

Abstract
Untimely activation of nicotinic acetylcholine receptor (nAChR) by nicotine results in short- and long-term consequences on learning and behavior. In this study, the aim was to determine how prenatal nicotine exposure affects components of glutamatergic signaling in the hippocampus during postnatal development. We investigated regulation of both nAChRs and glutamate receptors for α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and N-methyl-D-aspartate (NMDA), from postnatal day (P) 1 to P63 after a temporally restricted exposure to saline or nicotine for 14 days in utero. We analyzed postsynaptic density components associated with AMPAR and NMDAR signaling: Calcium/calmodulin-dependent protein kinase II α (CaMKIIα), Calmodulin (CaM), and postsynaptic density-95 (PSD95), as well as presynaptically localized synaptosomal-associated protein 25 (SNAP25). At P1, there was significantly heightened expression of AMPAR subunit GluR1 but not GluR2, and of NMDAR subunits NR1, NR2a and NR2d but not NR2b. NR2c was not detectable. At P1, the postsynaptic proteins CaMKIIα, CaM, and PSD95 were also significantly upregulated, together with presynaptic SNAP25. This enhanced expression of glutamate receptors and signaling proteins was concomitant with elevated levels of [3H] Epibatidine (EB) binding in prenatal nicotine-exposed hippocampus, indicating that α4β2 nAChR may influence glutamatergic function in the hippocampus at P1. By P14, neither [3H]EB binding nor the expression levels of subunits GluR1, GluR2, NR1, NR2a, NR2b, NR2c, or NR2d seemed changed with prenatal nicotine. However, CaMKIIα was significantly upregulated with nicotine treatment while CaM showed downregulation at P14. The effects of nicotine persisted in young adult brains at P63. They exhibited significantly downregulated GluR2, NR1, and NR2c expression levels in hippocampal homogenates and a considerably muted overall distribution of [3H]AMPA binding in areas CA1, CA2, CA3, and the dentate gyrus. Our results suggest that prenatal nicotine exposure can regulate the glutamatergic signaling system throughout postnatal development by enhancing or inhibiting availability of AMPAR and NMDAR or their signaling components. The persistent depression, in adults, of the requisite NR1 subunit for NMDAR assembly, and of GluR2, important for assembly, trafficking, and biophysical properties of AMPAR, indicates that nicotine may alter ionotropic glutamate receptor stoichiometry and functional properties in adults after prenatally restricted exposure.
Keywords: prenatal nicotine, developing hippocampus, AMPAR, NMDAR, nAChR, postsynaptic signaling
Developmental plasticity in the cerebral cortex and the hippocampus depends, in part, on afferent cholinergic input to modulate glutamatergic neuronal activity. Disruption of afferent cholinergic projections or acetylcholine secretory mechanisms during development reduces plasticity (Hohmann and Berger-Sweeney, 1998). In disease states, including Alzheimer's disease, damage to cholinergic basal forebrain neurons or their projections is an initiating event leading to extensive memory loss with accompanying neurodegeneration (Auld et al., 2002). Cholinergic projections to the hippocampus originate from the medial septal nuclei and the vertical limb of the nucleus of the diagonal band of Broca in the basal forebrain (Yoshida and Oka, 1995). These cholinergic neurons send afferents to the prefrontal cortex and other parts of the cerebral cortex (Mesulam et al., 1983, Yoshida and Oka, 1995, Auld et al., 2002) and form functional synapses with pyramidal neurons as well as granules cells, interneurons, and neurons in the hippocampal hilus during the first week after birth (Frotscher and Leranth, 1985, Rami et al., 1989, Linke and Frotscher, 1993). Developmental consequences of premature activation of these cholinergic fibers and the subsequent stimulation of nicotinic acetylcholine receptors (nAChRs) in the hippocampus by cholinergic agonists which are packaged and trafficked within synaptic vesicles (Park et al., 2011) have not been well studied, although there are many reports that gestational exposure to nicotine can lead to impaired learning and decreased brain function (Gingras and O'Donnell, 1998).
In fact, studies addressing postnatal, adolescent or adult intake of nicotine show that stimulation of nAChRs can influence not only cholinergic, but also serotonergic (King et al., 1991, Muneoka et al., 1997, Xu et al., 2001), dopaminergic (Carr et al., 1989, Muneoka et al., 1999, Eddins et al., 2009), as well as glutamatergic neurotransmitter systems (Ferrari and Fior-Chadi, 2007). These aminergic systems are all primarily upregulated in the presence of nicotine. Given that glutamate is a major modulator of learning, memory and cognition, and its synapses are the prevailing ones for most functional processes in hippocampus, we have chosen to study how gestational nicotine exposure regulates hippocampal glutamatergic synapses.
In a majority of hippocampal synapses, glutamate binds to the two principal postsynaptic ionotropic glutamate receptors (iGluRs), the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors (AMPARs) and the N-methyl-D-aspartate (NMDA) receptors (NMDARs), located on postsynaptic somatodendritic membranes and concentrated in the dendritic spines (Bliss and Collingridge, 1993, Koh et al., 1995, Ritter et al., 2001). The AMPAR is a heterotetramer composed of various combinations of four different subunits (GluR1–GluR4), and mediates fast excitatory synaptic transmission. In the hippocampus, AMPARs are primarily composed of iGluR subunits GluR1/2 or GluR2/3 (Hanley, 2010). The NMDAR is also a heterotetramer, a ligand gated ion channel composed of two requisite NR1 subunits together with two or three modulatory NR2 (NR2a–NR2d) and NR3 (NR3a–NR3b) subunits (Kohr, 2006). Like nAChRs, both AMPARs and NMDARs are functionally expressed at the surface of hippocampal neurons from the time of differentiation to the period of synaptogenesis (Cottrell et al., 2000), which in rodents occurs mostly during the first postnatal weeks.
Here, we focus our in vivo studies on how excitation of nAChRs, primarily the α4β2- and α7-containing nAChRs, by prenatal nicotine, might influence expression of AMPAR and NMDAR subunits and other proteins involved in postsynaptic signaling during development of hippocampal neurons. It has been reported in humans that gestational nicotine exposure results in sensorimotor as well as cognitive deficits (Ernst et al., 2001) and that in rats, a chronic dose of nicotine at 6mg/kg/ml throughout pregnancy results in long-lasting effects in performance of reflex, behavioral, and learning and retention tasks (Ajarem and Ahmad, 1998, Vaglenova et al., 2008, Chistyakov et al., 2010) .
In order to better understand the regulatory role that gestational exposure to nicotine has on nAChRs and the consequent effects on postsynaptic signaling in the hippocampus, we measured protein expression levels of AMPA and NMDA receptor subunits and associated synaptic proteins. Receptors, signaling molecules, scaffolding proteins, as well as presynaptic proteins are highly regulated during critical developmental periods. We report that just as high affinity α4β2 nAChRs are upregulated with prenatal nicotine at postnatal day 1 (P1), glutamatergic AMPARs and NMDARs are also upregulated, and this increased expression is concomitant with that of multiple synaptic proteins. Even when glutamate receptor levels were unchanged by gestational nicotine at P14, signaling molecules associated with iGluRs were still differentially regulated. Moreover, long-term regulation of AMPARs and NMDARs was also observed, with expression of GluR2, NR1, and NR2c subunits being significantly reduced during early adulthood at P63. Even functional AMPAR distribution was significantly decreased as evidenced by [3H]AMPA binding studies. Thus, our study represents an important advancement in identifying prenatal nicotine-induced regulation of iGluR expression and signaling during development and in adult function of hippocampal synapses.
Experimental Procedures
Animals and Drug treatment
All procedures were conducted according to an approved protocol and guidelines set forth by the Howard University Institutional Animal Care and Use Committee (HU-IACUC) and the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academic Press, Washington, DC, 1996). Timed-pregnant Sprague–Dawley rats weighing 250–300g (Harlan Laboratories, Frederick, MD) were housed under 12 h light/dark cycle, with free access to food and water. On gestational day 7 (G7), pregnant dams were implanted between the scapulae with a mini-osmotic infusion pump (Alzet, Cupertino, CA), containing either 0.9% saline or nicotine hydrogen tartrate (Sigma-Aldrich, St. Louis, MO) which was delivered at a rate of 4 mg /kg/day as previously published (Davila-Garcia et al., 2003). This dose is commonly used to mimic circulating blood nicotine levels of a 2-4 packs/day smoker (Murrin et al., 1987). All pumps hold a volume of ~ 200μl and have a flow rate of ~0.5μl/hr. Animal weights were monitored daily until birth. On G21, pumps were removed by aseptic surgery. Brains were then harvested and dissected from offspring at P1, P14 or P63 for further analysis. Within each age group, numbers (n) represent an approximately equal number of males and females.
