36 | 0.54 | 0.54 | Activity-center time | 0.67 | 0.04 |
| 2 | 1.48 | 0.24 | 0.78 | Activity-distance | 0.94 | −0.05 |
| 3 | 0.66 | 0.11 | 0.89 | Activity-stereotypy | 0.93 | 0.23 |
| 4 | 0.42 | 0.07 | 0.95 | Activity-vertical | 0.78 | −0.17 |
| 5 | 0.12 | 0.02 | 0.97 | EPS | −0.38 | 0.18 |
| 6 | 0.09 | 0.01 | 0.99 | VCM-chew overt | 0.18 | 0.48 |
| 7 | 0.06 | 0.01 | 1.00 | VCM-chew subtle | −0.34 | 0.54 |
| 8 | 0.02 | 0.00 | 1.00 | VCM-tongue protrude | 0.42 | 0.64 |
| 9 | 0.00 | 0.00 | 1.00 | VCM-tremor | −0.25 | 0.66 |
| Heritability | 0.86 | 0.96 | ||||
DISCUSSION
The purpose of this report was to initiate investigation of murine VCMs with the eventual goal of improving understanding of the genomics of human TD. If VCMs are a reasonable analogue of TD, then it might be possible to accelerate discovery by using MTH (mouse-then-human) designs whereby mouse genetic mapping resources are used to screen the genomic search space to derive high-probability targets whose orthologs can be studied in human samples. In this way, the multiple testing burden is paid in a relatively inexpensive and experimentally tractable system and precious human samples are used only for the most crucial targets.
To achieve this end, we pursued a number of systematic goals. The initial consideration was the ability to deliver human-like concentrations of haloperidol in a sustained, consistent, and reliable manner as haloperidol exposure is a critical risk factor for TD 14. A literature review 27 and pilot experiments (Table S2) demonstrated that implantable pellets yielded lower coefficients of variation for haloperidol levels in plasma (20.6%) and brain (11.6%) than other methods of drug delivery (>34%). Additional dose-ranging experiments showed that dosing mice with implantable pellets at 3.0 mg/kg/day (60 day sustained release) reliably yielded human-like plasma concentrations. The data that form the backbone of this report (Figure 1 and Table S3) indicate that implantable pellets yield low coefficients of variation in plasma haloperidol concentrations (median 0.19) and nearly always achieved human-like plasma concentrations (>10 nM in 98.1% of mice). Note that two months of haloperidol exposure is an appreciable fraction of the ~1.5–2 year lifespan of a laboratory mouse and thus constitutes chronic exposure.
Second, we demonstrated that haloperidol concentrations are highly variable between inbred strains. The observed variability was not influenced by potential confounders such as the dose implanted or body mass. Moreover, within- was considerably less than between-strain variation leading to heritability estimates for haloperidol concentrations of ~0.7 during the times when the drug was being actively released. Future work will attempt to identify the genetic determinants of haloperidol concentrations in a genome-wide and unbiased manner.
Third, we developed a battery of tests to assess the observable effects of haloperidol. Following careful rater training and calibration, assessment of VCMs in randomly assigned tapes by raters blinded to experimental date was reproducible. An immediate observation was that exposure to haloperidol yielded marked average changes across multiple domains. Four measures of activity in the open field, one measure of EPS, and four measures of orofacial movement all exhibited, on average, marked changes following haloperidol exposure (Figure 1). These changes did not return to baseline values by the end of the study (120 days). Crucially, these measures were independent of haloperidol plasma level and other covariates. Strain was again the major predictor of phenotypic variation.
Fourth, we observed that the three domains we assessed – orofacial movements, activity in the open field, and EPS – were not discrete constructs but rather loaded onto two factors. Analysis of human data from a large randomized clinical trial (CATIE) 28,show a similar pattern: subjects with TD by consensus criteria 29 had greater EPS and akathisia (P values < 0.0001) 30. To investigate this with an approach closer to that in Table 1, we analyzed the CATIE baseline data for total Abnormal Involuntary Movement scale (AIMS) score 31, EPS measured by the Abbreviated Simpson–Angus Side Effect Scale 32, and the Barnes Akathisia Scale 33. Similar to the analyses in Table 1, two principal components accounted for 78% of the variance (see Table S5), and the first component had similar loadings: AIMS total (0.61), EPS (0.56), and akathisia (0.56). Therefore, the human and mouse data converge and suggest that each of these scales is an imprecise but informative assessment of a more fundamental latent construct.