Pharmacological agents
Pharmacological agents used included (-)- nicotine hydrogen tartrate, cytisine, and A85380 (Sigma-Aldrich, St. Louis, MO). Nicotine was prepared fresh on the day of pump implantation in 0.9% saline, and the pH was adjusted to 7.4-7.6. For binding and autoradiography studies, the radioligands [3H]AMPA, [3H]Epibatidine ([3H]EB), and [125I]α-Bungarotoxin ([125I]α-BTX) were purchased from Perkin Elmer Life Sciences, Boston, MA.
nAChR Binding of [3H]EB in hippocampal membrane homogenates
Individual hippocampi from saline or nicotine treated groups were weighed, pooled, and homogenized with a Brinkmann Polytron homogenizer (Brinkman Instruments, Westbury, CT) in ice-cold 50 mM Tris-HCl buffer (pH 7.4 at 4°C ) and centrifuged at 32,000 × g for 20 min at 4°C to collect membrane fractions. The pellets were resuspended in fresh 50mM Tris-HCl buffer , pH 7.4, and binding studies were carried out as previously described with a few modifications (Houghtling et al., 1995). Aliquots of tissue homogenates equivalent to 10 mg tissue per 100μl were incubated in buffer for 2 h at RT with 300 pM [3H]EB with or without 15nM A85380 or 200nM Cytisine. The addition of these nicotinic ligands affords us the ability to differentiate between different nAChR subtypes (Perry et al., 2002). Binding reactions were started by the addition of tissue and terminated by vacuum filtration through Whatman GF/C filters, which were mounted on a Brandel cell harvester and primed with 0.5% polyethylenimine to reduce binding to the filter. The filters were washed with 3 × 4ml of buffer and then counted in a scintillation counter. Protein was determined using the BCA reagent (ThermoFisher - Pierce, Rockford, IL) and measured at 562 nm. Non-specific binding in all assays was determined in the presence of 300μM nicotine hydrogen tartrate, which displaces almost 100% of the binding. Specific binding was defined as the difference between total binding and non-specific binding. Drug specific binding for A85380 and cytisine was calculated by substracting the non-specific binding from total binding in the presence of each ligand. Specific binding data were analyzed by a studentg's t-test between control and nicotine-treated groups for each age separately and graphed using Graphpad Prism 5 (Graphpad Software, Inc, San Diego, CA). A p-value smaller than 0.05 was considered statistically significant.
Western blotting
The hippocampi were dissected from pup brains at P1, P14 or P63, immediately frozen on dry ice and stored at -80°C until needed. Frozen hippocampi were homogenized with a PRO200 homogenizer (PRO Scientific Inc., Oxford, CT) at 0°C in 1-2ml of ice-cold lysis buffer containing 20mM Tris, 150mM NaCl, 1mM EDTA, 1% Tx-100, 0.1% SDS, 1mM DTT, and 1:25 protease inhibitor cocktail (Roche Diagnostics Corp, Indianapolis, IN). The detergent-soluble fraction was removed by centrifugation at 13,000 × g for 20min at 4°C. The protein concentration in the supernatant was determined using the BCA reagent and read at a wavelength of 562nm with a Unicam Heλios Beta spectrophotometer (Spectronic Unicam, Cambridge, U.K.). Equivalent concentration of protein was electrophoresed by SDS-PAGE 4-12% NuPAGE® Bis-Tris gels (Invitrogen, Inc., Carlsbad, CA) and electroblotted onto 0.2μm nitrocellulose membranes (Invitrogen, Inc., Carlsbad, CA). Gel loading and protein transfer efficiency were tested with SimplyBlue™ SafeStain (Invitrogen, Inc., Carlsbad, CA) and Ponceau S staining of blots. Membranes were blocked in 10mM Tris-buffered saline, pH 7.6, 0.1% Tween 20, 5% nonfat milk (Bio-Rad, Hercules, CA) for 1h at room temperature, washed with 10mM Tris buffer/ 0.1% Tween 20 (TBS-T), and incubated overnight at 4°C with primary antibody diluted in blocking buffer. The next day, blots were washed 3 × 10 min in TBS–T, and incubated for 1 h with HRP-conjugated mouse or rabbit secondary antibody diluted in blocking buffer to 0.07μg/ml. After 3× 10 min washes in TBS–T, the signal was detected using Super Signal West Pico detection reagent (ThermoFisher - Pierce, Rockford, IL). The signal was captured on blue film (PHENIX Research Products, Candler, NC) below saturation levels, scanned using a CanoScan Lide 90 Scanner (Canon, Lake Success, NY), and relative pixel intensity values obtained with AlphaEaseFC software (Alpha Innotech, Santa Clara, CA). A subset of data was scanned and pixel intensities retrieved directly from blots using the ImageQuant LAS 4010 (GE Healthcare Biosciences Corp., Piscataway, NJ). Relative pixel intensity values were analyzed with Microsoft Excel utilizing an unpaired two-tail t-test to determine statistical significance between groups and compared to the loading control proteins GAPDH (for P1) or β-actin (for P14 or P63) for each band. P-values less than 0.05 were regarded as denoting statistical significance. Summarized data are presented as means ± SEM, and were graphed using GraphPad Prism5. Each bar represents pups from 5-10 different dams implanted with pumps at various times during the study, and for a given antibody, each sample was run on 3 different blots and the pixel intensity for each band on the 3 blots were averaged. Comparison between the 3 age groups was not possible due to a several fold difference (and oversaturation of signals) between developmental age groups when assessing them under identical conditions.
Antibodies used for Western Blot Analysis
Fourteen antibodies against the following proteins were used: β-actin (used at 0.07 μg/mL, Cat # ab8226), Calcium/calmodulin kinase IIα (CaMKIIα) (0.1μg/mL, ab2725) and GAPDH (0.25μg/mL, ab9485) were purchased from Abcam, Inc., Cambridge, MA. Calmodulin (CaM) (0.07μg/mL, 05-173), GluR1 (0.17μg/mL, AB1504), GluR2 (0.17μg/mL, AB1768), NR1 (1:10000, AB9864), NR2a and NR2b (0.05μg/ml, AB1555P and AB1557P), NR2c (0.07ug/mL, AB1592P), NR2d (1:3000, MAB35578) and α-tubulin (0.1μg/mL, #05-829) were obtained from Millipore, Inc., Bedford, Massachussets. SNAP-25 (0.25μg/ml, SL3730) was from Enzo Life Sciences – Biomol, Plymouth Meeting, PA, and PSD95(1/3000, #2507) from Cell Signaling Technology, Boston, MA.
[3H]AMPA, [3H]EB, [125I]α-BTX Binding and Autoradiography
The binding to AMPARs was performed according to published methods (Ekonomou et al., 2001, Pandis et al., 2006). Binding protocols for high affinity nAChRs (Houghtling et al., 1995) and α7-containing nAChRs were also performed as previously published (Tizabi et al., 2000). Briefly, sections were cut at 20 μm in a frozen cryostat, and collected onto gelatinized slides. Sections were then pre-washed and incubated with either 25nM [3H]AMPA, or 300pM [3H]EB, or 1μM [125I]α-BTX in 50mM Tris-HCl buffer (pH 7.2) for 1h (with 2.5mM CaCl2 at 4°C for [3H]AMPA) or 2hrs (at 4°C for [3H]EB) or 30min (at 37°C for [125I]α-BTX). Nonspecific binding was determined as binding in the presence of 1mM glutamic acid for [3H]AMPA or 300μM nicotine for [3H]EB and 1mM [125I]α-BTX in adjacent sections. Sections were washed in cold Tris-HCl buffer (with 2.5mM CaCl2 for [3H]AMPA ) and dried in a stream of cold air. Finally, dried sections were exposed to Amersham Hyperfilm-3H (RPN535B, GE Healthcare) for [3H]AMPA and [3H]EB along with tritium standards and stored in light tight film cassettes for 10-24 weeks. For [125I]α-BTX, Biomax MR film (Kodak) and [125I] standards were used. After 10 days incubation with [125I]α-BTX, or 10 weeks for [3H]EB or 24 weeks for [3H] AMPA, the films were developed using GBX Developer and Fixer (Kodak), and images were captured using an MCID system (Interfocus Imaging, Mering, Germany).
RESULTS
nAChRs Expression is briefly upregulated in developing hippocampus
The most abundant nAChRs in brain are composed of the homomeric α7 or the heteromeric α4β2 nAChRs subtype. We investigated the temporal pattern of nAChR expression in developing postnatal rat hippocampus with or without gestational exposure to nicotine by using radioligand binding to membrane homogenates. We used the radioligand [3H]EB, a potent agonist for non-α7 high affinity nAChRs (Gerzanich et al., 1995, Houghtling et al., 1995), in the presence or absence of A85380, a β2-selective agonist (Sullivan et al., 1996), or cytisine, a full agonist for α3β4- and α7-containing nAChRs but with low efficacy (Picciotto et al., 1995, Papke and Porter Papke, 2002) and a partial agonist with high efficacy for α4β2 nAChRs (Papke and Heinemann, 1994, Mineur et al., 2007). At P1, [3H]EB showed specific binding values of 57.39 ± 2.05 fmol/mg protein for nicotine treated groups which were significantly elevated compared to 45.88 ± 2.83 fmol/mg protein for control (Fig. 1A, 1B, **p≤0.01), an increased binding of 25%. In contrast, by P14, there was no statistical difference between the two groups, and by P63, any disparity was eliminated (Fig. 1A). In the presence of either 15nM A85380 or 200nM cytisine, [3H]EB binding was significantly reduced, indicating that during early development the binding sites were primarily α4-containing nAChRs, very likely α4β2. Additionally, in the presence of either A85380 or cytisine, the previously observed upregulation of [3H]EB binding for nicotine-exposed P1 animals was not present (Fig. 1A, 1B). This data indicates that at P1, specific upregulation of α4β2 nAChRs is a prominent component of the nicotine-induced regulation of nAChR expression and binding in hippocampus.
Figure 1. [3H]Epibatidine binding is elevated in hippocampus of rat neonates after nicotine exposure in utero.

(A) 300pM [3H]EB specific binding in pups prenatally exposed to saline (open circle, square, triangle) or nicotine (closed circle, square, triangle) in the presence of 15nM A85380 (squares) or 200nM cytisine (triangles), competitive agonists for the β2 or β2/β4 receptor, respectively. Specific binding of [3H]EB was defined as the difference between total and nonspecific binding, which was measured in the presence of 100μM nicotine. (B) The graph is a representation of the binding data from (A) at P1 – the only age at which [3H]EB binding in nicotine treated hippocampi was significantly higher than the saline treated ones (**p< 0.01 compared to control). Values are the mean ± SEM of the binding of sub-saturating concentrations of [3H]EB (n=5 for each group). CTL=control, NIC=nicotine-treated, n= the number of pups used.
Prenatal nicotine exposure alters the expression of β-actin
It has been reported that disease states and perturbation of differentiating cells during development can have an effect on expression of housekeeping or cytoskeletal proteins normally used as internal controls (Dittmer and Dittmer, 2006, Aldridge et al., 2008). Because the developing hippocampus is actively transcribing or translating genes into proteins during those crucial early postnatal days, we reasoned that there could be perturbations in the expression of general or cytoskeletal proteins with prenatal nicotine exposure. Thus, we first measured whether nicotine affected expression of β-actin, GAPDH or α-tubulin, used widely as housekeeping genes for western blot analysis. We demonstrated that prenatal nicotine exposure induced a 25±5% (n=8 for control and n=14 for nicotine group) increase for β-actin expression at P1 (Fig. 2A, **p<0.01), an augmentation that was not seen at P14 or P63 (Fig. 2A, p>0.05). However, GAPDH and α-tubulin expression levels remained comparable between nicotine treated and saline treated groups at all ages surveyed (Fig. 2B, 2C). This suggests that nicotine regulates cytoskeletal proteins such as β-actin during early neuronal growth and would not be an appropriate control at P1 under our experimental conditions.
Figure 2. Prenatal nicotine treatment influences β-actin expression during early postnatal development.
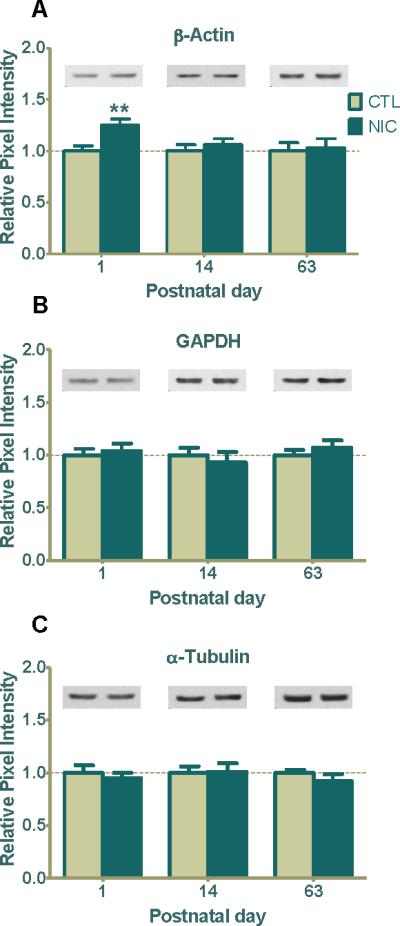
(A) Western blot analysis of β-actin expression in saline and nicotine treated rats at P1, P14 and P63. β-actin expression was significantly enhanced at P1 (**p < 0.01; n=8 CTL, 14 NIC) but not at P14 ( n=12 CTL, 11 NIC) or at P63 (n=8 CTL, 9 NIC). (B) GAPDH and (C) α-tubulin expression did not change at any age investigated (P1-P63; n=5-8, CTL; n=9-12, NIC). Values are reported as the mean ± SEM relative to saline-treated controls. Typical western blots for each treatment and age group are shown in band pairs above the bars (left band: control; right band: nicotine). CTL=control, NIC=nicotine-treated, n= the number of pups used.
Glutamate Receptor Expression: Enhanced during development and repressed in adults when exposed prenatally to nicotine
In order to understand how chronic gestational nicotine exposure might affect neuronal communication and result in attention and learning deficits in humans and rodent models of nicotine abuse, we investigated the expression of AMPARs and NMDARs. These receptors are present on the postsynaptic membrane and are necessary for strengthening synapses and to facilitate long-term potentiation of synaptic inputs (Malinow and Malenka, 2002, Malenka and Bear, 2004, Corera et al., 2009). Thus, we investigated the expression levels of GluR1 and GluR2 subunits, which are important for assembly and membrane insertion of functional AMPARs, and NR1, NR2a, NR2b, NR2c, and NR2d subunits, essential for functional NMDARs.
At P1, prenatal nicotine exposure significantly upregulated the abundantly expressed GluR1 by 31±4% (Fig. 3A, ***p<0.001), whereas the GluR2 subunit, although elevated by 16±6%, was not statistically different from control (Fig. 3B). By P14, GluR1 levels returned to comparatively normal and remained unchanged at P63 (Fig. 3A). Similar to its pattern at P1, the GluR2 subunit was also elevated at P14 by 17±9% but was not statistically significant (Fig. 3B). However, surprisingly, GluR2 expression levels decreased by 11±3% in P63 adult animals treated with nicotine (Fig. 3B, *p=0.04), demonstrating a direct or indirect long-term effect by nicotine.
Figure 3. Prenatal nicotine exposure regulates GluR1/GluR2 AMPAR subunits expression in developing rat hippocampus.
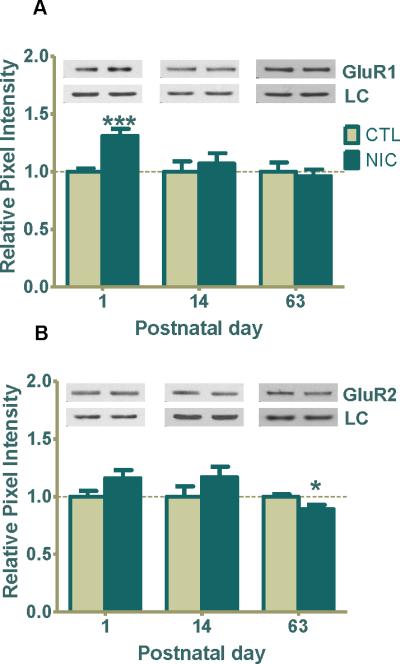
(A) GluR1 expression was 31% greater in nicotine treated pups at P1 (***p<0.001; n=6 CTL, 10 NIC), and no significant changes were seen at P14 (n=12 CTL, 11 NIC) or P63 (n=8 CTL, 9 NIC). (B) GluR2 expression was 16% greater at P1 (n=6 CTL, 11 NIC) and 17% greater at P14 (n=12, CTL; n=11, NIC) but neither were statistically significant. Importantly, the level of GluR2 was significantly decreased by 11% at P63 (*p<0.05; n=8 CTL, 9 NIC). Data are shown as the mean ± SEM relative to saline-treated controls. Typical western blots for each treatment and age group are shown in band pairs above the bars (left band: control; right band: nicotine). LC= loading control (GAPDH for P1 or β-actin for P14 & P63), CTL=control, NIC=nicotine-treated, n= the number of pups used.