Fifth, we estimated heritability for haloperidol-induced effects on VCMs, activity in the open field, and EPS. Figure 3 provides the results at successive levels of processing. Given that our design was inherently longitudinal, analysis of the response trajectories of individual mice is superior to individual time points and, given the inter-relatedness of the phenotype domains, analysis of factors from factor analysis of response trajectories is superior to trajectory analysis. Thus, we believe heritability estimates in Figure 3D provide the best representation of our study and of the phenotypes themselves. The heritability of factor 1 was 0.86 and 0.96 for factor 2. To our knowledge, these are the first estimates of heritability for VCMs. Variation in behavioral responses to chronic haloperidol exposure is predominantly determined by genetic influences. These data strongly support efforts to identify the specific genetic basis of variation in these traits and augment the case for the study of genetic influences on human TD.
A major assumption of the mouse-then-human approach is that murine VCMs are an adequate model for the human adverse drug reaction TD. Three criteria are generally applied to establish the validity of a mouse model of a human phenotype 34. The pun is unavoidable: VCMs have face validity for human TD. Key behavioral markers that define the predominant type of TD (repetitive and purposeless movements of the mouth and jaw) are highly similar to VCMs (purposeless jaw movements in the vertical plane). A board-certified neurologist (KW) with considerable experience in the diagnosis and treatment of TD reviewed tapes of mice with VCMs and also noted the face validity of VCMs for TD. The face validity of VCMs is augmented by multiple additional lines of argument (summarized in Table S1), and their persistence over large fractions of the mouse lifespan is compelling (Figure S1).
Evaluation of the remaining two criteria for VCMs as a valid model of human TD are for future studies. Construct validity requires a conceptual analogy to the cause of human TD. This criterion is partially fulfilled given the crucial role of typical antipsychotics in the etiology of both VCMs and TD. However, fully evaluating this criterion is a fundamental goal of the MTH design – if susceptibility loci can be identified in mouse, do the orthologous regions in human contain susceptibility loci for TD? The final criterion – predictive validity – assesses (in part) specificity of treatment response. Although this could be studied in the absence of precise pathophysiology, it is far more interesting to conduct such studies with the greater insight afforded by a clear understanding of how genes and haloperidol exposure interact to produce VCMs.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. Andrea Pinheiro and Randy Nonneman for helpful discussions. The mice used in this study were acquired as part of the Mouse Phenome Project, an ongoing international collaborative effort headquartered at The Jackson Laboratory (Bar Harbor, ME, USA). This work was supported by the Pharmacogenetics Research Network (U01 GM63340, PI Dr. McLeod), a NIMH/NHGRI Center of Excellence for Genome Sciences grant (P50 MH90338, PIs Drs. Fernando Pardo-Manuel de Villena and Sullivan), and the Mouse Behavioral Phenotyping Laboratory (NICHD P30 HD03110, PI Dr. Joseph Piven). Dr Sullivan was supported by MH080403, MH077139, and MH074027.
Footnotes
Supplementary information is available at The Pharmacogenomics Journal's website.
Conflicts of Interest: Dr. Sullivan reports receiving unrestricted research funding from Eli Lilly for genetic research in schizophrenia. The other authors report no conflicts.
On-Line Resources: Phenotypic data from this project will be available via the Mouse Phenome Database (MPD; http://www.jax.org/phenome) on 1 April 2011.