When the expression of NMDAR subunits was evaluated, the nicotine exposed hippocampus exhibited significant upward regulation at P1 by 38±9%, for NR1 (Fig. 4A,*p=0.03), 38±8% for NR2a (Fig. 4B, **p=0.01) and 34±12% for NR2d (Fig. 4E, *p=0.04), compared to controls. NR2c was not detected at P1 (Fig. 4D). There was no differential regulation of expression at P14 for any of the glutamate receptor subunits investigated (Fig. 4A, 4B, 4D, 4E). However, at P63, both NR1 and NR2c showed significant 18±6% (Fig. 4A, *p<0.05) and 31±9% (Fig. 4D, **p=0.01) respective downregulation by nicotine, a trend similar to GluR2 (Fig. 3B). NR2b levels did not fluctuate in the presence of nicotine at any age investigated (Fig. 4C), and NR2d was not detected in the adult (Fig. 4E). The low/undetectable expression of NR2c at P1 and that of NR2d at P63 is consistent with their expected developmental expression profiles (Monyer et al., 1994, Wenzel et al., 1996).
Figure 4. Prenatal nicotine exposure influences NMDAR subunits (NR1, NR2a, NR2b, NR2c, and NR2d) expression in developing rat hippocampus.

(A) Western blot analysis shows a significant nicotine-induced upregulation of NR1 at P1 (*p<0.05; n=6 CTL, 8 NIC), and no significant change at P14 (n=6 CTL, 8 NIC). However, at P63 the level of NR1 was significantly downregulated (*p<0.05; n=8 CTL, 9 NIC) whereas (B) NR2a expression was 38% increased from control (**p<0.01; n=7 CTL, 11 NIC) with no change seen at P14 (n=6 CTL, 6 NIC) or P63 (n=8 CTL, 9 NIC). (C) NR2b expression shows no significant regulation at any age in the presence of nicotine (n=6-8, CTL, 6-12 NIC) (D) NR2c expression was not detected at P1 (n=6 CTL, 9 NIC). Although NR2c shows no significant regulation at P14 (n=8 CTL 7 NIC), it was downregulated by 27% at P63 (**P<0.01; n=8 CTL, 9 NIC). (E) NR2d was upregulated at P1 (*P<0.05; n=6 CTL, 9 NIC) and was not changed at P14 (n=13 CTL, 13 NIC). It was not detected at P63 (n=5, CTL, 6 NIC). Values are the mean ± SEM relative to saline-treated controls. Typical western blots for each treatment and age group are shown in band pairs above the bars (left band: control; right band: nicotine). Abbreviation LC=loading control (GAPDH for P1 or β-actin for P14 & P63), CTL=control, NIC=nicotine-treated, n= the number of pups used for each bar.
Our data show a differential regulation of glutamate receptor subunits due to gestational nicotine exposure such that in P1 neonates, most receptor subunits were up-regulated whereas by the time they reach adulthood at P63 there was sustained down-regulation of NR1, NR2c and GluR2, but not other NMDAR or AMPAR subunits. These results suggest that nicotine may regulate the stoichiometric composition of both NMDARs and AMPARs during development to affect ligand binding and channel conductance throughout life.
Presynaptic and postsynaptic density proteins are modulated by prenatal nicotine exposure
To further understand the molecular changes occurring at the synapse, we next investigated SNAP25, a membrane t-SNARE protein involved in docking of synaptic vesicles at the terminal for exocytosis (Chen and Scheller, 2001). We reasoned that the observed nicotine-induced changes in glutamate receptor expression likely involve enhanced exocytosis of glutamate. Indeed, SNAP25 expression was significantly increased by 42±10% over saline-treated controls at P1 (Fig. 5A, **p<0.01) but not at P14 or P63 (Fig. 5A). Therefore, the increased exocytic events likely involved in glutamate release only occurred in neonatal animals which may still have circulating nicotine to influence presynaptic neurotransmitter release.
Figure 5. Age-related regulation of hippocampal presynaptic and postsynaptic markers (SNAP25, PSD95, CaM, and CaMKIIα) after prenatal nicotine exposure.

(A) Western blot analysis shows SNAP25 expression was significantly upregulated by 42% at P1 following prenatal nicotine exposure (**p<0.01, n=8 CTL, 8 NIC) but not at P14 (n=12 CTL, 11 NIC) or P63 (n=8 CTL, 9 NIC). (B) The level of PSD95 expression in the nicotine group was significantly increased by 29% at P1 (*p<0.05; n=6 CTL, 10 NIC) but there was no significant change at P14 (n=11 CTL, 11 NIC) or P63 (n=5 CTL, 6 NIC). (C) CaM expression was significantly upregulated at P1 (*p<0.05; n=11 CTL, 17 NIC) and downregulated at P14 (*p<0.05; n=12 CTL, 11 NIC), but remained normal at P63 (n=8 CTL, 9 NIC). (D) CaMKIIα expression was significantly upregulated at P1 (**p<0.01; n=8 CTL, 13 NIC), remained elevated at P14 (*p<0.05, n=10 CTL, 9 NIC), but showed no change relative to control at P63 (n=8 CTL, 9 NIC). Values are the mean ± SEM relative to saline-treated controls. Typical western blots for each treatment and age group are shown in band pairs above the bars (left band: control; right band: nicotine). Abbreviation LC=loading control (GAPDH for P1 or β-actin for P14 & P63). CTL=control, NIC=nicotine-treated, n= the number of pups used for each bar.
In order to be effective in transducing signals, glutamate receptors require functional proteins at the postsynaptic density. CaM and CaMKIIα are required for stabilization and maturation of dendritic arbors and to activate glutamate receptors via phosphorylation of specific sites in their subunits (Wu and Cline, 1998, Shen and Meyer, 1999), and PSD95 is required to stabilize numerous accessory proteins and AMPARs when not in the membrane (Kennedy, 1998). Therefore, determining if their functional status is affected in the presence of prenatal nicotine would be important in deciphering whether components of the postsynaptic density are competent for efficient signal transduction. Like SNAP25, PSD95 showed an upregulated expression of 29 ± 9% at P1 (Fig. 5B, *p<0.05) but no change at P14 or P63 (Fig. 5B) in nicotine-exposed hippocampi which suggests that at these later stages of development PSD95 may have stabilized and show no direct effect from the prenatal nicotine.
In order for CaMKIIα to become competent to phosphorylate other proteins, it must become active itself. This happens when Ca2+/CaM binds to CaMKIIα and induces a conformational change that allows autophosphorylation of the kinase (Fukunaga, 1993). In nicotine-exposed P1 animals, both CaM and CaMKIIα were increased at P1 by 22 ± 7% and 30 ± 7% respectively (Fig. 5C, 5D, *p<0.05 for CaM and **p<0.01 for CaMKIIα). However, at P14, there was an inverse regulation of expression where CaM levels were downregulated by 19 ± 6% (Fig. 5C, *p<0.05) and CaMKIIα was upregulated by 24 ± 3% (Fig. 5D, *p<0.05). At P63 neither CaM nor CaMKIIα showed any regulation by nicotine (Fig. 5C, 5D). These data show that at P1, a critical developmental point, SNAP25, PSD95, CaM, and CaMKIIα expression and recruitment were all enhanced by prenatal nicotine exposure. The heightened expression for CaMKIIα lasted through P14, whereas at this same period there was less CaM available for binding to its substrates. Thus even at times when glutamate receptors are seemingly not changed between saline or nicotine treated hippocampi, other signaling molecules associated with them may still be differentially affected to overactivate or compromise synaptic efficacy.
Functional AMPAR distribution is downregulated in prenatal nicotine exposed adults – comparison with normal adult binding of nAChRs
Next, we identified the relative spatial localization of nicotinic and glutamatergic receptors in the hippocampus, in order to better understand how nicotine might mediate glutamatergic synaptic activity. We employed receptor autoradiography with [3H]AMPA to label AMPARs, [3H]EB to label non-α7 nAChRs, and [125I]α-BTX to label α7 nAChR in normal adult rat hippocampus.
Our data show that [3H]AMPA binding was abundant throughout the hippocampus both in the cell body and dendrites of pyramidal neurons in field CA1 and CA3 and in granule neurons of the dentate gyrus (DG) (Fig. 6A, 6E). α7 nAChRs was minimally expressed in the granular cell layer of DG, as well as the stratum oriens (Or) and radiatum (Rad) of CA1, but highly abundant in those same synapse-rich apically oriented dendritic regions of CA3 (Fig. 6C, 6G). Therefore, the level of AMPAR binding closely corresponds with that of α7 nAChRs in Or & Rad layers in field CA2/CA3 in hippocampus (Fig. 6C, 6G), but not in CA1 where only AMPARs seem to be present at high density in most layers (Fig. 6A, 6E). By contrast, non-α7 nAChRs, most likely α4β2 nAChRs, are virtually absent in nearly all areas of the hippocampus and their distribution does not resemble that of either [3H]AMPA or [125I]α-BTX in the ventral lip of the molecular layer of DG in hippocampus where [3H]EB (Fig. 6B, 6F) and [3H]AMPA (Fig. 6A, 6E) binding are high. CA1 and CA3 localization of AMPARs is consistent with immunohistochemical distribution of GluR1 and GluR2 in the hippocampus where GluR1 is abundantly expressed in all neuronal layers of hippocampus including CA1/CA3/DG, as well as Or, Rad and molecular layers. GluR2 seems restricted to the neuronal cell bodies and proximal dendrites with less intense staining of distal dendritic arbors (data not shown).
Figure 6. Representative autoradiographic images of [3H]AMPA, [3H]EB, [125I]α-BTX binding to the dorsal hippocampus of a normal adult rat brain.
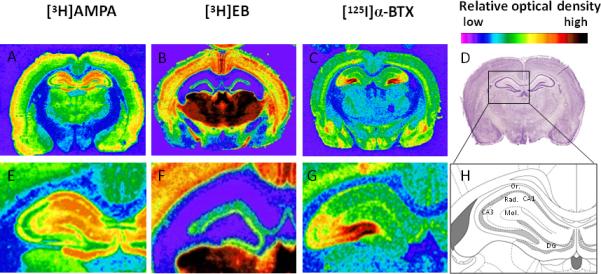
Receptor autoradiography of [3H]AMPA binding to determine the distribution of AMPAR (A, E), [3H]EB to label all high affinity nAChRs (B, F), and [125I]α-BTX to label low affinity α7 nAChRs (C,G). Nissl stained sections are shown at the same level of the hippocampus for comparison (D), as is a schematic line drawing (H) to distinguish the anatomical landmarks according to The Rat Brian in Stereotaxic Coordinates by George Paxinos and Charles Watson, 5th Ed, 2005. E, F, G, H are high magnification images of hippocampus regions from A, B, C, D. Color changes from light blue, purple, green, yellow, orange, red, to black represent low to high binding density.
Finally, to investigate whether functional AMPAR binding in the adult hippocampus can be affected by nicotine exposure temporally restricted to the prenatal period, we again used [3H]AMPA to label AMPARs by receptor autoradiography. Figure 7 shows a distinct and significant downregulation of [3H]AMPA binding of AMPARs throughout the hippocampus of prenatal nicotine-exposed animals (upper right panel), compared to saline-exposed controls (upper left panel), especially in CA1, CA3 and the dentate gyrus (Fig. 7). Because dorsal CA1, CA3, and DG subregions of the hippocampus are implicated in spatiotemporal processing of information, memory, and patterns, respectively (Hunsaker and Kesner, 2008), we speculate that prenatal nicotine may induce functional repression in these areas in an AMPAR-dependent manner, impairing the learning of new information.
Figure 7. Prenatal nicotine exposure continues to regulate [3H]AMPA binding to AMPAR in the young adult.
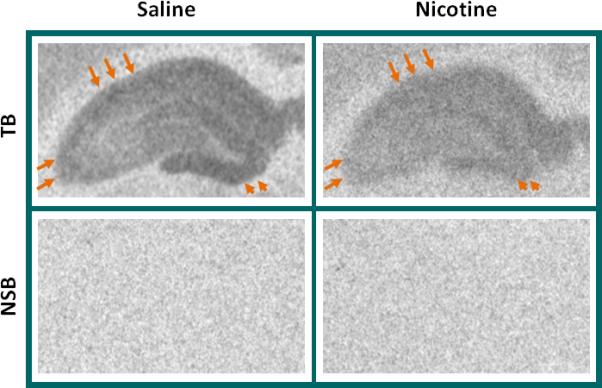
Rats were treated with saline (control) or 4mg/kg/day nicotine in utero and on P63, tissues were processed for autoradiography of [3H]AMPA binding, used to detect the distribution of functional AMPAR. A lower density of [3H]AMPA binding is evident throughout the hippocampus of nicotine-exposed pups (right) compared to those exposed to saline (left). Arrows point to CA1 (top) and CA3 (left) areas that show particularly high binding of [3H]AMPA in controls (left) but that were depressed after prenatal nicotine (right). Arrowheads indicate the dentate gyrus. Nonspecific binding (NSB) was assayed in adjacent sections and is shown as background directly below the total binding (TB) image for that animal. All images are from the same film.
Discussion
Here, we report the novel finding that exposure to chronic nicotine, even when limited to the gestational period between G7 to just before birth, can have profound effects on the hippocampal glutamatergic postsynaptic machinery in brains of young and adult offspring. There is an upregulation of glutamate receptor subunits and postsynaptic density proteins in early postnatal brains, followed by a sustained downregulation of NR1, NR2c and GluR2 subunit in adults. These findings are evidence that premature activation of nAChRs in the very plastic developing brain, during specification of neuronal architecture, induces a neuronal abnormality that alters expression of mediators of excitatory neurotransmission and synaptic plasticity necessary for encoding learning and memory in the hippocampus. We propose a dual mechanism whereby a sustained and continued exposure to circulating nicotine induces changes at an early age, which triggers an upregulation of synaptic components causing silent synapses to become prematurely active, and thus establishing synaptic contacts earlier than normal; however, this heightened activation is not sustained and leads to a compensatory decrease in activity of some synaptic components in more mature neurons that lasts into adulthood.
Cholinergic Projections to Hippocampus
In developing rats, cholinergic projections from the medial septum and lateral band of Broca onto prospective CA3 and CA1 pyramidal neurons are detected in CA1 and CA3 regions by embryonic day 20, concurrent with the birth and development of principle cells of the hippocampus proper (Linke and Frotscher, 1993) although synaptogenesis and maturation occurs throughout the first prenatal week and reaches an adult pattern by P10 of the second postnatal week (Rami et al., 1989, Linke and Frotscher, 1993, Nyakas et al., 1994). Comparatively, cholinergic projections to the granule-cell rich DG first reach their target by P2-P3. Late embryonic/early postnatal stages are thus critical periods for establishing the neuroarchitecture of the hippocampus. Neuronal precursors are competent to receive cholinergic stimulation as early as E15 because high affinity nAChR subunits, α4 and β2 are already expressed in the hippocampal mantle at that age, while α7 mRNA is present by E13 in the hippocampus (Adams et al., 2002). Thus, exposure of acetylcholine-responsive neurons during gestation to chronic nicotine, the main ingredient in tobacco and a strong cholinergic agonist, can perturb the developmental programme of neurons. In the absence of nicotine stimulation, nAChRs normally exhibit heightened expression during the first two weeks of postnatal development (Fig. 1) (Shacka and Robinson, 1998), consistent with an important role for cholinergic stimulation during neuronal maturation. Our results indicate that exposure to nicotine, which binds nAChRs with the same or higher affinity as the endogenous ligand acetylcholine, induces heightened expression of primarily α4β2 receptors (Fig. 1). We cannot exclude the possibility that other nAChRs such as the α3-containing receptors which peak at E13 in hippocampus (Zoli et al., 1995) may have been regulated during early embryonic periods, and this could still have consequences at various postnatal stages. We also cannot exclude a role for α5-containing nAChR (Winzer-Serhan and Leslie, 2005) or α7 homomeric receptor involvement during the first two weeks of development. Nevertheless, our results are strong evidence that nAChRs may act in concert with AMPARs, NMDARs, to coordinate the development of hippocampal neuronal connections.