REFERENCES
- 1.Harrill AH, Watkins PB, Su S, Ross PK, Harbourt DE, Stylianou IM, et al. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res. 2009;19(9):1507–1515. doi: 10.1101/gr.090241.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen BJ, et al. A customized and versatile high-density genotyping array for the mouse. Nat Methods. 2009;6(9):663–666. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson GM. Long-acting, antipsychotic agents and extrapyramidal side effects. Dis Nerv Syst. 1970;31(Suppl):12–14. [PubMed] [Google Scholar]
- 4.Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451–1462. doi: 10.1517/14656566.9.9.1451. [DOI] [PubMed] [Google Scholar]
- 5.Soares-Weiser K, Fernandez HH. Tardive dyskinesia. Seminars in neurology. 2007;27(2):159–169. doi: 10.1055/s-2007-971169. [DOI] [PubMed] [Google Scholar]
- 6.Crane GE. Tardive dyskinesia in patients treated with major neuroleptics: a review of the literature. Am J Psychiatry. 1968;124(Suppl)(8):40–48. doi: 10.1176/ajp.124.8S.40. [DOI] [PubMed] [Google Scholar]
- 7.Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and the Ser9Gly polymorphism in the DRD3 gene: a meta analysis. Schizophrenia research. 2006;83(2–3):185–192. doi: 10.1016/j.schres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Basile VS, Masellis M, Badri F, Paterson AD, Meltzer HY, Lieberman JA, et al. Association of the MscI polymorphism of the dopamine D3 receptor gene with tardive dyskinesia in schizophrenia. Neuropsychopharmacology. 1999;21(1):17–27. doi: 10.1016/S0893-133X(98)00114-6. [DOI] [PubMed] [Google Scholar]
- 9.Basile VS, Ozdemir V, Masellis M, Meltzer HY, Lieberman JA, Potkin SG, et al. Lack of association between serotonin-2A receptor gene (HTR2A) polymorphisms and tardive dyskinesia in schizophrenia. Mol Psychiatry. 2001;6(2):230–234. doi: 10.1038/sj.mp.4000847. [DOI] [PubMed] [Google Scholar]
- 10.de Leon J, Susce MT, Pan RM, Koch WH, Wedlund PJ. Polymorphic variations in GSTM1, GSTT1, PgP, CYP2D6, CYP3A5, and dopamine D2 and D3 receptors and their association with tardive dyskinesia in severe mental illness. J Clin Psychopharmacol. 2005;25(5):448–456. doi: 10.1097/01.jcp.0000177546.34799.af. [DOI] [PubMed] [Google Scholar]
- 11.Eichhammer P, Albus M, Borrmann-Hassenbach M, Schoeler A, Putzhammer A, Frick U, et al. Association of dopamine D3-receptor gene variants with neuroleptic induced akathisia in schizophrenic patients: a generalization of Steen's study on DRD3 and tardive dyskinesia. Am J Med Genet. 2000;96(2):187–191. doi: 10.1002/(sici)1096-8628(20000403)96:2<187::aid-ajmg13>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 12.Patsopoulos NA, Ntzani EE, Zintzaras E, Ioannidis JP. CYP2D6 polymorphisms and the risk of tardive dyskinesia in schizophrenia: a meta-analysis. Pharmacogenetics and genomics. 2005;15(3):151–158. doi: 10.1097/01213011-200503000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Turrone P, Remington G, Nobrega JN. The vacuous chewing movement (VCM) model of tardive dyskinesia revisited: is there a relationship to dopamine D(2) receptor occupancy? Neuroscience and biobehavioral reviews. 2002;26(3):361–380. doi: 10.1016/s0149-7634(02)00008-8. [DOI] [PubMed] [Google Scholar]
- 14.Hsin-Tung E, Simpson G. Medication-induced movement disorders. In: Kaplan HI, Sadock BJ, editors. Comprehensive Textbook of Psychiatry. Philadephia, PA: Lippincott, Williams and Wilkins; 2000. [Google Scholar]
- 15.Fleischmann N, Christ G, Sclafani T, Melman A. The effect of ovariectomy and long-term estrogen replacement on bladder structure and function in the rat. J Urol. 2002;168(3):1265–1268. doi: 10.1016/S0022-5347(05)64637-X. [DOI] [PubMed] [Google Scholar]