Ionotropic Glutamate receptors in Development
Nascent hippocampal synapses consist primarily of silent glutamatergic synapses, i.e., they are not functional at rest until AMPARs are inserted into postsynaptic sites (Liao et al., 1995, Durand et al., 1996, Hanse et al., 2009). Upon nicotine administration, silent synapses of CA1 neurons can be induced to become functional at P1-P5, as demonstrated by the presence of miniature excitatory postsynaptic currents (mEPSCs), and this exposure also persistently enhances synaptic efficacy (Maggi et al., 2003). These actions of nicotine occur presumably by activating both presynaptic and postsynaptic mechanisms (Zhang and Berg, 2007). Consistent with this finding, our results show that at P1 prenatal nicotine indeed enhanced expression of the presynaptically localized SNARE protein, SNAP-25, indicative of increased glutamate vesicular docking and release. There was also a nicotine-induced differential regulation of expression of all postsynaptic proteins investigated, inclusive of AMPAR subunits (GluR1 and GluR2), NMDAR subunits (NR1, NR2a, and NR2d), and downstream postsynaptic regulatory proteins (CaM, CaMKIIα, and PSD95). β-Actin, normally used as a loading control for comparison with other proteins, was also significantly upregulated at P1 (Fig. 2A), possibly in response to the need for cytoskeletal rearrangements in neurons due to nicotine exposure. Although β-actin is widely used as a loading control, its expression levels can change during perturbation of development or in disease systems as demonstrated in this report and elsewhere (Dittmer and Dittmer, 2006, Aldridge et al., 2008). The massive upregulation of synaptic components in conjunction with the cytoskeletal protein, β-actin, is likely to contribute to enhanced synaptic efficacy but could also contribute to glutamate-induced excitotoxicity which could cause induction of compensatory rectifying mechanisms. Indeed, this effect may be directly modulated by the presence of recirculating nicotine since our data shows upregulation of α4β2 nAChRs during early postnatal ages. Thus, nicotine enhancement of nAChRs expression and the parallel amplification of the glutamatergic synaptic machinery occur at a time when neurons are normally sending out their projections to interact with their targets and extending their dendrites to form the hippocampal matrix and thus are most sensitive, or more vulnerable.
Nicotine's Effect on Mature(ing) Brain
Recordings of mEPSCs from CA1 neurons in adult hippocampal slices showed marked reduction in the amplitude and frequency of AMPAR-mediated EPSCs after nicotine treatment, indicating that there may be selective loss or downregulation of AMPARs as a whole at those synapses between P60 to P70 (Vaglenova et al., 2008). Potential excitotoxicity due to specific regulation of calcium impermeable GluR2 containing AMPAR-mediated signaling has been reported in other models of brain insult, including ischemia (Gorter et al., 1997, Liu et al., 2006) and traumatic brain injury (Spaethling et al., 2008). In both cases, GluR2 can be downregulated in favor of the calcium-permeable AMPAR subunits made up of GluR1. Further, decreased expression of the GluR2 subunit component of AMPAR in developing brain, can confer susceptibility of the young, immature hippocampus to epileptic seizures (Sanchez et al., 2001). Likewise, the downregulation of NR1 subunit will decrease the number of NMDARs available for synaptic transmission in the mature brain. Indeed we find that NR1 and NR2c were respectively downregulated by 18% and 31% in P63 animals (Fig. 4A, 4D), and GluR2 levels, which is a major component of the heteromeric AMPAR tetramers (Fig. 3B), were diminished by 11%, p<0.05. Our data strongly suggest a diminished function of synapses in adult brain either through fewer overall synaptic contacts or by reducing the number of NMDAR-mediated AMPAR incorporation into membranes. Further, if the GluR2 subunit is diminished, then AMPARs may be primarily made up of GluR1 homomers which may further contribute to excitotoxicity (Sanchez et al., 2001, Spaethling et al., 2008) or altered signaling. This is because during normal development of pyramidal neurons, there is a developmental switch that occurs from calcium permeable GluR2-lacking AMPARs in immature dendrites to calcium impermeable GluR2-containing AMPARs, dominant for efficient neurotransmission between glutamatergic neurons and neuroprotection throughout the nervous system (Kumar et al., 2002, Ho et al., 2007, Brill and Huguenard, 2008).
In our studies, the long-term changes in functional protein expression observed in the adult may have begun as early as P14 when CaMKIIα expression was enhanced and CaM expression was repressed in prenatal nicotine-exposed animals (Fig. 5D and 5C). CaMKIIα has been extensively studied for its role in stabilization and maturation of dendritic spines during neuronal differentiation and synaptic plasticity (Pettit et al., 1994, Griffith et al., 2003). CaMKIIα requires cooperative Ca2+/CaM binding to autophosphorylate and become competent to phosphorylate its substrates, including the AMPAR and their accessory proteins. CaM is a multifunctional protein with many binding partners in addition to the CaMK family of cytoplasmic or nuclear proteins, which include phosphatases, protein kinase C, and transcription factors. The downregulation of CaM at P14 could also be indicative of an effect of nicotine on gene transcription or abnormal function of its other substrates such that the metabolic balance necessary for efficient synaptic responses extends beyond a specific effect on CaMKIIα only. Moreover, the early regulation of these downstream postsynaptic proteins may be influential in GluR2, NR1 and NR2c expression, assembly and conductivity later in development and in adults. Further studies need to be conducted to correlate these changes. Although overall levels of a number of iGluR subunits at P14 and P63 seem similar to control (figures 3 and 4), it is improbable that this is due to a simple “normalization” of receptor expression levels in nicotine-treated brains. Rather, our data and ongoing research support possible alterations in distribution and signaling of these proteins at the cellular and tissue levels.
Finally, we compared the spatial distribution of AMPARs using [3H]AMPA, with that of the α7 and α4β2 containing nAChRs in adult hippocampus using [125I]α-BTX and [3H]EB respectively. A direct comparison of the distribution of AMPARs and nAChRs in the adult suggests a significant presence of both AMPARs and α7 nAChRs in dendrites of non-pyramidal granular cells and interneurons of the stratum radiatum (Fig. 6A, 6E, 6C, and 6G). All three radioligand binding sites were moderately expressed in the lacunosum molecular layer (Fig. 6). These data reveal a coincident distribution of the α7 nAChRs and AMPAR subtypes (Li et al., 2004) and may partially explain the nicotine-mediated mechanistic convergence of regulatory effects on AMPARs and nAChRs in the hippocampus. α7 nAChRs are primarily responsible for presynaptic modulation of NMDARs and glutamate release (Lin et al., 2010). Moreover, receptor binding for [3H]AMPA was qualitatively reduced throughout the hippocampus in young adults at P63 (Fig. 7). This is in strong agreement with P63 data presented in figures 3 and 4 which show a downregulation of protein levels of both AMPAR subunit GluR2 and NMDAR subunits NR1 and NR2c. The overall depression of functional AMPAR in nicotine-exposed P63 brains, as in Fig. 7, can be attributed to downregulation of GluR2 subunit expression; however, it is possible that downregulation of NR1 and NR2c are also responsible for the observed AMPAR downregulation in hippocampus. NMDAR function and activation is required for the insertion of AMPAR into the membrane during long-term potentiation (LTP), therefore the decreased availability of NMDAR can also affect AMPAR assembly and signaling. In NMDAR-dependent long-term potentiation (LTP) of synaptic inputs in adults, NMDAR activation allows calcium influx to the postsynaptic density, which leads CaMKIIα to become competent to phosphorylate AMPAR which allows enhanced depolarization of the postsynaptic cell (Malenka and Bear, 2004). Thus the activity of AMPAR is directly correlated with the functional activation of NMDAR. It cannot be ruled out that the effects observed at P63 may be related to LTD, the long term depression of synaptic inputs. LTD is characterized by a functional loss of AMPARs at the synaptic membrane, causing a more muted response to stimulation (Malinow and Malenka, 2002, Malenka and Bear, 2004). These theories can be tested further with electrophysiological paradigms.
Conclusions
The data presented here constitute the first longitudinal developmental study where iGluR subunits from AMPARs and NMDARs, together with postsynaptic components necessary for synaptic plasticity and memory formation have been investigated in a prenatal nicotine model. Most iGluRs investigated were upregulated at P1, their levels returned to normal at P14, but levels of CaMKIIα and CaM, responsible for iGluR phosphorylation and activation, remained inversely regulated at P14 such that CaMKIIα levels were increased and CaM levels showed a decrease. Interestingly, in brains of P63 animals, NR1, NR2c and GluR2 expression levels were significantly downregulated (summarized in table 1). This extensive analysis of the regulation of nAChR, AMPAR and NMDAR receptors by gestational nicotine will help guide future studies on the mechanisms involved in nicotine modulation of glutamatergic signaling.
Table 1.
Summary of nicotine-induced regulation of various hippocampal synaptic proteins at three different postnatal ages after prenatal nicotine exposure.
| Protein | P1 | P14 | P63 | Protein | P1 | P14 | P63 |
|---|---|---|---|---|---|---|---|
| B-actin | ↑ ** | - | - | GluR1 | ↑ *** | - | - |
| GAPDH | - | - | - | GluR2 | - | - | ↓ * |
| α-tubulin | - | - | - | NR1 | ↑ * | - | ↓ * |
| SNAP25 | ↑ ** | - | - | NR2a | ↑ ** | - | - |
| PSD95 | ↑ * | - | - | NR2b | - | - | - |
| CaM | ↑ * | ↓ * | - | NR2c | --- | - | ↓ ** |
| CaMKHa | ↑ ** | ↑ * | - | NR2d | ↑ * | - | --- |
(↑) up-regulation, (↓) down-regulation, (-) no statistically significant change, (---) undetected
p<0.001
p<0.01
p<0.05 compared to control.
Pediatric neurologists and behaviorists have reported attention deficits, low IQs and delayed development in reading and writing skills in susceptible children who were chronically exposed to nicotine during gestation (Eskenazi and Castorina, 1999, Herrmann et al., 2008). Developmental events occurring in a cortical or hippocampal neuron of a human fetus during the 3rd trimester has been equated with those occurring in the rat brain during early postnatal development (Dobbing, 1971, Quinn, 2005). In normal rat P1 neonates, pyramidal neurons are born and are extending their first dendritic processes. nAChRs are significantly expressed soon after, at P4 (Zoli et al., 1995). By P14, dendritic arborization is largely complete, synaptic targets have been reached and synaptogenesis is robust with the majority of synapses having been formed. At P63, in early adulthood, neuronal architecture and connections are established and connections have stabilized and achieved their mature form (Zancanaro et al., 2001). We hypothesize that the malleable prenatal period and the early postnatal period, from birth to P14, represent critical times when prenatal nicotine exposure could perturb synaptic gene expression and protein localization and function to effect long-term regulation of glutamatergic synaptic transmission.
Acknowledgments
This work was supported by NIH grant # NS065385, ARRA funding, and a Howard University intramural SEED Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the alpha7 nicotinic cholinergic receptor in rat hippocampal formation. Brain research Developmental brain research. 2002;139:175–187. doi: 10.1016/s0165-3806(02)00547-3. [DOI] [PubMed] [Google Scholar]
- Ajarem JS, Ahmad M. Prenatal nicotine exposure modifies behavior of mice through early development. Pharmacology, biochemistry, and behavior. 1998;59:313–318. doi: 10.1016/s0091-3057(97)00408-5. [DOI] [PubMed] [Google Scholar]
- Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods. 2008;172:250–254. doi: 10.1016/j.jneumeth.2008.05.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Progress in neurobiology. 2002;68:209–245. doi: 10.1016/s0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brill J, Huguenard JR. Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J Neurosci. 2008;28:13918–13928. doi: 10.1523/JNEUROSCI.3229-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr LA, Rowell PP, Pierce WM., Jr. Effects of subchronic nicotine administration on central dopaminergic mechanisms in the rat. Neurochemical research. 1989;14:511–515. doi: 10.1007/BF00964911. [DOI] [PubMed] [Google Scholar]
- Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol. 2001;2:98–106. doi: 10.1038/35052017. [DOI] [PubMed] [Google Scholar]
- Chistyakov V, Patkina N, Tammimaki A, Talka R, Salminen O, Belozertseva I, Galankin T, Tuominen R, Zvartau E. Nicotine exposure throughout early development promotes nicotine self-administration in adolescent mice and induces long-lasting behavioural changes. Eur J Pharmacol. 2010;640:87–93. doi: 10.1016/j.ejphar.2010.04.044. [DOI] [PubMed] [Google Scholar]
- Corera AT, Doucet G, Fon EA. Long-term potentiation in isolated dendritic spines. PLoS One. 2009;4:e6021. doi: 10.1371/journal.pone.0006021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell JR, Dube GR, Egles C, Liu G. Distribution, density, and clustering of functional glutamate receptors before and after synaptogenesis in hippocampal neurons. Journal of neurophysiology. 2000;84:1573–1587. doi: 10.1152/jn.2000.84.3.1573. [DOI] [PubMed] [Google Scholar]
- Davila-Garcia MI, Musachio JL, Kellar KJ. Chronic nicotine administration does not increase nicotinic receptors labeled by [125I]epibatidine in adrenal gland, superior cervical ganglia, pineal or retina. J Neurochem. 2003;85:1237–1246. doi: 10.1046/j.1471-4159.2003.01774.x. [DOI] [PubMed] [Google Scholar]
- Dittmer A, Dittmer J. Beta-actin is not a reliable loading control in Western blot analysis. Electrophoresis. 2006;27:2844–2845. doi: 10.1002/elps.200500785. [DOI] [PubMed] [Google Scholar]
- Dobbing J. Undernutrition and the developing brain: the use of animal models ot elucidate the human problem. Psychiatr Neurol Neurochir. 1971;74:433–442. [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Eddins D, Petro A, Williams P, Cerutti DT, Levin ED. Nicotine effects on learning in zebrafish: the role of dopaminergic systems. Psychopharmacology. 2009;202:103–109. doi: 10.1007/s00213-008-1287-4. [DOI] [PubMed] [Google Scholar]
- Ekonomou A, Smith AL, Angelatou F. Changes in AMPA receptor binding and subunit messenger RNA expression in hippocampus and cortex in the pentylenetetrazole-induced ‘kindling’ model of epilepsy. Brain research Molecular brain research. 2001;95:27–35. doi: 10.1016/s0169-328x(01)00230-3. [DOI] [PubMed] [Google Scholar]
- Ernst M, Moolchan ET, Robinson ML. Behavioral and neural consequences of prenatal exposure to nicotine. J Am Acad Child Adolesc Psychiatry. 2001;40:630–641. doi: 10.1097/00004583-200106000-00007. [DOI] [PubMed] [Google Scholar]
- Eskenazi B, Castorina R. Association of prenatal maternal or postnatal child environmental tobacco smoke exposure and neurodevelopmental and behavioral problems in children. Environ Health Perspect. 1999;107:991–1000. doi: 10.1289/ehp.99107991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MF, Fior-Chadi DR. Chronic nicotine administration. Analysis of the development of hypertension and glutamatergic neurotransmission. Brain research bulletin. 2007;72:215–224. doi: 10.1016/j.brainresbull.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Leranth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. The Journal of comparative neurology. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Fukunaga K. [The role of Ca2+/calmodulin-dependent protein kinase II in the cellular signal transduction]. Nippon Yakurigaku Zasshi. 1993;102:355–369. doi: 10.1254/fpj.102.355. [DOI] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48:774–782. [PubMed] [Google Scholar]
- Gingras JL, O'Donnell KJ. State control in the substance-exposed fetus. I. The fetal neurobehavioral profile: an assessment of fetal state, arousal, and regulation competency. Annals of the New York Academy of Sciences. 1998;846:262–276. [PubMed] [Google Scholar]
- Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MV, Connor JA, Zukin RS. Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith LC, Lu CS, Sun XX. CaMKII, an enzyme on the move: regulation of temporospatial localization. Mol Interv. 2003;3:386–403. doi: 10.1124/mi.3.7.386. [DOI] [PubMed] [Google Scholar]
- Hanley JG. Endosomal sorting of AMPA receptors in hippocampal neurons. Biochemical Society transactions. 2010;38:460–465. doi: 10.1042/BST0380460. [DOI] [PubMed] [Google Scholar]
- Hanse E, Taira T, Lauri S, Groc L. Glutamate synapse in developing brain: an integrative perspective beyond the silent state. Trends Neurosci. 2009;32:532–537. doi: 10.1016/j.tins.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Herrmann M, King K, Weitzman M. Prenatal tobacco smoke and postnatal secondhand smoke exposure and child neurodevelopment. Curr Opin Pediatr. 2008;20:184–190. doi: 10.1097/MOP.0b013e3282f56165. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann CF, Berger-Sweeney J. Cholinergic regulation of cortical development and plasticity. New twists to an old story. Perspect Dev Neurobiol. 1998;5:401–425. [PubMed] [Google Scholar]
- Houghtling RA, Davila-Garcia MI, Kellar KJ. Characterization of (+/-)(-)[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Molecular pharmacology. 1995;48:280–287. [PubMed] [Google Scholar]
- Hunsaker MR, Kesner RP. Evaluating the differential roles of the dorsal dentate gyrus, dorsal CA3, and dorsal CA1 during a temporal ordering for spatial locations task. Hippocampus. 2008;18:955–964. doi: 10.1002/hipo.20455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB. Signal transduction molecules at the glutamatergic postsynaptic membrane. Brain Res Brain Res Rev. 1998;26:243–257. doi: 10.1016/s0165-0173(97)00043-x. [DOI] [PubMed] [Google Scholar]