- 16.Tomiyama K, McNamara FN, Clifford JJ, Kinsella A, Koshikawa N, Waddington JL. Topographical assessment and pharmacological characterization of orofacial movements in mice: dopamine D(1)-like vs. D(2)-like receptor regulation. Eur J Pharmacol. 2001;418(1–2):47–54. doi: 10.1016/s0014-2999(01)00908-6. [DOI] [PubMed] [Google Scholar]
- 17.Crawley JN. Exploratory behavior models of anxiety in mice. Neurosci Biobehav Rev. 1985;9(1):37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- 18.Barnes DE, Robinson B, Csernansky JG, Bellows EP. Sensitization versus tolerance to haloperidol-induced catalepsy: multiple determinants. Pharmacol Biochem Behav. 1990;36(4):883–887. doi: 10.1016/0091-3057(90)90094-x. [DOI] [PubMed] [Google Scholar]
- 19.Gueorguieva R, Krystal JH. Move over ANOVA: progress in analyzing repeated-measures data and its reflection in papers published in the Archives of General Psychiatry. Arch Gen Psychiatry. 2004;61(3):310–317. doi: 10.1001/archpsyc.61.3.310. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein H. Multilevel statistical models. In: Searle S, Casella G, McCulloch C, editors. Variance Components. New York: Wiley; 1995. [Google Scholar]
- 21.Joreskog K. A general approach to confirmatory maximum likelihood factor analysis. Psychometrika. 1969;34:183–202. [Google Scholar]
- 22.Muthén B, Muthén L. Traditional latent variable modeling using Mplus: Mplus Short course notes. Los Angeles, CA: 2003. [Google Scholar]
- 23.Van Prooijen J, Van Der Kloot WA. Confirmatory analysis of exploratively obtained factor structures. Educational and Psychological Measurement. 2001;51:777–792. [Google Scholar]
- 24.Baile JS, Grabowski-Boas L, Steff BM, Wiltshire T, Churchil GA, Tarantino LM. Identification of quantitative trait loci for locomotor activation and anxiety using closely related inbred strains. Genes Brain Behav. 2008;7(7):761–769. doi: 10.1111/j.1601-183x.2008.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kas MJ, de Mooij-van Malsen JG, de Krom M, van Gassen KL, van Lith HA, Olivier B, et al. High-resolution genetic mapping of mammalian motor activity levels in mice. Genes Brain Behav. 2009;8(1):13–22. doi: 10.1111/j.1601-183X.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- 26.Crocker L, Algina J. Introduction to Classical and Modern Test Theory. New York: Wadsworth Publishing; 1986. [Google Scholar]
- 27.Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther. 2003;305(2):625–631. doi: 10.1124/jpet.102.046987. [DOI] [PubMed] [Google Scholar]
- 28.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 29.Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486–487. doi: 10.1001/archpsyc.1982.04290040080014. [DOI] [PubMed] [Google Scholar]
- 30.Miller D, McEvoy JP, Davis SM, Caroff SN, Saltz BL, Chakos MH, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res. 2005;80(1):33–43. doi: 10.1016/j.schres.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 31.Guy W. ECDEU Assessment Manual for Psychopharmacology-Revised. Bethesda, MD: US Deptartment of Health, Education, and Welfare; 1976. (DHEW Publication No. ADM 534–537) [Google Scholar]
- 32.Tracy K, Adler LA, Rotrosen J, Edson R, Lavori P. Interrater reliability issues in multicenter trials, Part I: Theoretical concepts and operational procedures used in Department of Veterans Affairs Cooperative Study #394. Psychopharmacol Bull. 1997;33(1):53–57. [PubMed] [Google Scholar]
- 33.Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672–676. doi: 10.1192/bjp.154.5.672. [DOI] [PubMed] [Google Scholar]
- 34.Chadman KK, Yang M, Crawley JN. Criteria for validating mouse models of psychiatric diseases. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(1):1–11. doi: 10.1002/ajmg.b.30777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.