- King JA, Davila-Garcia M, Azmitia EC, Strand FL. Differential effects of prenatal and postnatal ACTH or nicotine exposure on 5-HT high affinity uptake in the neonatal rat brain. Int J Dev Neurosci. 1991;9:281–286. doi: 10.1016/0736-5748(91)90048-q. [DOI] [PubMed] [Google Scholar]
- Koh DS, Geiger JR, Jonas P, Sakmann B. Ca(2+)-permeable AMPA and NMDA receptor channels in basket cells of rat hippocampal dentate gyrus. The Journal of physiology 485. 1995;(Pt 2):383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–446. doi: 10.1007/s00441-006-0273-6. [DOI] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, Huguenard JR. A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J Neurosci. 2002;22:3005–3015. doi: 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC, Soffe SR, Roberts A. Glutamate and acetylcholine corelease at developing synapses. Proc Natl Acad Sci U S A. 2004;101:15488–15493. doi: 10.1073/pnas.0404864101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lin H, Vicini S, Hsu FC, Doshi S, Takano H, Coulter DA, Lynch DR. Axonal alpha7 nicotinic ACh receptors modulate presynaptic NMDA receptor expression and structural plasticity of glutamatergic presynaptic boutons. Proc Natl Acad Sci U S A. 2010;107:16661–16666. doi: 10.1073/pnas.1007397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke R, Frotscher M. Development of the rat septohippocampal projection: tracing with DiI and electron microscopy of identified growth cones. The Journal of comparative neurology. 1993;332:69–88. doi: 10.1002/cne.903320106. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6). Neuroscience. 1983;10:1185–1201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Mineur YS, Somenzi O, Picciotto MR. Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology. 2007;52:1256–1262. doi: 10.1016/j.neuropharm.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Muneoka K, Nakatsu T, Fuji J, Ogawa T, Takigawa M. Prenatal administration of nicotine results in dopaminergic alterations in the neocortex. Neurotoxicology and teratology. 1999;21:603–609. doi: 10.1016/s0892-0362(99)00028-8. [DOI] [PubMed] [Google Scholar]
- Muneoka K, Ogawa T, Kamei K, Muraoka S, Tomiyoshi R, Mimura Y, Kato H, Suzuki MR, Takigawa M. Prenatal nicotine exposure affects the development of the central serotonergic system as well as the dopaminergic system in rat offspring: involvement of route of drug administrations. Brain research Developmental brain research. 1997;102:117–126. doi: 10.1016/s0165-3806(97)00092-8. [DOI] [PubMed] [Google Scholar]
- Murrin LC, Ferrer JR, Zeng WY, Haley NJ. Nicotine administration to rats: methodological considerations. Life sciences. 1987;40:1699–1708. doi: 10.1016/0024-3205(87)90020-8. [DOI] [PubMed] [Google Scholar]
- Nyakas C, Buwalda B, Kramers RJ, Traber J, Luiten PG. Postnatal development of hippocampal and neocortical cholinergic and serotonergic innervation in rat: effects of nitrite-induced prenatal hypoxia and nimodipine treatment. Neuroscience. 1994;59:541–559. doi: 10.1016/0306-4522(94)90176-7. [DOI] [PubMed] [Google Scholar]
- Pandis C, Sotiriou E, Kouvaras E, Asprodini E, Papatheodoropoulos C, Angelatou F. Differential expression of NMDA and AMPA receptor subunits in rat dorsal and ventral hippocampus. Neuroscience. 2006;140:163–175. doi: 10.1016/j.neuroscience.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Papke RL, Heinemann SF. Partial agonist properties of cytisine on neuronal nicotinic receptors containing the beta 2 subunit. Mol Pharmacol. 1994;45:142–149. [PubMed] [Google Scholar]
- Papke RL, Porter Papke JK. Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol. 2002;137:49–61. doi: 10.1038/sj.bjp.0704833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JJ, Gondre-Lewis MC, Eiden LE, Loh YP. A distinct trans-Golgi network subcompartment for sorting of synaptic and granule proteins in neurons and neuroendocrine cells. J Cell Sci. 2011;124:735–744. doi: 10.1242/jcs.076372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Davila-Garcia MI, Kellar KJ. Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82:468–481. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Perlman S, Malinow R. Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science. 1994;266:1881–1885. doi: 10.1126/science.7997883. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, Le Novere N, Vincent P, Pich EM, Brulet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Quinn R. Comparing rat's to human's age: how old is my rat in people years? Nutrition. 2005;21:775–777. doi: 10.1016/j.nut.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Rami A, Rabie A, Clos J. The time course of hippocampal cholinergic innervation in the developing hypothyroid rat. A combined histochemical and biochemical study of acetylcholinesterase activity. Int J Dev Neurosci. 1989;7:301–308. doi: 10.1016/0736-5748(89)90035-x. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Unis AS, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Brain research Developmental brain research. 2001;127:123–133. doi: 10.1016/s0165-3806(01)00126-2. [DOI] [PubMed] [Google Scholar]
- Sanchez RM, Koh S, Rio C, Wang C, Lamperti ED, Sharma D, Corfas G, Jensen FE. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J Neurosci. 2001;21:8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Robinson SE. Postnatal developmental regulation of neuronal nicotinic receptor subunit alpha 7 and multiple alpha 4 and beta 2 mRNA species in the rat. Brain research Developmental brain research. 1998;109:67–75. doi: 10.1016/s0165-3806(98)00058-3. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Spaethling JM, Klein DM, Singh P, Meaney DF. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J Neurotrauma. 2008;25:1207–1216. doi: 10.1089/neu.2008.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Piattoni-Kaplan M, Campbell JE, McKenna DG, Molinari E, Hettinger AM, Garvey DS, Wasicak JT, Holladay MW, Williams M, Arneric SP. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity alpha 4 beta 2 nicotinic acetylcholine receptor ligand. Neuropharmacology. 1996;35:725–734. doi: 10.1016/0028-3908(96)84644-2. [DOI] [PubMed] [Google Scholar]
- Tizabi Y, Russell LT, Nespor SM, Perry DC, Grunberg NE. Prenatal nicotine exposure: effects on locomotor activity and central [125I]alpha-BT binding in rats. Pharmacology, biochemistry, and behavior. 2000;66:495–500. doi: 10.1016/s0091-3057(00)00171-4. [DOI] [PubMed] [Google Scholar]
- Vaglenova J, Parameshwaran K, Suppiramaniam V, Breese CR, Pandiella N, Birru S. Long-lasting teratogenic effects of nicotine on cognition: gender specificity and role of AMPA receptor function. Neurobiology of learning and memory. 2008;90:527–536. doi: 10.1016/j.nlm.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Villa M, Mohler H, Benke D. Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain. J Neurochem. 1996;66:1240–1248. doi: 10.1046/j.1471-4159.1996.66031240.x. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol. 2005;481:19–30. doi: 10.1002/cne.20357. [DOI] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Xu Z, Seidler FJ, Ali SF, Slikker W., Jr. Slotkin TA (Fetal and adolescent nicotine administration: effects on CNS serotonergic systems. Brain research. 2001;914:166–178. doi: 10.1016/s0006-8993(01)02797-4. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Oka H. Topographical projections from the medial septum-diagonal band complex to the hippocampus: a retrograde tracing study with multiple fluorescent dyes in rats. Neuroscience research. 1995;21:199–209. doi: 10.1016/0168-0102(94)00852-7. [DOI] [PubMed] [Google Scholar]
- Zancanaro C, Bolner A, Righetti C. NMR spectroscopic analysis of rat brain development: in vitro proton and carbon studies of whole tissue and its phospholipid fraction. Dev Neurosci. 2001;23:107–112. doi: 10.1159/000048702. [DOI] [PubMed] [Google Scholar]
- Zhang J, Berg DK. Reversible inhibition of GABAA receptors by alpha7-containing nicotinic receptors on the vertebrate postsynaptic neurons. The Journal of physiology. 2007;579:753–763. doi: 10.1113/jphysiol.2006.124578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Le Novere N, Hill JA, Jr., Changeux JP. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J Neurosci. 1995;15:1912–1939. doi: 10.1523/JNEUROSCI.15-03-01912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]